New Insights on Genetic Diagnostics in Cardiomyopathy and Arrhythmia Patients Gained by Stepwise Exome Data Analysis

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Evaluation
2.2. Exome Sequencing
2.3. Bioinformatics and Workflow of Genetic Data Analysis
3. Results
3.1. Patient Characteristics
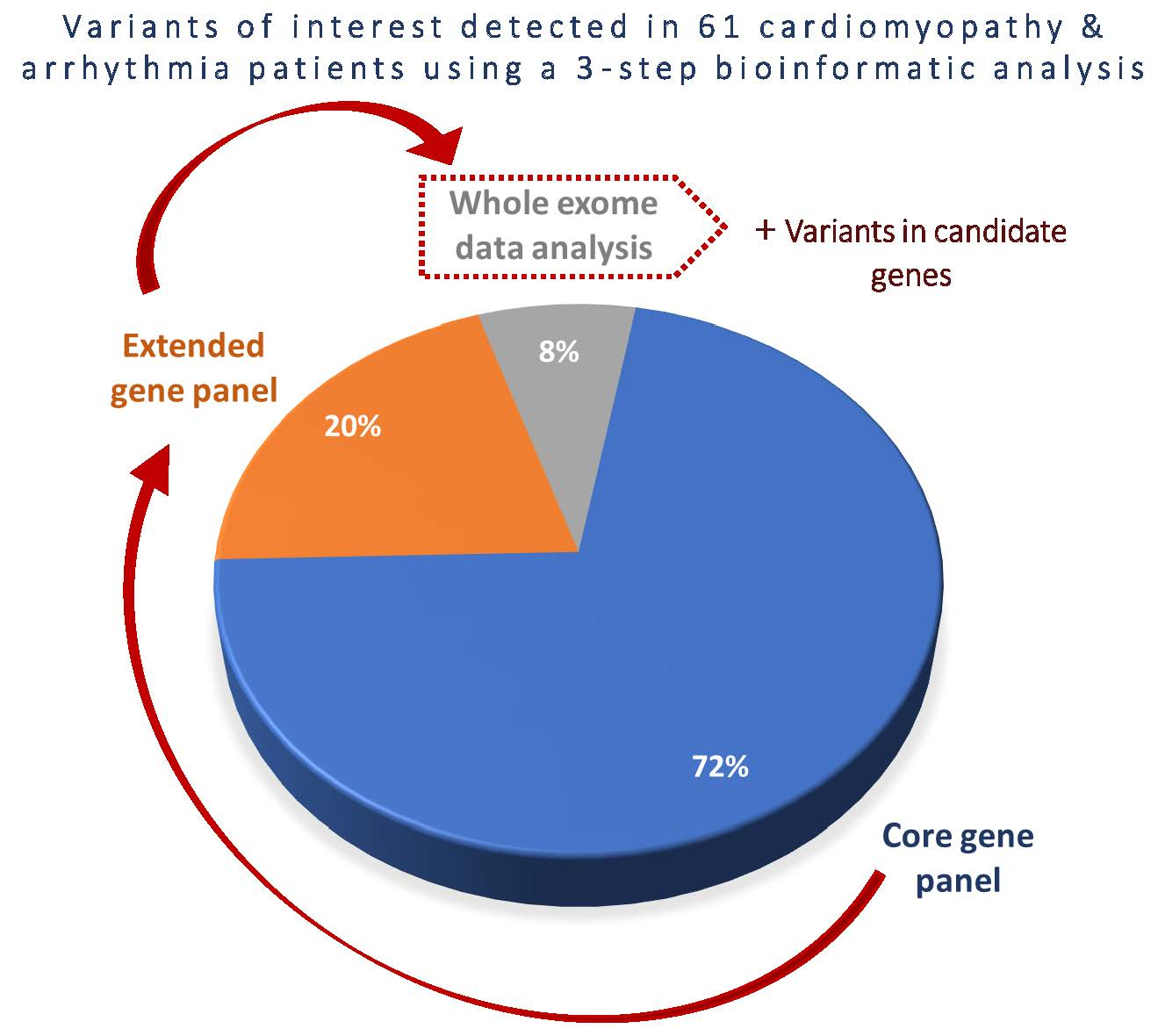
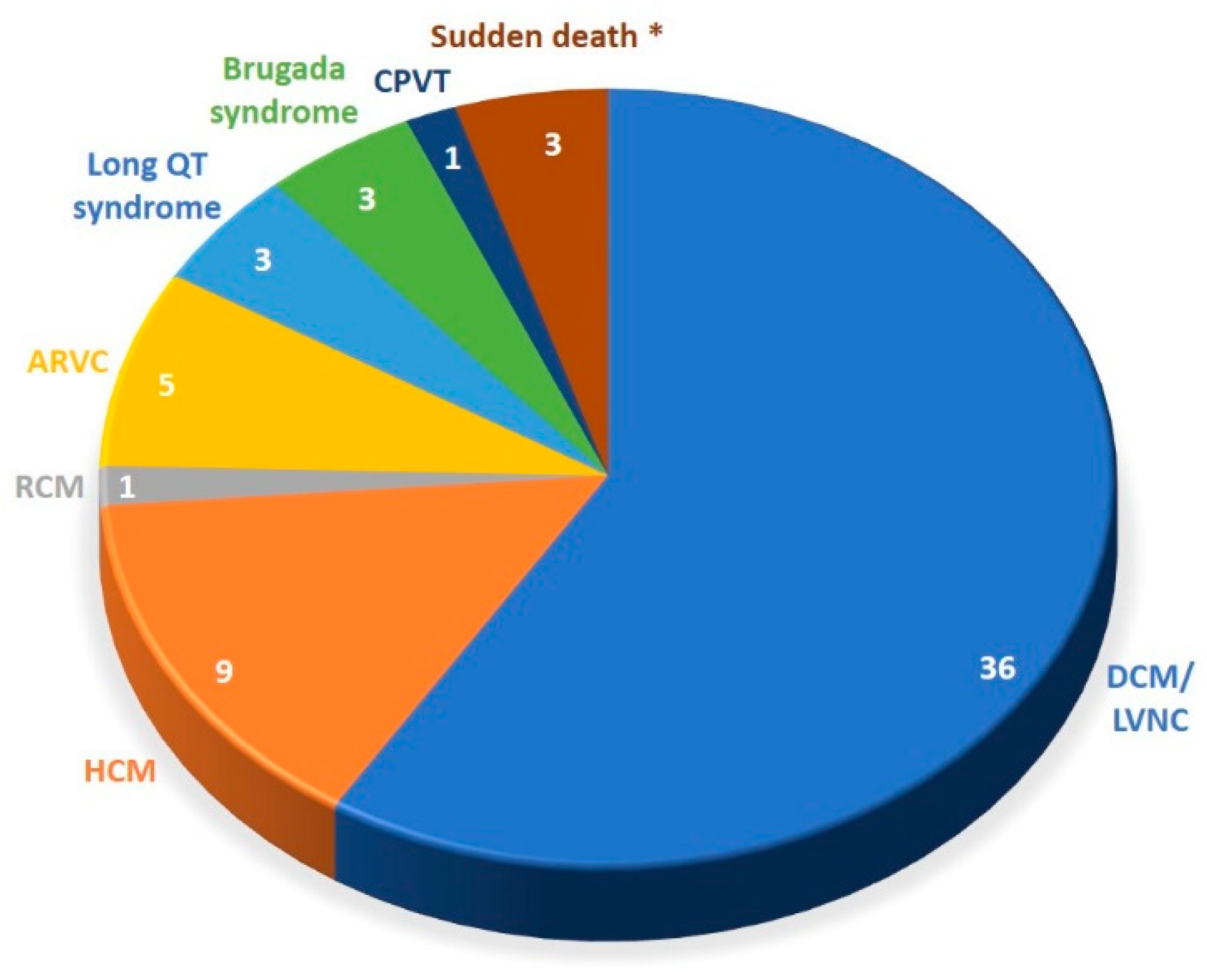
3.2. Detection Rates of Variants of Interest and Spectrum of Incolved Genes
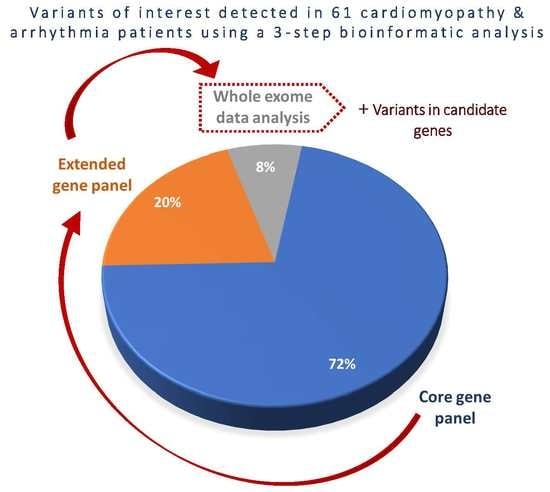
3.3. Diagnostic Yield of Core Gene Panel, Extended Gene Panel and Exome Analysis.
3.4. Novel Variants in Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef]
- Jacoby, D.; McKenna, W.J. Genetics of inherited cardiomyopathy. Eur Heart J. 2012, 33, 296–304. [Google Scholar] [CrossRef]
- Nair, N.U.; Das, A.; Amit, U.; Robinson, W.; Park, S.G.; Basu, M.; Lugo, A.; Leor, J.; Ruppin, E.; Hannenhalli, S. Putative functional genes in idiopathic dilated cardiomyopathy. Sci Rep. 2018, 8, 66. [Google Scholar] [CrossRef]
- Mak, T.S.H.; Lee, Y.K.; Tang, C.S.; Hai, J.S.H.; Ran, X.; Sham, P.C.; Tse, H.F. Coverage and diagnostic yield of Whole Exome Sequencing for the Evaluation of Cases with Dilated and Hypertrophic Cardiomyopathy. Sci. Rep. 2018, 8, 10846. [Google Scholar] [CrossRef]
- Cirino, A.L.; Lakdawala, N.K.; McDonough, B.; Conner, L.; Adler, D.; Weinfeld, M.; O’Gara, P.; Rehm, H.L.; Machini, K.; Lebo, M.; et al. A Comparison of Whole Genome Sequencing to Multigene Panel Testing in Hypertrophic Cardiomyopathy Patients. Circ. Cardiovasc. Genet. 2017, 10, e001768. [Google Scholar] [CrossRef]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, S.; et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270ra6. [Google Scholar] [CrossRef]
- van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.; et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef]
- Elliott, P.; O’Mahony, C.; Syrris, P.; Evans, A.; Rivera Sorensen, C.; Sheppard, M.N.; Carr-White, G.; Pantazis, A.; McKenna, W.J. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 314–322. [Google Scholar] [CrossRef]
- Hartmannova, H.; Kubanek, M.; Sramko, M.; Piherova, L.; Noskova, L.; Hodanova, K.; Stranecky, V.; Pristoupilova, A.; Sovova, J.; Marek, T.; et al. Isolated X-linked hypertrophic cardiomyopathy caused by a novel mutation of the four-and-a-half LIM domain 1 gene. Circ. Cardiovasc. Genet. 2013, 6, 543–551. [Google Scholar] [CrossRef][Green Version]
- Kolokotronis, K.; Kuhnisch, J.; Klopocki, E.; Dartsch, J.; Rost, S.; Huculak, C.; Mearini, G.; Stork, S.; Carrier, L.; Klaassen, S.; et al. Biallelic mutation in MYH7 and MYBPC3 leads to severe cardiomyopathy with left ventricular noncompaction phenotype. Hum. Mutat. 2019, 40, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Jokela, M.; Baumann, P.; Huovinen, S.; Penttila, S.; Udd, B. Homozygous Nonsense Mutation p.Q274X in TRIM63 (MuRF1) in a Patient with Mild Skeletal Myopathy and Cardiac Hypertrophy. J. Neuromuscul. Dis. 2019, 6, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Abdul-Hussein, S.; Oldfors, A.; Gonzalez-Costello, J.; van der Ven, P.F.; Furst, D.O.; Gonzalez, L.; Moreno, D.; Torrejon-Escribano, B.; Alio, J.; et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 2015, 24, 3638–3650. [Google Scholar] [CrossRef]
- Luxan, G.; Casanova, J.C.; Martinez-Poveda, B.; Prados, B.; D’Amato, G.; MacGrogan, D.; Gonzalez-Rajal, A.; Dobarro, D.; Torroja, C.; Martinez, F.; et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 2013, 19, 193–201. [Google Scholar] [CrossRef]
- Tobita, T.; Nomura, S.; Morita, H.; Ko, T.; Fujita, T.; Toko, H.; Uto, K.; Hagiwara, N.; Aburatani, H.; Komuro, I. Identification of MYLK3 mutations in familial dilated cardiomyopathy. Sci. Rep. 2017, 7, 17495. [Google Scholar] [CrossRef]
- Poon, K.L.; Tan, K.T.; Wei, Y.Y.; Ng, C.P.; Colman, A.; Korzh, V.; Xu, X.Q. RNA-binding protein RBM24 is required for sarcomere assembly and heart contractility. Cardiovasc. Res. 2012, 94, 418–427. [Google Scholar] [CrossRef]
- Liu, J.; Kong, X.; Lee, Y.M.; Zhang, M.K.; Guo, L.Y.; Lin, Y.; Lim, T.K.; Lin, Q.; Xu, X.Q. Stk38 Modulates Rbm24 Protein Stability to Regulate Sarcomere Assembly in Cardiomyocytes. Sci. Rep. 2017, 7, 44870. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medical History | All n = 61 | All CMP 1 n = 51 | Primary Arrhythmias n = 10 | DCM/LVNC 2 n = 36 |
|---|---|---|---|---|
| Mean age at presentation, years (±SD) | 42 ± 15.3 | 42 ± 15.1 | 39 ± 15.5 | 42 ± 15.4 |
| Mean age of first diagnosis, years (±SD) | 37 ± 15.6 | 37 ± 15.6 | 35 ± 15.1 | 37 ± 15.7 |
| Males, n (%) | 39 (64) | 36 (70) | 3 (30) | 26 (72) |
| Family history of CMP1 or arrhythmia, n (%) | 29 (47.5) | 25 (49) | 4 (40) | 20 (55.5) |
| Family history of sudden death, n (%) | 19 (31) | 16 (31) | 3 (30) | 15 (42) |
| Presentation with ventricular arrhythmia or sudden death, n (%) | 16 (26) | 11 (22) | 5 (50) | 3 (8) |
| Presentation chronic heart failure, n (%) | 18(29.5) | 18 (35) | 0 | 17 (47) |
| Presentation acute heart failure, n (%) | 18 (29.5) | 18 (35) | 0 | 16 (44) |
| Detected Variant | ||||
| Pathogenic/likely pathogenic, n (%) | 28 (46) | 26 (51) | 2 (20) | 16 (44) |
| VUS 3 favor pathogenic, n (%) | 11 (18) | 9 (17) | 2 (20) | 9 (25) |
| VUS, n (%) | 11 (18) | 8 (16) | 3 (30) | 5 (14) |
| No variant found, n (%) | 11 (18) | 8 (16) | 3 (30) | 6 (17) |
| Variant of interest (VOI), n (%) | 39 (64) | 35 (68) | 4 (40) | 25 (69) |
| Pat. # | Diagnosis | Gene | REFSEQ Transcript | HGVSc | HGVSp | Consequence | gnomAD AF | Popmax Filtering AF | ACMG Class | ACMG Rules | Missing Criterium for Classification as Likely Pathogenic § |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ARVC | HCN4 | NM_005477.2 | c.2266G>A | p.(Ala756Thr) | missense variant | 0.00004923 | 0.00004798 | variant of unknown significance | PP3 | |
| 2 | ARVC | CAV3 | NM_033337.2 | c.233C>T | p.(Thr78Met) | missense variant | 0.002667 | 0.003500 | variant of unknown significance | PP2 PP3 PP5 BP6 | |
| 3 | ARVC | PKP2 | NM_004572.3 | c.1237C>T | p.(Arg413 *) | nonsense variant | 0.00001415 | Pathogenic | PVS1 PP5 | ||
| PKP2 | NM_004572.3 | c.2636T>C | p.(Leu879Pro) | missense variant | 0.00001591 | 0.000007010 | variant of unknown significance | PM2 PP3 | |||
| DSP | NM_004415.3 | c.5383T>A | p.(Ser1795Thr) | missense variant | 0.00001062 | variant of unknown significance | PM1 PM2 | ||||
| 4 | ARVC | PKP2 | NM_004572.3 | c.369G>A | p.(Trp123 *) | nonsense variant | 0.000004025 | Pathogenic | PVS1 PP5 | ||
| 5 | DCM | TNNT2 | NM_001001430.2 | c.621G>C | p.(Lys207Asn) | missense variant | variant of unknown significance * | PM1 PM2 PP3 | literature/database report (PP5) | ||
| 6 | DCM | TTN | NM_001267550.2 | c.71083G>T | p.(Glu23695 *) | nonsense variant | Pathogenic | PVS1 PM2 PP4 | |||
| 7 | DCM | TTN | NM_001267550.2 | c.101996G>A | p.(Trp33999 *) | nonsense variant | Pathogenic | PVS1 PM2 PP4 | |||
| TNNT2 | NM_001001430.2 | c.83C>T | p.(Ala28Val) | missense variant | 0.0004420 | 0.0005631 | variant of unknown significance | PP5 BP6 | |||
| 8 | DCM | TTN | NM_001267550.2 | c.5383A>T | p.(Lys1795 *) | nonsense variant | Pathogenic | PVS1 PM2 PP4 | |||
| 9 | DCM | FLNC | NM_001458.4 | c.2504dup | p.(Pro836Thrfs * 84) | frameshift variant | Pathogenic | PVS1 PM2 PP4 | |||
| 10 | DCM | TTN | NM_001267550.2 | c.75546C>A | p.(Tyr25182 *) | nonsense variant | Pathogenic | PVS1 PM2 PP4 | |||
| 11 | DCM | TNNT2 | NM_001001430.2 | c.406G>A | p.(Glu136Lys) | missense variant | 0.00001066 | 0.000002930 | variant of unknown significance * | PM1 PP3 PP5 PP4 | variant not absent from controls(PM2) |
| 12 | DCM | RBM20 | NM_001134363.2 | c.1901G>T | p.(Arg634Leu) | missense variant | Pathogenic | PS3 PM2 PM5 PP3 PP5 | |||
| 13 | DCM | DSP | NM_004415.3 | c.7570_7573del | p.(Thr2524Alafs*36) | frameshift variant | Pathogenic | PVS1 PM2 PP4 | |||
| 14 | DCM | VCL | NM_014000.2 | c.1382C>A | p.(Ala461Asp) | missense variant | variant of unknown significance | PM2 | |||
| 15 | DCM | FLNC | NM_001458.4 | c.4192A>G | p.(Lys1398Glu) | missense variant | variant of unknown significance | PM1 PM2 PP3 | |||
| 16 | DCM | TTN | NM_001267550.2 | c.79684C>T | p.(Arg26562*) | nonsense variant | Pathogenic | PVS1 PM2 PP4 PP5 | |||
| 17 | DCM | SCN5A | NM_198056.2 | c.1538G>C | p.(Arg513Pro) | missense variant | 0.000004331 | variant of unknown significance | PM2 BP4 | ||
| 18 | DCM | SCN5A | NM_198056.2 | c.2441G>A | p.(Arg814Gln) | missense variant | 0.00002507 | 0.000007170 | likely pathogenic | PM1 PM4 PP3 PP5 | |
| 19 | DCM | LMNA | NM_170707.3 | c.555_556del | p.(Asp185Glufs * 9) | frameshift variant | Pathogenic | PVS1 PM2 PP4 | |||
| 20 | DCM | EMD | NM_000117.2 | c.153dup | p.(Ser52Glnfs * 9) | frameshift variant | Pathogenic | PVS1 PM2 PP4 PP5 | |||
| 21 | DCM | RYR2 | NM_001035.2 | c.2026G>A | p.(Glu676Lys) | missense variant | variant of unknown significance * | PM1 PM2 PP3 | literature/database report (PP5) | ||
| 22 | DCM | RBM20 | NM_001134363.2 | c.686A>G | p.(Tyr229Cys) | missense variant | variant of unknown significance | PM2 PP3 | |||
| MYH7 | NM_000257.3 | c.3866G>A | p.(Arg1289Gln) | missense variant | 0.00001061 | 0.000002920 | variant of unknown significance * | PM1 PP2 PP3 | variant not absent from controls(PM2) | ||
| 23 | HCM | MYH7 | NM_000257.3 | c.1207C>T | p.(Arg403Trp) | missense variant | Pathogenic | PS3 PM1 PM2 PM5 PP1 PP3 PP4 PP5 | |||
| MYH7 | NM_000257.3 | c.1000-1G>A | p.? | splice variant | 0.000007073 | likely pathogenic | PM2 PP3 PP4 | ||||
| 24 | HCM | TNNT2 | NM_001001430.2 | c.281G>T | p.(Arg94Leu) | missense variant | likely pathogenic | PS3 PM2 PP3 PP5 | |||
| 25 | HCM | FHL1 | NM_001449.4 | c.501G>C | p.(Lys167Asn) | missense variant | likely pathogenic | PM1 PM2 PP1 PP3 | |||
| 26 | HCM | MYBPC3 | NM_000256.3 | c.1440_1441delinsC | p.(Glu480Aspfs * 8) | frameshift variant | likely pathogenic | PVS1 PM2 | |||
| 27 | HCM | DSG2 | NM_001943.4 | c.593A>G | p.(Tyr198Cys) | missense variant | 0.00001781 | 0.00002233 | likely pathogenic | PM1 PM2 PP3 PP5 | |
| 28 | DCM/Arrhythmias | TTN | NM_001267550.2 | c.81341dup | p.(Asn27115Glufs * 10 | frameshift variant | Pathogenic | PVS1 PM2 PP4 | |||
| RYR2 | NM_001035.2 | c.322G>A | p.(Gly108Ser) | missense variant | 0.00002271 | 0.000004630 | variant of unknown significance | PM1 PM2 PP3 | |||
| 29 | LVNC | MYH7 | NM_000257.3 | c.3286G>T | p.(Asp1096Tyr) | missense variant | 0.0001414 | 0.00005238 | variant of unknown significance * | PM1 PP2 PP3 PP5 | variant not absent from controls (PM2) |
| 30 | LVNC | TTN | NM_001267550.2 | c.59848C>T | p.(Arg19950*) | nonsense variant | Pathogenic | PVS1 PM2 PP4 | |||
| MYH7 | NM_000257.3 | c.5735T>A | p.(Ile1912Asn) | missense variant | variant of unknown significance | PM2 PP2 PP3 | |||||
| 31 | RCM | FLNC | NM_001458.4 | c.6031G>A | p.(Gly2011Arg) | missense variant | Likely pathogenic | PM1 PM2 PP1 PP3 | |||
| 32 | Long-QT/Arrhythmias | KCNH2 | NM_000238.3 | c.944T>C | p.(Leu315Ser) | missense variant | variant of unknown significance * | PM2 PP2 PP3 PP4 | literature/database report (PP5) | ||
| 33 | Long-QT | KCNQ1 | NM_000218.2 | c.785T>C | p.(Leu262Pro) | missense variant | Likely pathogenic | PM1 PM2 PM5 PP3 PP5 | |||
| 34 | Brugada syndrome | SCN5A | NM_198056.2 | c.4747C>T | p.(Arg1583Cys) | missense variant | 0.000008026 | 0.000002940 | Likely pathogenic | PM1 PM5 PP3 PP5 | |
| 35 | DCM | FLNC | NM_001458.4 | c.3275_3278delinsAAGA | p.(Thr1092_Gly1093delinsLysAsp) | in-frame delins | variant of unknown significance * | PM2 PP2 PP3 PP4 | literature/database report (PP5) | ||
| 36 | Long-QT | KCNH2 | NM_000238.3 | c.526C>T | p.(Arg176Trp) | missense variant | 0.0003237 | 0.0004289 | variant of unknown significance | PP5 | |
| 37 | DCM | TNNI3K | NM_015978.2 | c.500T>C | p.(Phe167Ser) | missense variant | 0.000007084 | variant of unknown significance | PM1 | ||
| 38 | DCM | MYBPC3 | NM_000256.3 | c.2381C>T | p.(Pro794Leu) | missense variant | 0.0001365 | 0.00002245 | variant of unknown significance * | PM1 PP3 PP5 | variant not absent from controls (PM2) |
| 39 | DCM | MYH7 | NM_000257.3 | c.1565A>T | p.(Asp522Val) | missense variant | variant of unknown significance * | PM1 PM2 PP2 | literature/database report (PP5) | ||
| 40 | Survived sudden cardiac death | CACNB2 | NM_201590.2 | c.165A>T | p.(Lys55Asn) | missense variant | variant of unknown significance * | PM1 PM2 PP3 | literature/database report (PP5) | ||
| 41 | DCM | DSG2 | NM_001943.4 | c.2533del | p.(Ile845 *) | frameshift variant | Likely pathogenic | PVS1 PM2 PP5 | |||
| 42 | HCM | MYBPC3 | NM_000256.3 | c.2454G>A | p.(Trp818 *) | nonsense variant | Pathogenic | PVS1 PM2 PP5 | |||
| 43 | DCM | TTN | NM_001267550.2 | c.68022del | p.(Glu22675Lysfs * 7) | frameshift variant | Pathogenic | PVS1 PM2 PP4 | |||
| 44 | DCM | MIB1 | NM_020774.3 | c.1111C>T | p.(Arg371 *) | nonsense variant | 0.00008856 | 0.00006398 | Likely pathogenic | PVS1 PP1 PP4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolokotronis, K.; Pluta, N.; Klopocki, E.; Kunstmann, E.; Messroghli, D.; Maack, C.; Tejman-Yarden, S.; Arad, M.; Rost, S.; Gerull, B. New Insights on Genetic Diagnostics in Cardiomyopathy and Arrhythmia Patients Gained by Stepwise Exome Data Analysis. J. Clin. Med. 2020, 9, 2168. https://doi.org/10.3390/jcm9072168
Kolokotronis K, Pluta N, Klopocki E, Kunstmann E, Messroghli D, Maack C, Tejman-Yarden S, Arad M, Rost S, Gerull B. New Insights on Genetic Diagnostics in Cardiomyopathy and Arrhythmia Patients Gained by Stepwise Exome Data Analysis. Journal of Clinical Medicine. 2020; 9(7):2168. https://doi.org/10.3390/jcm9072168
Chicago/Turabian StyleKolokotronis, Konstantinos, Natalie Pluta, Eva Klopocki, Erdmute Kunstmann, Daniel Messroghli, Christoph Maack, Shai Tejman-Yarden, Michael Arad, Simone Rost, and Brenda Gerull. 2020. "New Insights on Genetic Diagnostics in Cardiomyopathy and Arrhythmia Patients Gained by Stepwise Exome Data Analysis" Journal of Clinical Medicine 9, no. 7: 2168. https://doi.org/10.3390/jcm9072168
APA StyleKolokotronis, K., Pluta, N., Klopocki, E., Kunstmann, E., Messroghli, D., Maack, C., Tejman-Yarden, S., Arad, M., Rost, S., & Gerull, B. (2020). New Insights on Genetic Diagnostics in Cardiomyopathy and Arrhythmia Patients Gained by Stepwise Exome Data Analysis. Journal of Clinical Medicine, 9(7), 2168. https://doi.org/10.3390/jcm9072168