Master Regulator Analysis of the SARS-CoV-2/Human Interactome




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Interactome Model for SARS-CoV-2/Human Cell Interaction
2.2. Coexpression-Based Lung Network
2.3. Datasets of MERS and SARS Infection
2.4. Master Regulator Analysis
2.5. BLAST
2.6. Phylogenetic Analysis of Viral Genomes and ACE Orthologs
2.7. 3D Structural Analysis
3. Results
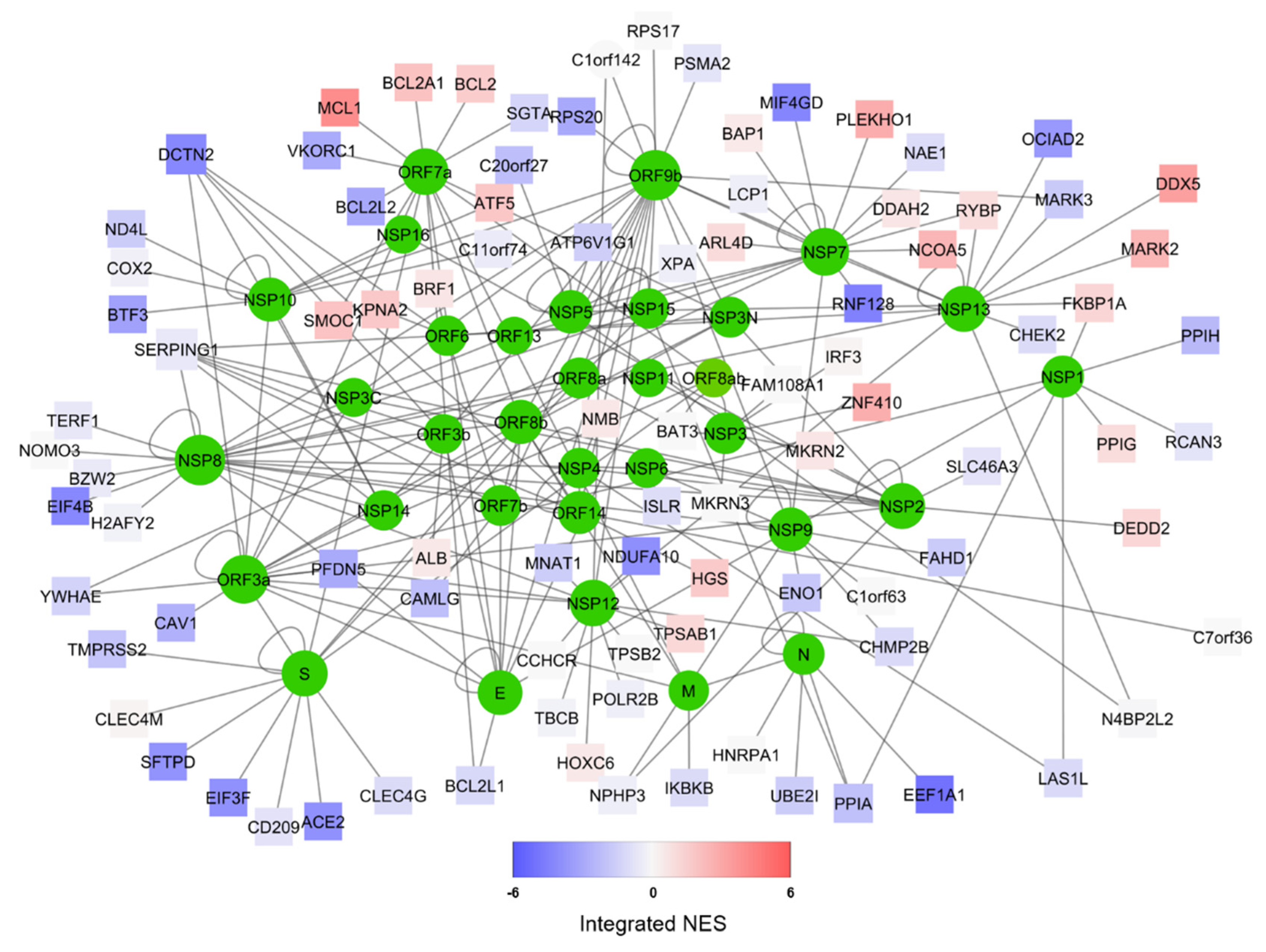
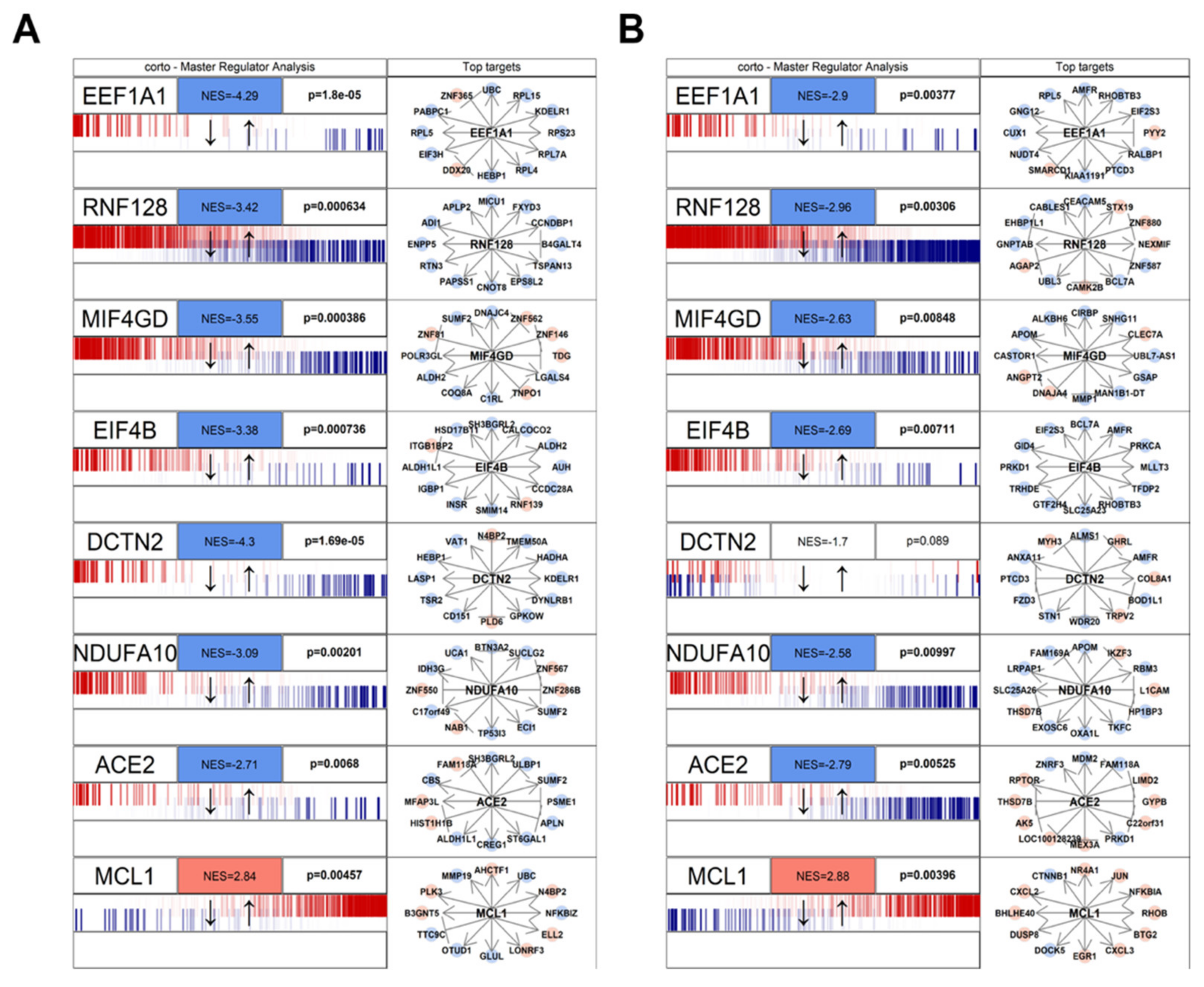
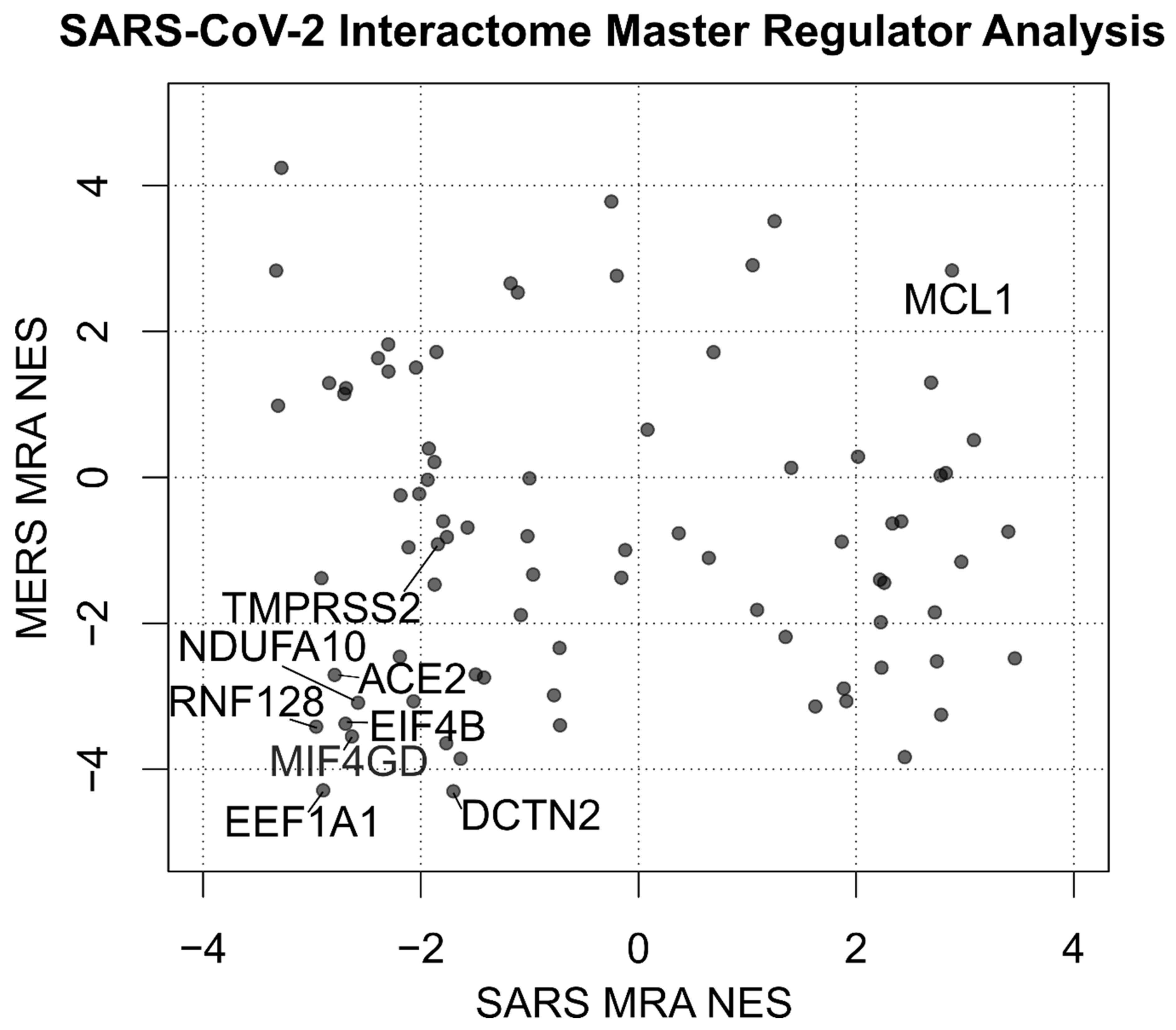
3.1. Human/COVID-19 Interactome Response to Coronavirus Infection
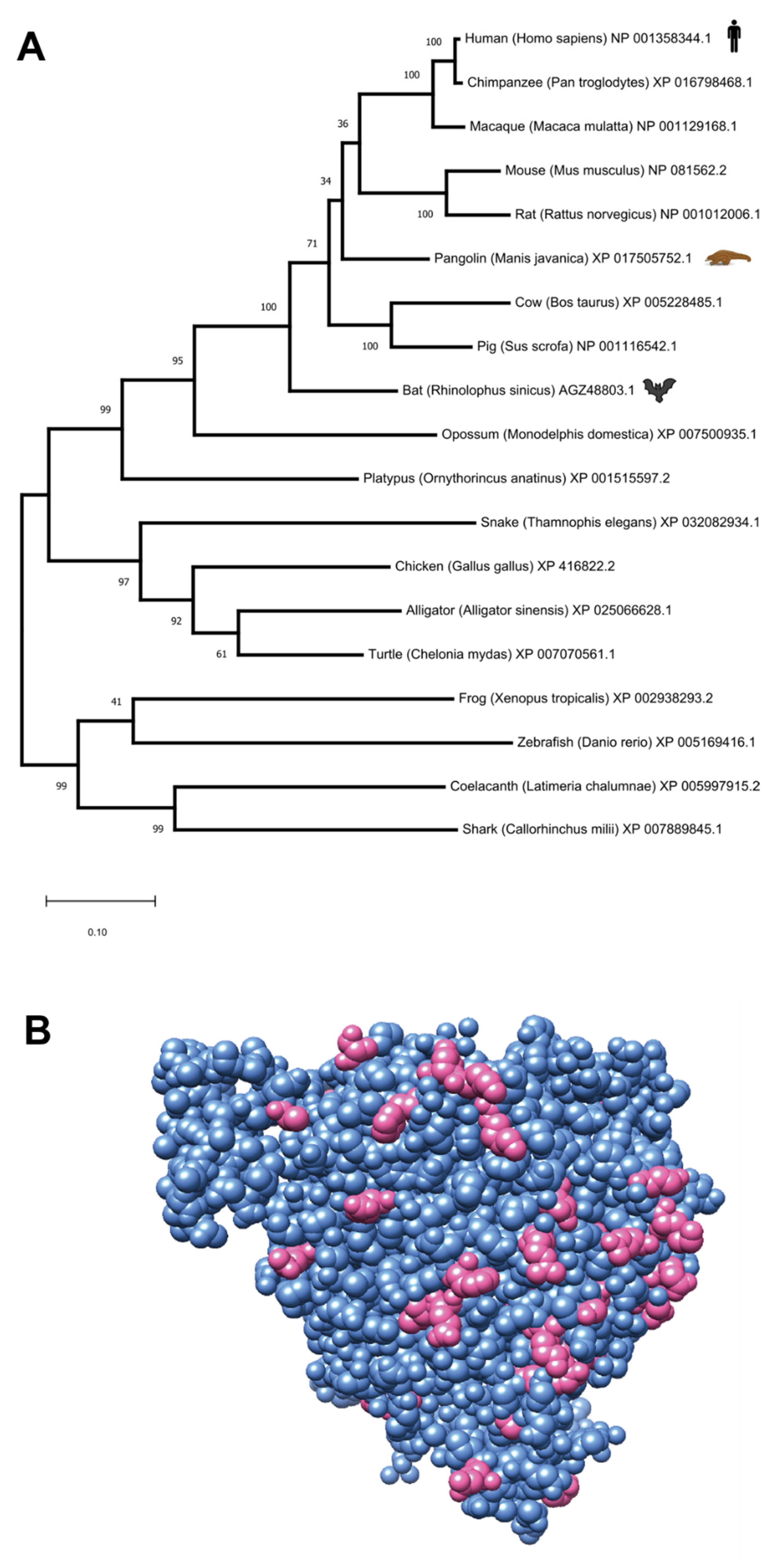
3.2. ACE2: Implications for Coronavirus Origin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2019; Volume 105, pp. 93–116. [Google Scholar]
- Zeng, Z.-Q.; Chen, D.-H.; Tan, W.-P.; Qiu, S.-Y.; Xu, D.; Liang, H.-X.; Chen, M.-X.; Li, X.; Lin, Z.-S.; Liu, W.-K.; et al. Epidemiology and clinical characteristics of human coronaviruses OC43, 229E, NL63, and HKU1: A study of hospitalized children with acute respiratory tract infection in Guangzhou, China. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Zheng, B.; Li, Y.; Poon, L.; Xie, Z.; Chan, K.; Li, P.; Tan, S.; Chang, Q.; Xie, J.; et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003, 362, 1353–1358. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections—More Than Just the Common Cold. JAMA 2020, 323, 707. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Park, Y.-J.; Walls, A.C.; Wang, Z.; Sauer, M.M.; Li, W.; Tortorici, M.A.; Bosch, B.-J.; DiMaio, F.; Veesler, D. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019, 26, 1151–1157. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Coronavirus: Covid-19 has killed more people than SARS and MERS combined, despite lower case fatality rate. BMJ 2020, 368, m641. [Google Scholar] [CrossRef] [PubMed]
- Ceraolo, C.; Giorgi, F.M. Genomic variance of the 2019-nCoV coronavirus. J. Med. Virol. 2020, 92, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Li, R.; Qiao, S.; Zhang, G. Analysis of angiotensin-converting enzyme 2 (ACE2) from different species sheds some light on cross-species receptor usage of a novel coronavirus 2019-nCoV. J. Infect. 2020, 80, 469–496. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef]
- Lakshmi, B.S.; Abillasha, M.L.; Kangueane, P. Emerging Technologies for Antiviral Drug Discovery. In Global Virology III: Virology in the 21st Century; Shapshak, P., Balaji, S., Kangueane, P., Chiappelli, F., Somboonwit, C., Menezes, L.J., Sinnott, J.T., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 59–71. [Google Scholar]
- Caldera, M.; Buphamalai, P.; Müller, F.; Menche, J. Interactome-based approaches to human disease. Curr. Opin. Syst. Biol. 2017, 3, 88–94. [Google Scholar] [CrossRef]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2019, 194430. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rajbhandari, P.; Alvarez, M.J.; Bandaru, P.; Lim, W.K.; Sato, M.; Wang, K.; Sumazin, P.; Kustagi, M.; Bisikirska, B.C.; et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol. Syst. Biol. 2010, 6, 377. [Google Scholar] [CrossRef]
- Giorgi, F.M.; Lopez, G.; Woo, J.H.; Bisikirska, B.; Califano, A.; Bansal, M. Inferring protein modulation from gene expression data using conditional mutual information. PLoS ONE 2014, 9, e109569. [Google Scholar] [CrossRef] [PubMed]
- Holding, A.N.; Giorgi, F.M.; Donnelly, A.; Cullen, A.E.; Nagarajan, S.; Selth, L.A.; Markowetz, F. VULCAN integrates ChIP-seq with patient-derived co-expression networks to identify GRHL2 as a key co-regulator of ERa at enhancers in breast cancer. Genome Biol. 2019, 20, 91. [Google Scholar] [CrossRef] [PubMed]
- Tovar, H.; García-Herrera, R.; Espinal-Enríquez, J.; Hernández-Lemus, E. Transcriptional master regulator analysis in breast cancer genetic networks. Comput. Biol. Chem. 2015, 59, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses 2020, 12, 360. [Google Scholar] [CrossRef] [PubMed]
- Vasilevski, A.; Giorgi, F.M.; Bertinetti, L.; Usadel, B. LASSO modeling of the Arabidopsis thaliana seed/seedling transcriptome: A model case for detection of novel mucilage and pectin metabolism genes. Mol. Biosyst. 2012, 8, 2566–2574. [Google Scholar] [CrossRef]
- Mercatelli, D.; Lopez-Garcia, G.; Giorgi, F.M. corto: A lightweight R package for Gene Network Inference and Master Regulator Analysis. Bioinformatics 2020. [Google Scholar] [CrossRef]
- The GTEx Consortium; Ardlie, K.G.; Deluca, D.S.; Segre, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Reverter, A.; Chan, E.K.F. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef]
- Mercatelli, D.; Ray, F.; Giorgi, F.M. Pan-Cancer and Single-Cell Modeling of Genomic Alterations through Gene Expression. Front. Genet. 2019, 10, 671. [Google Scholar] [CrossRef]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Hill, T.E.; Yoshikawa, N.; Popov, V.L.; Galindo, C.L.; Garner, H.R.; Peters, C.J.; Tseng, C.-T. (Kent) Dynamic Innate Immune Responses of Human Bronchial Epithelial Cells to Severe Acute Respiratory Syndrome-Associated Coronavirus Infection. PLoS ONE 2010, 5, e8729. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.M.; Bolger, A.M.; Lohse, M.; Usadel, B. Algorithm-driven artifacts in median polish summarization of microarray data. BMC Bioinform. 2010, 11, 553. [Google Scholar] [CrossRef] [PubMed]
- Dai, M. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed]
- Selinger, C.; Tisoncik-Go, J.; Menachery, V.D.; Agnihothram, S.; Law, G.; Chang, J.; Kelly, S.M.; Sova, P.; Baric, R.S.; Katze, M.G. Cytokine systems approach demonstrates differences in innate and pro-inflammatory host responses between genetically distinct MERS-CoV isolates. BMC Genom. 2014, 15, 1161. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R.; on behalf of the International Nucleotide Sequence Database Collaboration. The sequence read archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef]
- Liu, P.; Chen, W.; Chen, J.-P. Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis javanica). Viruses 2019, 11, 979. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2.0: Resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. In Protein Crystallography; Wlodawer, A., Dauter, Z., Jaskolski, M., Eds.; Springer: New York, NY, USA, 2017; Volume 1607, pp. 627–641. [Google Scholar]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kozopas, K.M.; Yang, T.; Buchan, H.L.; Zhou, P.; Craig, R.W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 1993, 90, 3516–3520. [Google Scholar] [CrossRef]
- Ren, J.; Shi, M.; Liu, R.; Yang, Q.-H.; Johnson, T.; Skarnes, W.C.; Du, C. The Birc6 (Bruce) gene regulates p53 and the mitochondrial pathway of apoptosis and is essential for mouse embryonic development. Proc. Natl. Acad. Sci. USA 2005, 102, 565–570. [Google Scholar] [CrossRef]
- Zhong, Y.; Liao, Y.; Fang, S.; Tam, J.P.; Liu, D.X. Up-Regulation of Mcl-1 and Bak by Coronavirus Infection of Human, Avian and Animal Cells Modulates Apoptosis and Viral Replication. PLoS ONE 2012, 7, e30191. [Google Scholar] [CrossRef]
- Bordi, L.; Castilletti, C.; Falasca, L.; Ciccosanti, F.; Calcaterra, S.; Rozera, G.; Di Caro, A.; Zaniratti, S.; Rinaldi, A.; Ippolito, G.; et al. Bcl-2 inhibits the caspase-dependent apoptosis induced by SARS-CoV without affecting virus replication kinetics. Arch. Virol. 2006, 151, 369–377. [Google Scholar] [CrossRef]
- Liu, C.; Xu, H.Y.; Liu, D.X. Induction of Caspase-Dependent Apoptosis in Cultured Cells by the Avian Coronavirus Infectious Bronchitis Virus. J. Virol. 2001, 75, 6402–6409. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Nara, K.; Yoshikawa, H.; Suzuki, N. Txk, a member of the non-receptor tyrosine kinase of the Tec family, forms a complex with poly(ADP-ribose) polymerase 1 and elongation factor 1α and regulates interferon-γ gene transcription in Th1 cells: Txk, PARP1 and EF-1α make a Th1 cell transcription factor. Clin. Exp. Immunol. 2006, 147, 164–175. [Google Scholar]
- Sikora, D.; Greco-Stewart, V.S.; Miron, P.; Pelchat, M. The hepatitis delta virus RNA genome interacts with eEF1A1, p54nrb, hnRNP-L, GAPDH and ASF/SF2. Virology 2009, 390, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, W.; Li, X.; Cao, H.; Wang, Y.; Zheng, S.J. Critical role of eukaryotic elongation factor 1 alpha 1 (EEF1A1) in avian reovirus sigma-C-induced apoptosis and inhibition of viral growth. Arch. Virol. 2015, 160, 1449–1461. [Google Scholar] [CrossRef]
- Loeffen, J.L.C.M.; Triepels, R.H.; van den Heuvel, L.P.; Schuelke, M.; Buskens, C.A.F.; Smeets, R.J.P.; Trijbels, J.M.F.; Smeitink, J.A.M. cDNA of Eight Nuclear Encoded Subunits of NADH:Ubiquinone Oxidoreductase: Human Complex I cDNA Characterization Completed. Biochem. Biophys. Res. Commun. 1998, 253, 415–422. [Google Scholar] [CrossRef]
- Munday, D.C.; Hiscox, J.A.; Barr, J.N. Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus subgroup B using SILAC coupled to LC-MS/MS. Proteomics 2010, 10, 4320–4334. [Google Scholar] [CrossRef]
- Anandasabapathy, N.; Ford, G.S.; Bloom, D.; Holness, C.; Paragas, V.; Seroogy, C.; Skrenta, H.; Hollenhorst, M.; Fathman, C.G.; Soares, L. GRAIL: An E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity 2003, 18, 535–547. [Google Scholar] [CrossRef]
- Song, G.; Liu, B.; Li, Z.; Wu, H.; Wang, P.; Zhao, K.; Jiang, G.; Zhang, L.; Gao, C. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63-linked ubiquitination of TBK1. Nat. Immunol. 2016, 17, 1342–1351. [Google Scholar] [CrossRef]
- Jalal, C.; Uhlmann-Schiffler, H.; Stahl, H. Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic Acids Res. 2007, 35, 3590–3601. [Google Scholar] [CrossRef]
- Li, C.; Ge, L.; Li, P.; Wang, Y.; Sun, M.; Huang, L.; Ishag, H.; Di, D.; Shen, Z.; Fan, W.; et al. The DEAD-box RNA helicase DDX5 acts as a positive regulator of Japanese encephalitis virus replication by binding to viral 3′ UTR. Antivir. Res. 2013, 100, 487–499. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Pöhlmann, S. Cellular entry of the SARS coronavirus. Trends Microbiol. 2004, 12, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020. [Google Scholar] [CrossRef]
- Liu, X.; Yang, N.; Tang, J.; Liu, S.; Luo, D.; Duan, Q.; Wang, X. Downregulation of angiotensin-converting enzyme 2 by the neuraminidase protein of influenza A (H1N1) virus. Virus Res. 2014, 185, 64–71. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential Downregulation of ACE2 by the Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, J.-Z.; Wan, X.-F.; Hua, Y.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; Chen, J. Are pangolins the intermediate host of the 2019 novel coronavirus (2019-nCoV)? bioRxiv 2020. [Google Scholar] [CrossRef]
- Lam, T.T.-Y.; Shum, M.H.-H.; Zhu, H.-C.; Tong, Y.-G.; Ni, X.-B.; Liao, Y.-S.; Wei, W.; Cheung, W.Y.-M.; Li, W.-J.; Li, L.-F.; et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 2020. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 2020, 395, 473–475. [Google Scholar] [CrossRef]
- Zhang, H.; Baker, A. Recombinant human ACE2: Acing out angiotensin II in ARDS therapy. Crit. Care 2017, 21, 305. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Batlle, D.; Wysocki, J.; Satchell, K. Soluble angiotensin-converting enzyme 2: A potential approach for coronavirus infection therapy? Clin. Sci. 2020, 134, 543–545. [Google Scholar] [CrossRef]
- Lei, C.; Fu, W.; Qian, K.; Li, T.; Zhang, S.; Ding, M.; Hu, S. Potent neutralization of 2019 novel coronavirus by recombinant ACE2-Ig. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef]
- Shi, C.-S.; Nabar, N.R.; Huang, N.-N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef]
- Boya, P.; Pauleau, A.-L.; Poncet, D.; Gonzalez-Polo, R.-A.; Zamzami, N.; Kroemer, G. Viral proteins targeting mitochondria: Controlling cell death. Biochim. Biophys. Acta BBA Bioenerg. 2004, 1659, 178–189. [Google Scholar] [CrossRef]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzzi, P.H.; Mercatelli, D.; Ceraolo, C.; Giorgi, F.M. Master Regulator Analysis of the SARS-CoV-2/Human Interactome. J. Clin. Med. 2020, 9, 982. https://doi.org/10.3390/jcm9040982
Guzzi PH, Mercatelli D, Ceraolo C, Giorgi FM. Master Regulator Analysis of the SARS-CoV-2/Human Interactome. Journal of Clinical Medicine. 2020; 9(4):982. https://doi.org/10.3390/jcm9040982
Chicago/Turabian StyleGuzzi, Pietro H., Daniele Mercatelli, Carmine Ceraolo, and Federico M. Giorgi. 2020. "Master Regulator Analysis of the SARS-CoV-2/Human Interactome" Journal of Clinical Medicine 9, no. 4: 982. https://doi.org/10.3390/jcm9040982
APA StyleGuzzi, P. H., Mercatelli, D., Ceraolo, C., & Giorgi, F. M. (2020). Master Regulator Analysis of the SARS-CoV-2/Human Interactome. Journal of Clinical Medicine, 9(4), 982. https://doi.org/10.3390/jcm9040982