Emergent Properties of the HNF4α-PPARγ Network May Drive Consequent Phenotypic Plasticity in NAFLD


Abstract

1. Introduction
2. Materials and Methods
2.1. RAndom CIrcuit PErturbation (RACIPE) Analysis:
2.1.1. RACIPE Simulations
2.1.2. Z-Score Normalizations of The Steady State Values
2.1.3. Density Plots, Bimodality Coefficients, and Clustering Analysis:
2.2. Relative Stability Analysis
2.3. Dynamic Simulations
2.3.1. Bifurcation Diagrams




2.3.2. Switching of States




2.4. Randomization of Networks
2.5. Jensen–Shannon Divergence (JSD) and Plasticity Scores
2.6. Clinical Data Analysis
2.7. Statistical Tests and Correlation Coefficients
3. Results
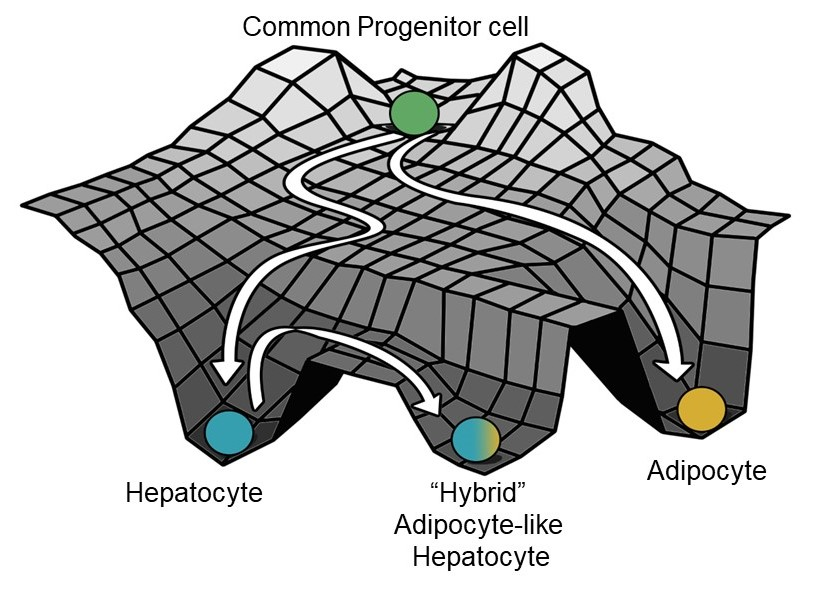
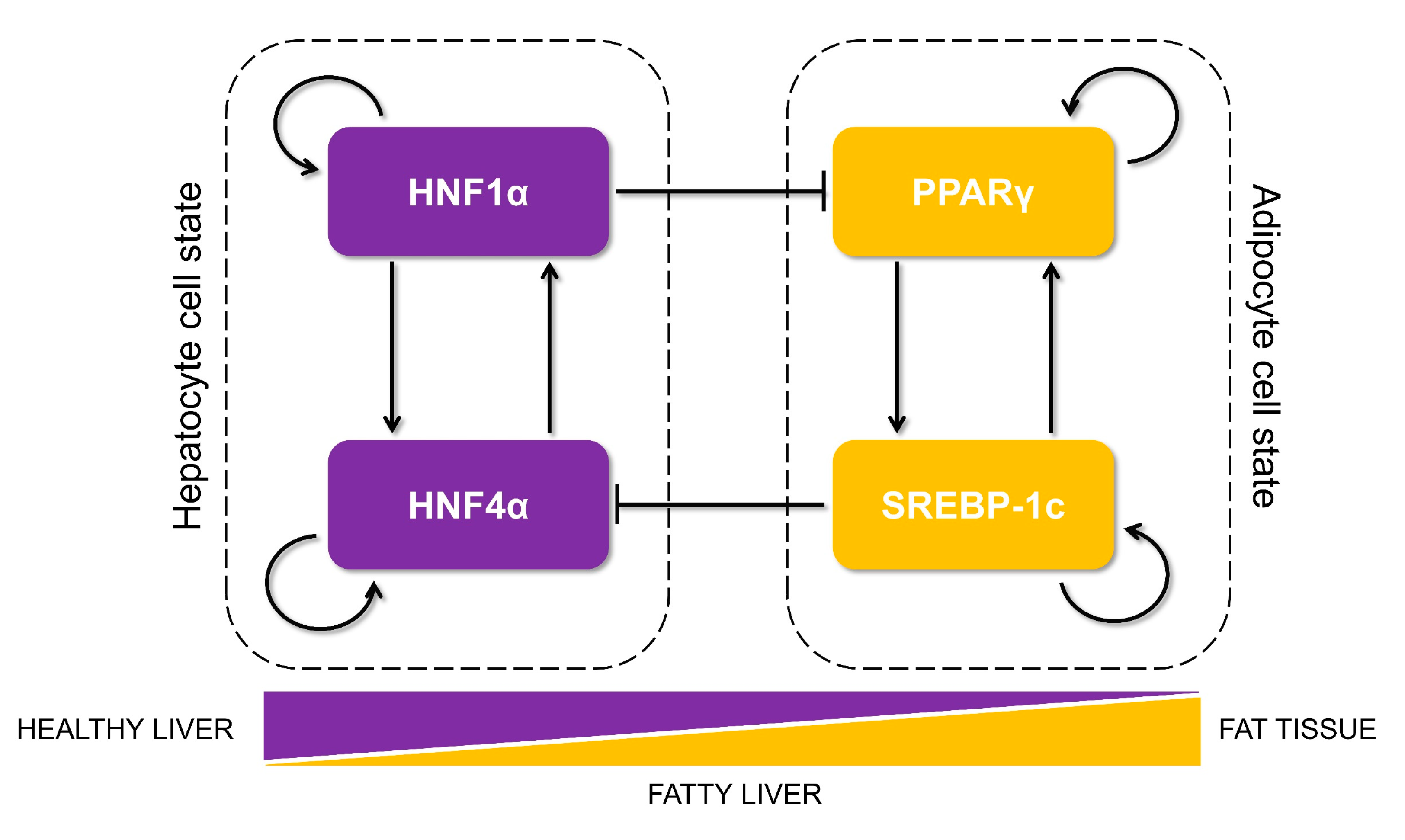
3.1. Identification of a Core HNF4α-PPARγ Network in Hepatocytes
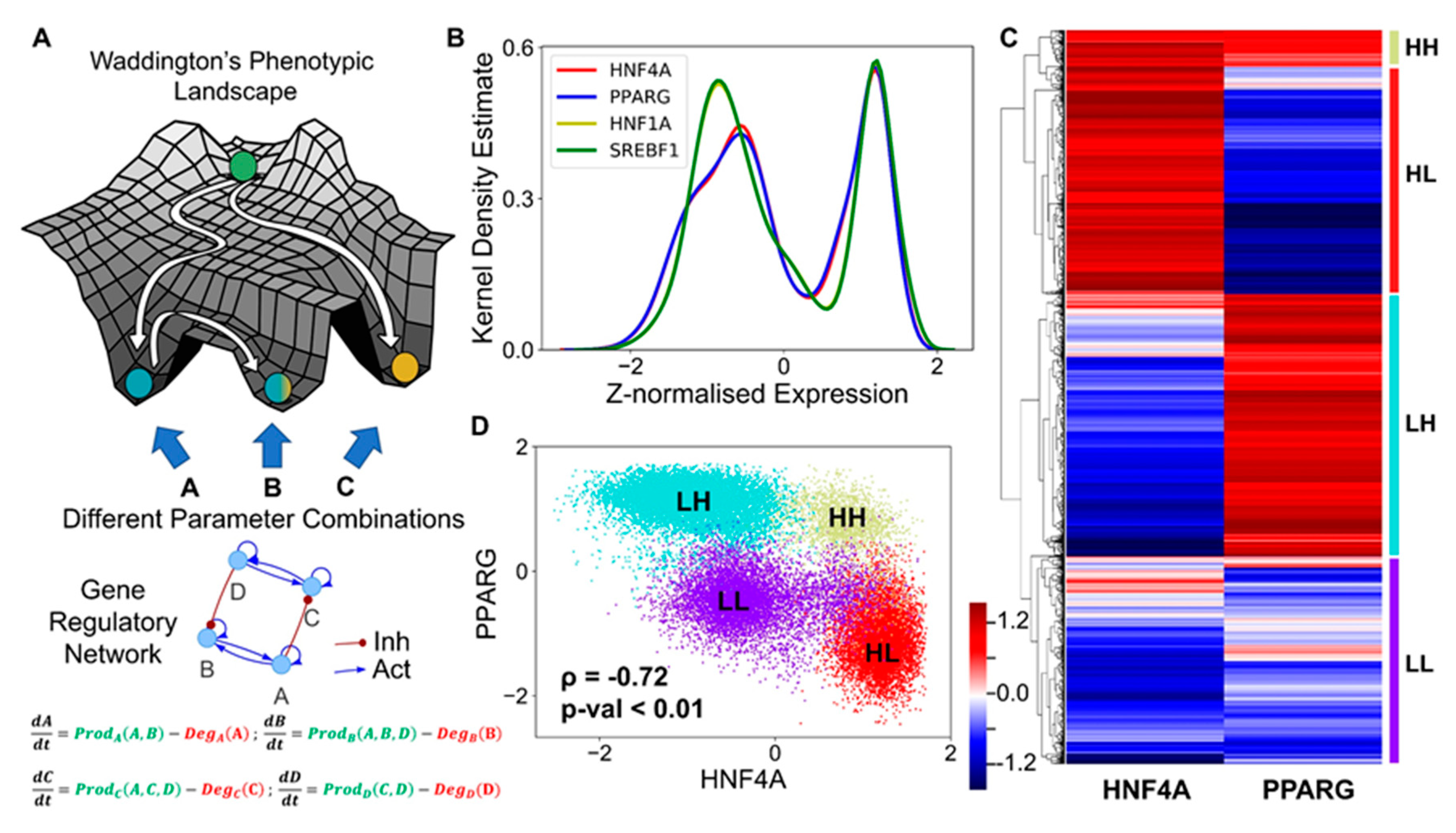
3.2. The Emergent Properties of This Core Regulatory Network Enable The Existence of Multiple Phenotypes
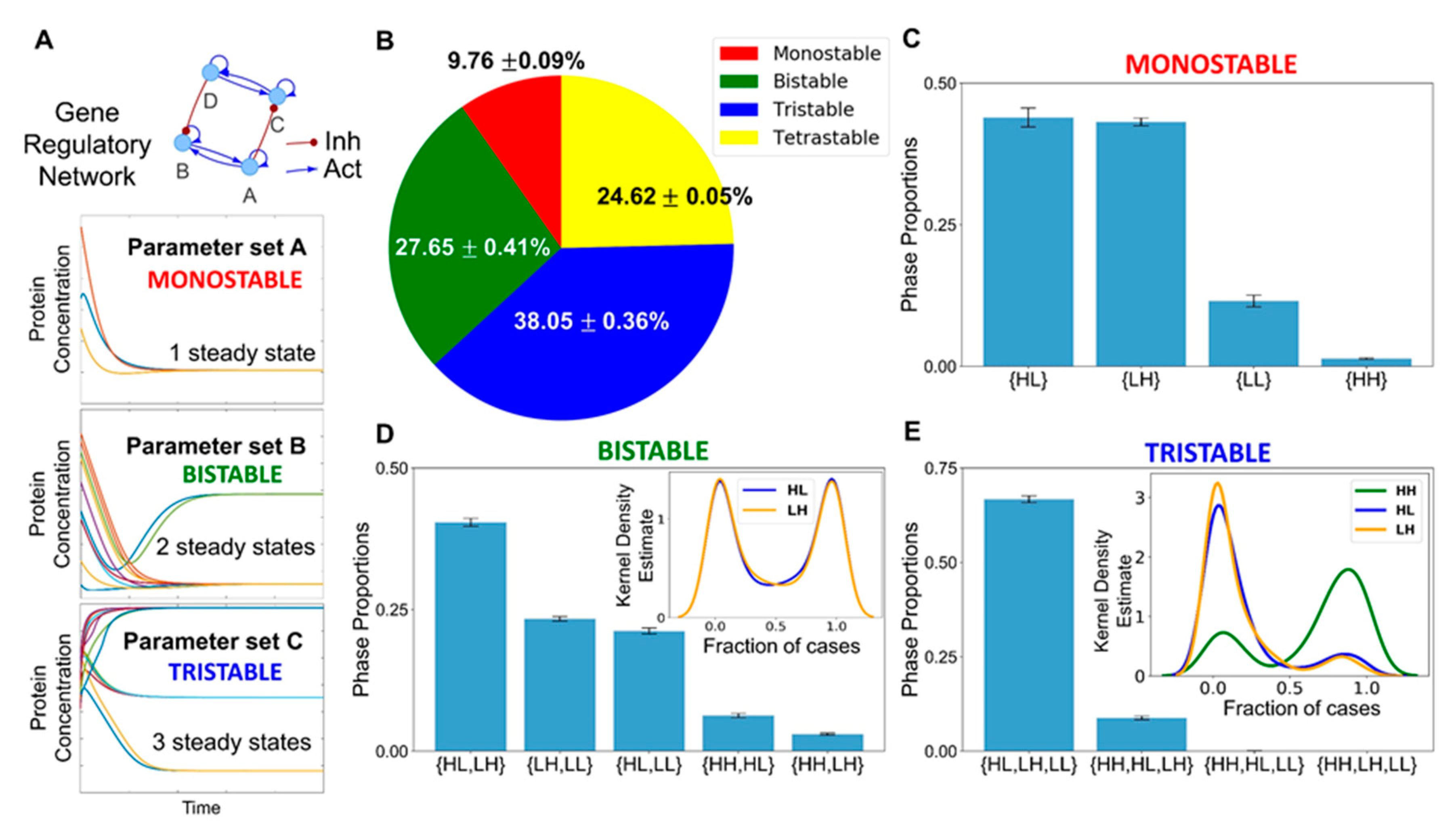
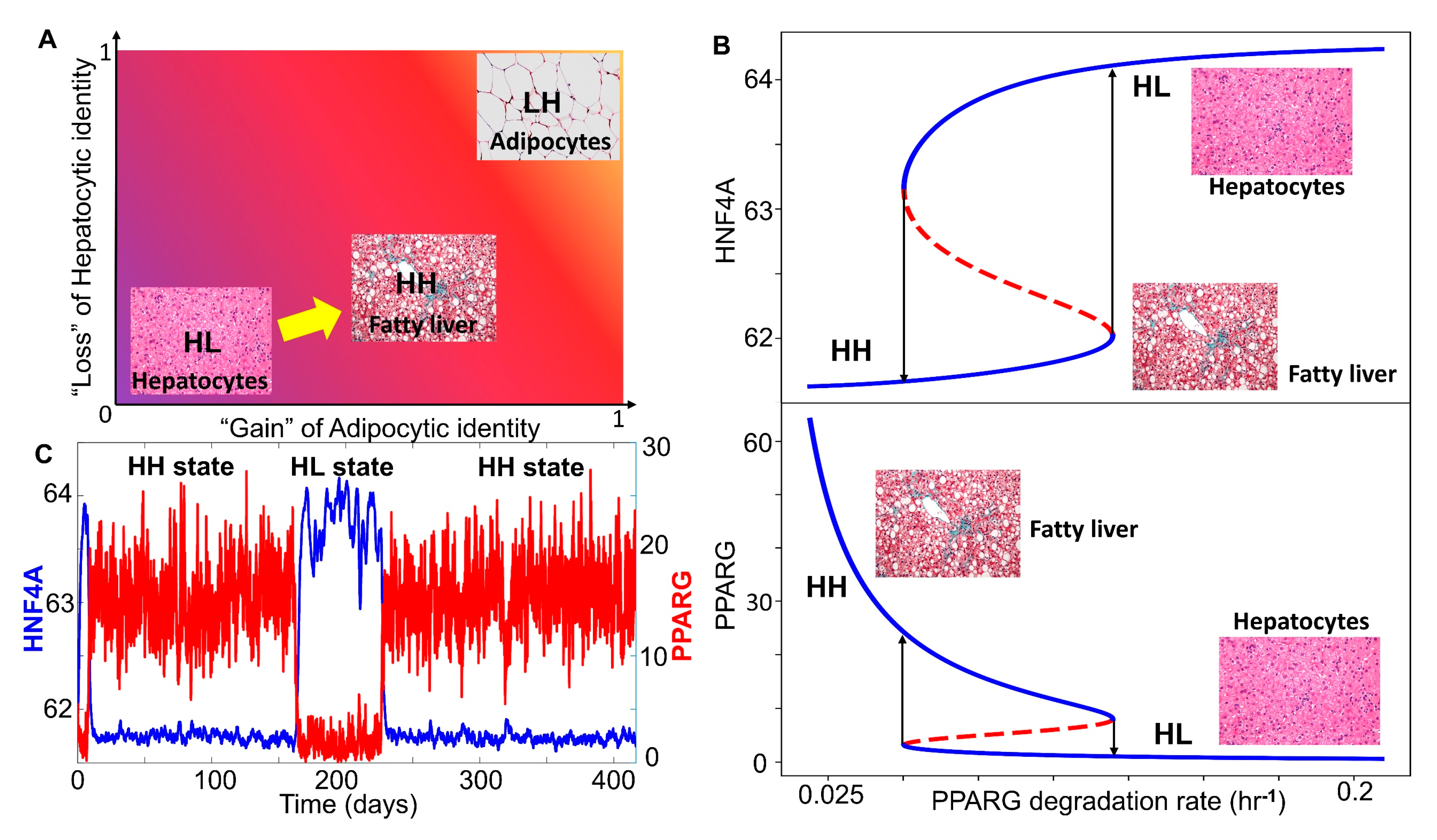
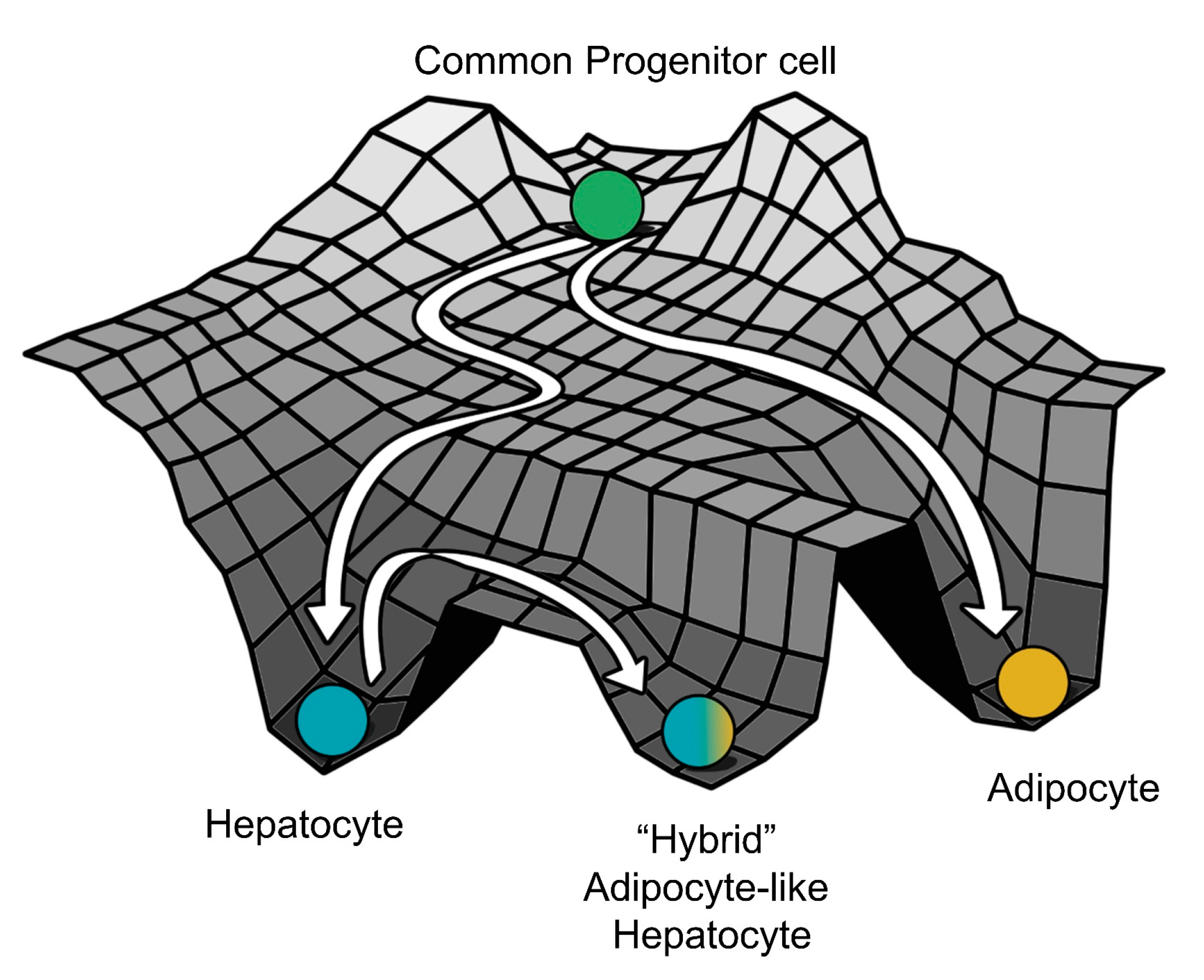
3.3. Multiple Stable States (Phenotypes) Can Co-Exist, Giving Rise To Phenotypic Plasticity
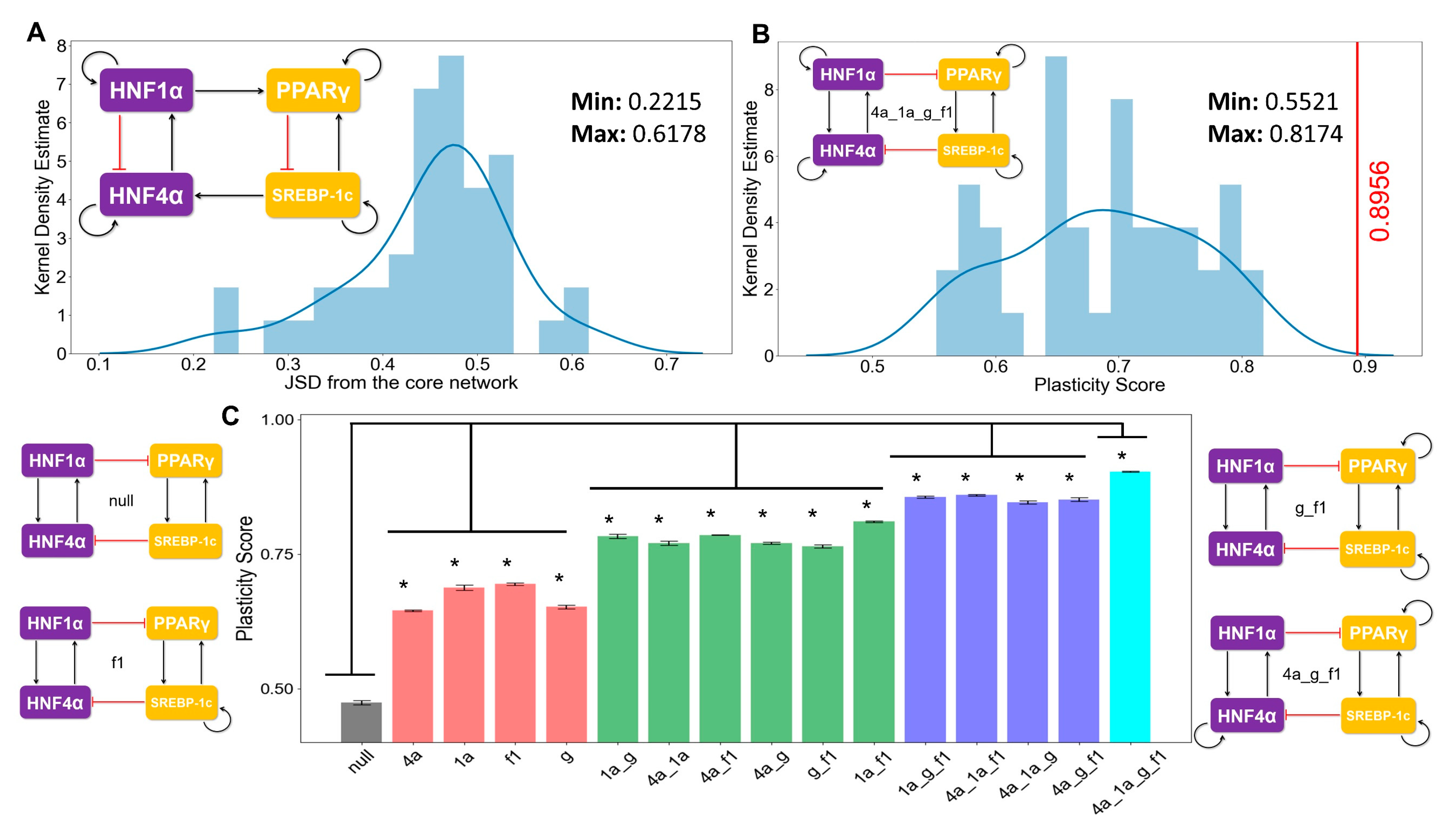
3.4. The Topology of the Core Regulatory Network is Designed to Enhance Phenotypic Plasticity
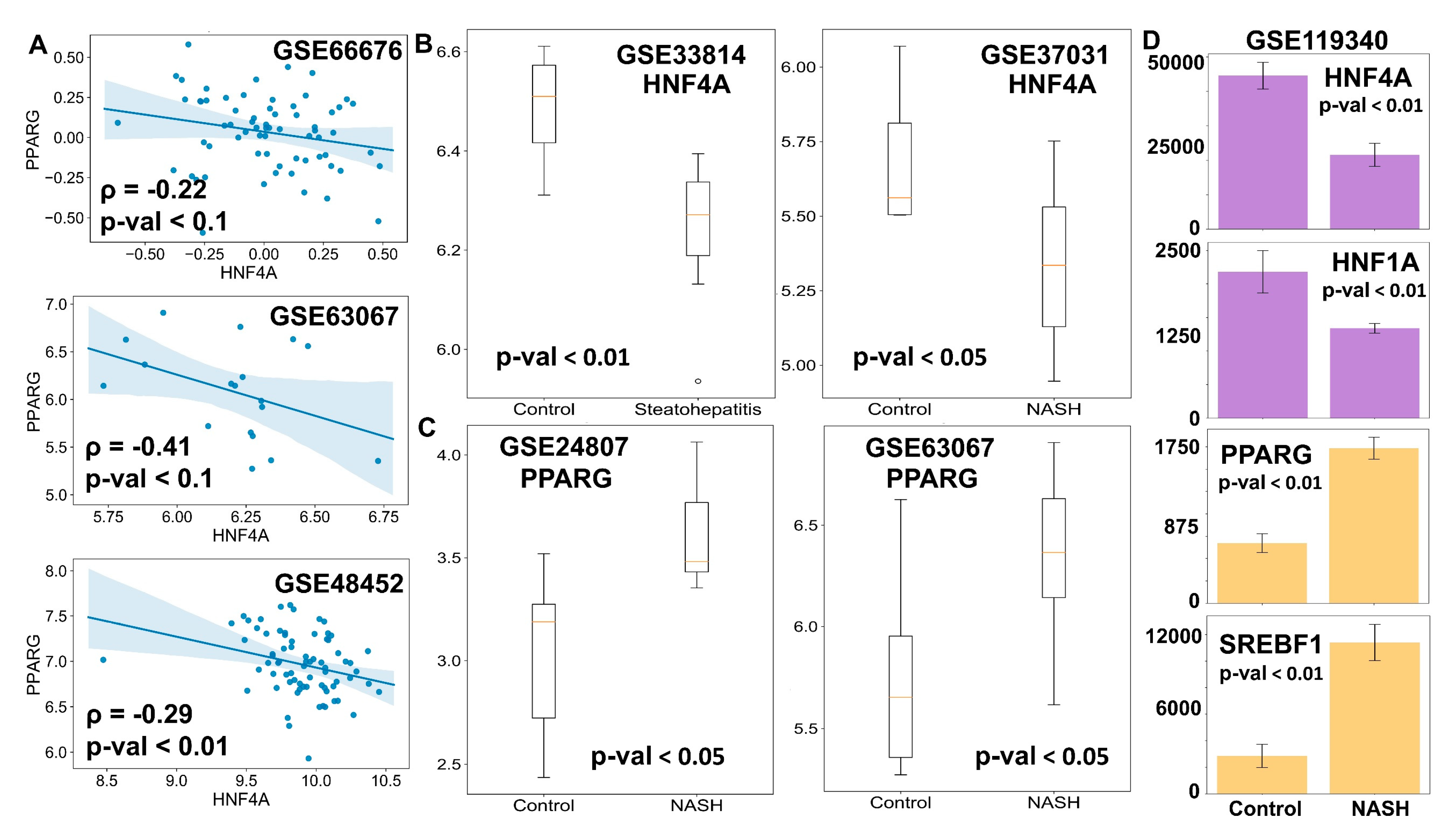
3.5. Clinical Data Support the Model Predictions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Walia, G.K.; Gupta, V.; Sachdeva, M.P. Genetics of nonalcoholic fatty liver disease in Asian populations. J. Genet. 2019, 98, 29. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, K.; Alazawi, W. Genetics of Non-Alcoholic Fatty Liver and Cardiovascular Disease: Implications for Therapy? Front. Pharmacol. 2020, 10, 1413. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Sarin, S.K.; Kumar, M.; Eslam, M.; George, J.; Al Mahtab, M.; Akbar, S.M.F.; Jia, J.; Tian, Q.; Aggarwal, R.; Muljono, D.H.; et al. Liver diseases in the Asia-Pacific region: A Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 2020, 5, 167–228. [Google Scholar]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatology 2019. [Google Scholar] [CrossRef]
- Duarte, S.M.B.; Stefano, J.T.; Miele, L.; Ponziani, F.R.; Souza-Basqueira, M.; Okada, L.S.R.R.; de Barros Costa, F.G.; Toda, K.; Mazo, D.F.C.; Sabino, E.C.; et al. Gut microbiome composition in lean patients with NASH is associated with liver damage independent of caloric intake: A prospective pilot study. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Yasutake, K.; Nakamuta, M.; Shima, Y.; Ohyama, A.; Masuda, K.; Haruta, N.; Fujino, T.; Aoyagi, Y.; Fukuizumi, K.; Yoshimoto, T.; et al. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: The significance of dietary cholesterol. Scand. J. Gastroenterol. 2009, 44, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Tang, W.J.; Yin, J.J.; Zhou, B.J. Signal transductions and nonalcoholic fatty liver: A mini-review. Int. J. Clin. Exp. Med. 2014, 7, 1624–1631. [Google Scholar] [PubMed]
- Wafer, R.; Tandon, P.; Minchin, J.E.N. The role of peroxisome proliferator-activated receptor gamma (PPARG) in adipogenesis: Applying knowledge from the fish aquaculture industry to biomedical research. Front. Endocrinol. 2017, 8, 102. [Google Scholar] [CrossRef]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef]
- Lake, A.D.; Chaput, A.L.; Novak, P.; Cherrington, N.J.; Smith, C.L. Transcription factor binding site enrichment analysis predicts drivers of altered gene expression in nonalcoholic steatohepatitis. Biochem. Pharmacol. 2016, 122, 62–71. [Google Scholar] [CrossRef][Green Version]
- Baciu, C.; Pasini, E.; Angeli, M.; Schwenger, K.; Afrin, J.; Humar, A.; Fischer, S.; Patel, K.; Allard, J.; Bhat, M. Systematic integrative analysis of gene expression identifies HNF4A as the central gene in pathogenesis of non-alcoholic steatohepatitis. PLoS ONE 2017, 12, e0189223. [Google Scholar] [CrossRef]
- Lau, H.H.; Ng, N.H.J.; Loo, L.S.W.; Jasmen, J.B.; Teo, A.K.K. The molecular functions of hepatocyte nuclear factors—In and beyond the liver. J. Hepatol. 2018, 68, 1033–1048. [Google Scholar] [CrossRef]
- Bonzo, J.A.; Ferry, C.H.; Matsubara, T.; Kim, J.H.; Gonzalez, F.J. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J. Biol. Chem. 2012, 287, 7345–7356. [Google Scholar] [CrossRef]
- Huck, I.; Gunewardena, S.; Espanol-Suner, R.; Willenbring, H.; Apte, U. Hepatocyte Nuclear Factor 4 Alpha Activation Is Essential for Termination of Liver Regeneration in Mice. Hepatology 2019, 70, 666–681. [Google Scholar] [CrossRef]
- Ni, Q.; Ding, K.; Wang, K.Q.; He, J.; Yin, C.; Shi, J.; Zhang, X.; Xie, W.F.; Shi, Y.Q. Deletion of HNF1α in hepatocytes results in fatty liver-related hepatocellular carcinoma in mice. FEBS Lett. 2017, 591, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, T.; Franken, H.; Kosinski, J.; Kurzawa, N.; Zinn, N.; Sweetman, G.; Poeckel, D.; Ratnu, V.S.; Schramm, M.; Becher, I.; et al. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Li, H.; Singh, A.B.; Cao, A.; Liu, J. Inhibition of PCSK9 transcription by Berberine involves down-regulation of hepatic HNF1α protein expression through the ubiquitin-proteasome degradation pathway. J. Biol. Chem. 2015, 290, 4047–4058. [Google Scholar] [CrossRef] [PubMed]
- Waite, K.J.; Floyd, Z.E.; Arbour-Reily, P.; Stephens, J.M. Interferon-γ-induced Regulation of Peroxisome Proliferator-activated Receptor γ and STATs in Adipocytes. J. Biol. Chem. 2001, 276, 7062–7068. [Google Scholar] [CrossRef]
- Hirano, Y.; Yoshida, M.; Shimizu, M.; Sato, R. Direct Demonstration of Rapid Degradation of Nuclear Sterol Regulatory Element-binding Proteins by the Ubiquitin-Proteasome Pathway. J. Biol. Chem. 2001, 276, 36431–36437. [Google Scholar] [CrossRef]
- Patitucci, C.; Couchy, G.; Bagattin, A.; Cañeque, T.; De Reyniès, A.; Scoazec, J.Y.; Rodriguez, R.; Pontoglio, M.; Zucman-Rossi, J.; Pende, M.; et al. Hepatocyte nuclear factor 1α suppresses steatosisassociated liver cancer by inhibiting PPARγ transcription. J. Clin. Investig. 2017, 127, 1873–1888. [Google Scholar] [CrossRef]
- Kim, J.B.; Wright, H.M.; Wright, M.; Spiegelman, B.M. ADD1/SREBP1 activates PPARγ through the production of endogenous ligand. Proc. Nat. Acad. Sci. USA 1998, 95, 4333–4337. [Google Scholar] [CrossRef]
- Xie, X.; Liao, H.; Dang, H.; Pang, W.; Guan, Y.; Wang, X.; Shyy, J.Y.J.; Zhu, Y.; Sladek, F.M. Down-regulation of Hepatic HNF4α Gene Expression during Hyperinsulinemia via SREBPs. Mol. Endocrinol. 2009, 23, 434–443. [Google Scholar] [CrossRef]
- Odom, D.T.; Dowell, R.D.; Jacobsen, E.S.; Nekludova, L.; Rolfe, P.A.; Danford, T.W.; Gifford, D.K.; Fraenkel, E.; Bell, G.I.; Young, R.A. Core transcriptional regulatory circuitry in human hepatocytes. Mol. Syst. Biol. 2006, 2, 0017. [Google Scholar] [CrossRef]
- Li, J.; Ning, G.; Duncan, S.A. Mammalian hepatocyte differentiation requires the transcription factor HNF-4α. Genes Dev. 2000, 14, 464–474. [Google Scholar]
- Hayhurst, G.P.; Lee, Y.-H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte Nuclear Factor 4 (Nuclear Receptor 2A1) Is Essential for Maintenance of Hepatic Gene Expression and Lipid Homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef]
- Martinez-Jimenez, C.P.; Kyrmizi, I.; Cardot, P.; Gonzalez, F.J.; Talianidis, I. Hepatocyte Nuclear Factor 4 Coordinates a Transcription Factor Network Regulating Hepatic Fatty Acid Metabolism. Mol. Cell. Biol. 2010, 30, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef]
- Pettinelli, P.; Videla, L.A. Up-regulation of PPAR-γ mRNA expression in the liver of obese patients: An additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.; Kwon, J.S.; Sandhu, J.; Tontonoz, P.; Lee, S.K.; Lee, S.; Lee, J.W. Critical Roles of the Histone Methyltransferase MLL4/KMT2D in Murine Hepatic Steatosis Directed by ABL1 and PPARγ2. Cell Rep. 2016, 17, 1671–1682. [Google Scholar] [CrossRef]
- Bahrami-Nejad, Z.; Zhao, M.; Tholen, S.; Hunerdosse, D.; Tkach, K.; van Schie, S.; Chung, M.; Teruel, M. A Transcriptional Circuit Filters Oscillating Circadian Hormonal Inputs to Regulate Fat Cell Differentiation. Cell Metab. 2018, 27, 854–868. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.; Wang, F.; Zhou, S.; Shangguan, M.; Xue, L.; Zhang, B.; Ding, F.; Hui, D.; Liang, A.; et al. Treatment with PPAR α agonist clofibrate inhibits the transcription and activation of srebps and reduces triglyceride and cholesterol levels in liver of broiler chickens. PPAR Res. 2015, 2015, 347245. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Schoonjans, K.; Gelman, L.; Kim, J.B.; Najib, J.; Martin, G.; Fruchart, J.-C.; Briggs, M.; Spiegelman, B.M.; Auwerx, J. Regulation of Peroxisome Proliferator-Activated Receptor γ Expression by Adipocyte Differentiation and Determination Factor 1/Sterol Regulatory Element Binding Protein 1: Implications for Adipocyte Differentiation and Metabolism. Mol. Cell. Biol. 1999, 19, 5495–5503. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Shimano, H.; Nakagawa, Y.; Ide, T.; Yahagi, N.; Matsuzaka, T.; Nakakuki, M.; Takahashi, A.; Suzuki, H.; Sone, H.; et al. SREBP-1 Interacts with Hepatocyte Nuclear Factor-4α and Interferes with PGC-1 Recruitment to Suppress Hepatic Gluconeogenic Genes. J. Biol. Chem. 2004, 279, 12027–12035. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.X.; Huang, S. Understanding gene circuits at cell-fate branch points for rational cell reprogramming. Trends Genet. 2011, 27, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jolly, M.K.; Tripathi, S.C.; Hollander, P.D.; Huang, B.; Lu, M.; Celiktas, M.; Ramirez-Pena, E.; Ben-Jacob, E.; Onuchic, J.N.; et al. Distinguishing Mechanisms Underlying EMT Tristability. Cancer Converg. 2017, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lu, M.; Jia, D.; Ben-Jacob, E.; Levine, H.; Onuchic, J.N. Interrogating the topological robustness of gene regulatory circuits by randomization. PLoS Comput. Biol. 2017, 13, e1005456. [Google Scholar] [CrossRef]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18174–18179. [Google Scholar] [CrossRef]
- Waddington, C.H. The Strategy of the Genes. A Discussion of Some Aspects of Theoretical Biology. With an Appendix by H. Kacser; George Allen and Unwin: London, UK, 1957. [Google Scholar]
- Ellison, A.M. Effect of Seed Dimorphism on the Density-Dependent Dynamics of Experimental Populations of Atriplex triangularis (Chenopodiaceae). Am. J. Bot. 1987, 74, 1280. [Google Scholar] [CrossRef]
- Khurana, S.; Jaiswal, A.K.; Mukhopadhyay, A. Hepatocyte nuclear factor-4α induces transdifferentiation of hematopoietic cells into hepatocytes. J. Biol. Chem. 2010, 285, 4725–4731. [Google Scholar] [CrossRef]
- Mooney, S.M.; Jolly, M.K.; Levine, H.; Kulkarni, P. Phenotypic plasticity in prostate cancer: Role of intrinsically disordered proteins. Asian J. Androl. 2016, 18, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Hybrid T-Helper Cells: Stabilizing the Moderate Center in a Polarized System. PLoS Biol. 2013, 11, e1001632. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Deshmukh, A.; Mani, S.A.; Jolly, M.K.; Levine, H. A possible role for epigenetic feedback regulation in the dynamics of the Epithelial-Mesenchymal Transition (EMT). Phys. Biol. 2019, 16, 066004. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Somarelli, J.A.; Hanash, S.M.; Levine, H. Epithelial-mesenchymal plasticity: How have quantitative mathematical models helped improve our understanding ? Mol. Oncol. 2017, 11, 739–754. [Google Scholar] [CrossRef]
- Huang, B.; Jolly, M.K.; Lu, M.; Tsarfaty, I.; Ben-Jacob, E.; Onuchic, J.N. Modeling the Transitions between Collective and Solitary Migration Phenotypes in Cancer Metastasis. Sci. Rep. 2015, 5, 17379. [Google Scholar] [CrossRef]
- Wheeler, M.C.; Gekakis, N. Hsp90 modulates PPARγ activity in a mouse model of nonalcoholic fatty liver disease. J. Lipid Res. 2014, 55, 1702–1710. [Google Scholar] [CrossRef]
- Dhooge, A.; Govaerts, W.; Kuznetsov, Y.A.; Meijer, H.G.E.; Sautois, B. New features of the software MatCont for bifurcation analysis of dynamical systems. Math. Comput. Model. Dyn. Syst. 2008, 14, 147–175. [Google Scholar] [CrossRef]
- Lakhani, H.V.; Sharma, D.; Dodrill, M.W.; Nawab, A.; Sharma, N.; Cottrill, C.L.; Shapiro, J.I.; Sodhi, K. Phenotypic alteration of hepatocytes in non-alcoholic fatty liver disease. Int. J. Med. Sci. 2018, 15, 1591–1599. [Google Scholar] [CrossRef]
- Carpino, G.; Renzi, A.; Onori, P.; Gaudio, E. Role of hepatic progenitor cells in nonalcoholic fatty liver disease development: Cellular cross-talks and molecular networks. Int. J. Mol. Sci. 2013, 14, 20112–20130. [Google Scholar] [CrossRef]
- Balázsi, G.; van Oudenaarden, A.; Collins, J.J.; Van Oudenaarden, A.; Collins, J.J. Cellular decision making and biological noise: From microbes to mammals. Cell 2011, 144, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.S.; Saraf, N.; Saigal, S.; Gautam, D.; Lipi, L.; Rastogi, A.; Goja, S.; Menon, P.B.; Bhangui, P.; Ramchandra, S.K.; et al. Rapid Reversal of Liver Steatosis With Life Style Modification in Highly Motivated Liver Donors. J. Clin. Exp. Hepatol. 2015, 5, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Lin, J. Divergence Measures Based on the Shannon Entropy. IEEE Trans. Inf. Theory 1991, 37, 145–151. [Google Scholar] [CrossRef]
- Hari, K.; Sabuwala, B.; Subramani, B.V.; Porta, C.L.; Zapperi, S.; Font-Clos, F.; Jolly, M.K. Identifying inhibitors of epithelial-mesenchymal plasticity using a network topology based approach. bioRxiv 2019, 854307. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and epigenetic regulation in nonalcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef]
- Ma, M.; Duan, R.; Zhong, H.; Liang, T.; Guo, L. The crosstalk between fat homeostasis and liver regional immunity in NAFLD. J. Immunol. Res. 2019, 2019, 10. [Google Scholar] [CrossRef]
- Blencowe, M.; Karunanayake, T.; Wier, J.; Hsu, N.; Yang, X. Network Modeling Approaches and Applications to Unravelling Non-Alcoholic Fatty Liver Disease. Genes (Basel). 2019, 10, 966. [Google Scholar] [CrossRef]
- Shubham, K.; Vinay, L.; Vinod, P.K. Systems-level organization of non-alcoholic fatty liver disease progression network. Mol. Biosyst. 2017, 13, 1898–1911. [Google Scholar] [CrossRef]
- Ashworth, W.B.; Davies, N.A.; Bogle, I.D.L. A Computational Model of Hepatic Energy Metabolism: Understanding Zonated Damage and Steatosis in NAFLD. PLOS Comput. Biol. 2016, 12, e1005105. [Google Scholar] [CrossRef]
- Maldonado, E.M.; Fisher, C.P.; Mazzatti, D.J.; Barber, A.L.; Tindall, M.J.; Plant, N.J.; Kierzek, A.M.; Moore, J.B. Multi-scale, whole-system models of liver metabolic adaptation to fat and sugar in non-alcoholic fatty liver disease. npj Syst. Biol. Appl. 2018, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Sookoian, S. Tackling the complexity of nonalcoholic steatohepatitis treatment: Challenges and opportunities based on systems biology and machine learning approaches. HepatoBiliary Surg. Nutr. 2018, 7, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genetic Insights for Drug Development in NAFLD. Trends Pharmacol. Sci. 2019, 40, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Sookoian, S. Multiomics biomarkers for the prediction of nonalcoholic fatty liver disease severity. World J. Gastroenterol. 2018, 24, 1601–1615. [Google Scholar] [CrossRef]
- Guantes, R.; Poyatos, J.F. Multistable decision switches for flexible control of epigenetic differentiation. PLoS Comput. Biol. 2008, 4, e1000235. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Lu, M.; Onuchic, J.; Clementi, C.; Ben-Jacob, E. Operating principles of Notch–Delta–Jagged module of cell–cell communication. New J. Phys. 2015, 17. [Google Scholar] [CrossRef]
- Ozbudak, E.M.; Thattai, M.; Lim, H.N.; Shraiman, B.I.; van Oudenaarden, A. Multistability in the lactose utilization network of Escherichia coli. Nature 2004, 427, 737–740. [Google Scholar] [CrossRef]
- Jolly, M.K.; Celia-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J Clin Med 2019, 8, 1542. [Google Scholar] [CrossRef]
- Parafati, M.; Kirby, R.J.; Khorasanizadeh, S.; Rastinejad, F.; Malany, S. A nonalcoholic fatty liver disease model in human induced pluripotent stem cell-derived hepatocytes, created by endoplasmic reticulum stress-induced steatosis. DMM Dis. Model. Mech. 2018, 11, dmm033530. [Google Scholar] [CrossRef]
- Tripathi, S.; Chakraborty, P.; Levine, H.; Jolly, M.K. A mechanism for epithelial-mesenchymal heterogeneity in a population of cancer cells. PLoS Comput Biol 2020, 16, e1007619. [Google Scholar] [CrossRef]
- Farrell, G.C.; Van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7, S771–S781. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, T.; Maniyadath, B.; Bagul, H.P.; Chakraborty, A.; Shukla, N.; Budnar, S.; Rajendran, A.; Shukla, A.; Kamat, S.S.; Kolthur-Seetharam, U. Spatiotemporal gating of SIRT1 functions by O-GlcNAcylation is essential for liver metabolic switching and prevents hyperglycemia. Proc. Nat. Acad. Sci. USA 2020. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Bashmakov, Y.; Shimomura, I.; Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Nat. Acad. Sci. USA 1998, 95, 5987–5992. [Google Scholar] [CrossRef] [PubMed]
- Billon, N.; Dani, C. Developmental Origins of the Adipocyte Lineage: New Insights from Genetics and Genomics Studies. Stem Cell Rev. Reports 2012, 8, 55–66. [Google Scholar] [CrossRef]
- Zaret, K.S. Hepatocyte differentiation: From the endoderm and beyond. Curr. Opin. Genet. Dev. 2001, 11, 568–574. [Google Scholar] [CrossRef]
- Li, H.; Zhu, L.; Chen, H.; Li, T.; Han, Q.; Wang, S.; Yao, X.; Feng, H.; Fan, L.; Gao, S.; et al. Generation of Functional Hepatocytes from Human Adipose-Derived MYC+ KLF4+ GMNN+ Stem Cells Analyzed by Single-Cell RNA-Seq Profiling. Stem Cells Transl. Med. 2018, 7, 792–805. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. MiRNA signature in NAFLD: A turning point for a non-invasive diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef]
- Cheng, Y.; Hou, T.; Ping, J.; Chen, G.; Chen, J. Quantitative succinylome analysis in the liver of non-alcoholic fatty liver disease rat model. Proteome Sci. 2016, 14, 3. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Production Rate (106 molecules hr−1) | Degradation Rate (hr−1) | References |
|---|---|---|---|
| HNF4A | 0.4540 | 0.0491 | [22] |
| HNF1A | 0.0570 | 0.0494 | [23] |
| PPARG | 0.0628 | 0.08 | [24] |
| SREBF1 | 1.1058 | 0.1598 | [25] |
| Description | Fold Change | Value | # of binding sites | Value | Threshold | Value | Reference (est: Estimated) |
|---|---|---|---|---|---|---|---|
| Self-Activation of HNF4A | 4 | 4 | 3 | est | |||
| Self-Activation of HNF1A | 2 | 4 | 0.8 | est | |||
| Self-Activation of PPARG | 9.514 | 5 | 6.320 | est | |||
| Self-Activation of SREBF1 | 4 | 2 | 5.25 | est | |||
| Activation of HNF4A by HNF1A | 4 | 3 | 0.8 | est | |||
| Activation of HNF1A by HNF4A | 5.328 | 4 | 5.108 | est, [26] | |||
| Activation of PPARG by SREBF1 | 3 | 2 | 5.25 | est, [27] | |||
| Activation of SREBF1 by PPARG | 3.729 | 2 | 9.283 | est | |||
| Inhibition of HNF4A by SREBF1 | 0.415 | 2 | 5 | [28] | |||
| Inhibition of PPARG by HNF1A | 0.68 | 4 | 0.674 | [26] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahoo, S.; Singh, D.; Chakraborty, P.; Jolly, M.K. Emergent Properties of the HNF4α-PPARγ Network May Drive Consequent Phenotypic Plasticity in NAFLD. J. Clin. Med. 2020, 9, 870. https://doi.org/10.3390/jcm9030870
Sahoo S, Singh D, Chakraborty P, Jolly MK. Emergent Properties of the HNF4α-PPARγ Network May Drive Consequent Phenotypic Plasticity in NAFLD. Journal of Clinical Medicine. 2020; 9(3):870. https://doi.org/10.3390/jcm9030870
Chicago/Turabian StyleSahoo, Sarthak, Divyoj Singh, Priyanka Chakraborty, and Mohit Kumar Jolly. 2020. "Emergent Properties of the HNF4α-PPARγ Network May Drive Consequent Phenotypic Plasticity in NAFLD" Journal of Clinical Medicine 9, no. 3: 870. https://doi.org/10.3390/jcm9030870
APA StyleSahoo, S., Singh, D., Chakraborty, P., & Jolly, M. K. (2020). Emergent Properties of the HNF4α-PPARγ Network May Drive Consequent Phenotypic Plasticity in NAFLD. Journal of Clinical Medicine, 9(3), 870. https://doi.org/10.3390/jcm9030870