Ponatinib Induces Vascular Toxicity through the Notch-1 Signaling Pathway


 ,
,  , ,
, ,  , ,
, ,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
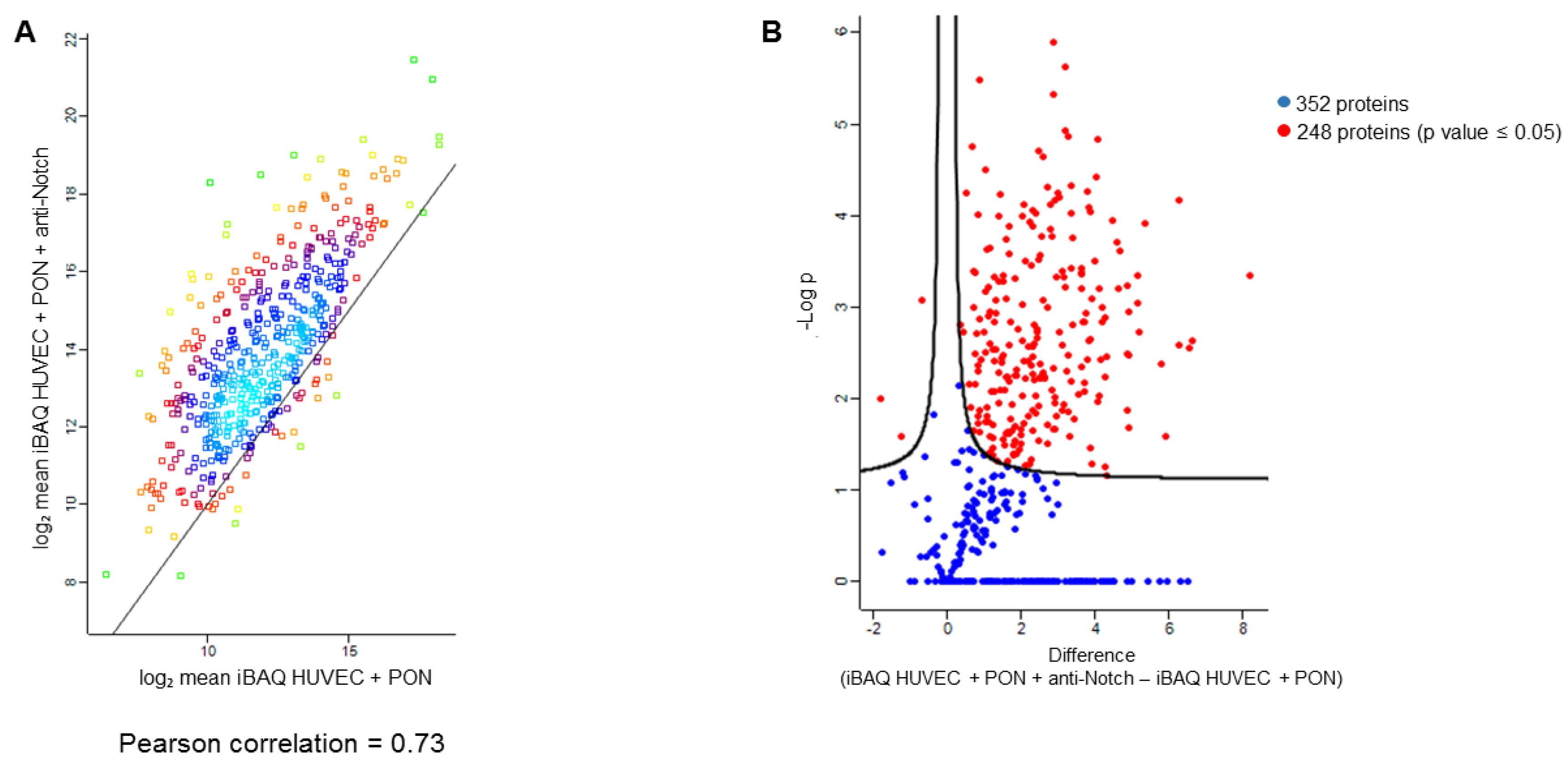{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. EPC Isolation from Peripheral Blood Mononuclear Cells
2.3. CFU-EC Isolation and Quantification
2.4. Cell Culture Treatments
2.5. Cell Proliferation Assay
2.6. Label Free Proteomics
2.7. Proteomics Data Processing
2.8. Tube Formation Assay
2.9. Senescence-Associated β-galactosidase Assay
2.10. Immunoblotting
2.11. Statistical Analysis
3. Results
3.1. Ponatinib Reduces the Viability of Endothelial Cell and Peripheral Blood Mononuclear Cells
3.2. Ponatinib Decreases Pro-Angiogenic Endothelial Function In Vitro
3.3. Ponatinib Induces Endothelial Senescence and Apoptosis and Reduces Endothelial Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Bovine Serum Albumin | BSA |
| Chronic myeloid leukemia | CML |
| Colony forming units-endothelial cells | CFU-ECs |
| Coronary artery disease | CAD |
| Dimethyl sulfoxide | DMSO |
| Endothelial progenitor cells | EPCs |
| Endothelial nitric oxide kinase | eNOS |
| Epidermal growth factor receptor | EGFR |
| Fibroblast growth factor | FGF |
| Filter Aided Sample Preparation | FASP |
| Human umbilical endothelial cells | HUVECs |
| Ice-cold RadioImmuno Precipitation Assay | RIPA |
| Ingenuity Pathway Analysis | IPA |
| Intensity-based absolute quantification | iBAQ |
| Intercellular adhesion molecular | ICAM-1 |
| Liquid chromatography tandem mass spectrometry | LC-MS/MS |
| NF-kappa-B inhibitor alpha | NFKBIA |
| Nitric oxide | NO |
| Peripheral arterial obstructive disease | PAOD |
| Peripheral blood | PB |
| Peripheral blood mononuclear cells | PBMNCs |
| Phosphate Buffered Saline | PBS |
| Platelet-derived growth factors | PDGF |
| Senescence-associated β-galactosidase | SA β-gal |
| Transforming Growth Factor-Beta1 | TGFB1 |
| Tyrosine kinase inhibitor | TKI |
| Vascular endothelial growth factor | VEGF |
| Vascular adverse events | VAEs |
| Vascular cell adhesion molecule | VCAM-1 |
Appendix A

References
- Kalidas, M.; Kantarjian, H.; Talpaz, M. Chronic myelogenous leukemia. JAMA 2001, 286, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Hehlmann, R. CML—Where do we stand in 2015? Ann. Hematol. 2015, 94, S103–S105. [Google Scholar] [CrossRef] [PubMed]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94, S107–S121. [Google Scholar] [CrossRef] [PubMed]
- Haberbosch, I.; Rafiei, A.; Oancea, C.; Ottmann, G.O.; Ruthardt, M.; Mian, A.A. BCR: A new target in resistance mediated by BCR/ABL-315I? Genes Cancer 2016, 7, 36–46. [Google Scholar] [PubMed]
- Shanmuganathan, N.; Hiwase, D.K.; Ross, D.M. Treatment of chronic myeloid leukemia: Assessing risk, monitoring response, and optimizing outcome. Leuk. Lymphoma 2017, 58, 2799–2810. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.X. Discontinuation of tyrosine kinase therapy in CML. Ann. Hematol. 2015, 94, S187–S193. [Google Scholar] [CrossRef]
- Tournaire, G.; Despas, F.; Huguet, F.; Montastruc, J.L.; Bondon-Guitton, E. Peripheral arterial occlusive disease during ponatinib therapy after failure of imatinib: A case report. J. Clin. Pharm. Ther. 2016, 41, 360–361. [Google Scholar] [CrossRef]
- Caldemeyer, L.; Dugan, M.; Edwards, J.; Akard, L. Long-Term Side Effects of Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2016, 11, 71–79. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.; Jabbour, E.; Gonzalez, G.N.; Borthakur, G.; Pemmaraju, N.; Daver, N.; Gachimova, E.; Ferrajoli, A.; Kornblau, S.; et al. Ponatinib as first-line treatment for patients with chronic myeloid leukaemia in chronic phase: A phase 2 study. Lancet Haematol. 2015, 2, e376–e383. [Google Scholar] [CrossRef]
- Moslehi, J.J.; Deininger, M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 4210–4218. [Google Scholar] [CrossRef]
- Cortes, J.; Digumarti, R.; Parikh, P.; Wetzler, M.; Lipton, J.; Hochhaus, A.; Craig, A.; Benichou, A.-C.; Nicolini, F.; Kantarjian, H.; et al. Phase 2 study of subcutaneous omacetaxine mepesuccinate for chronic-phase chronic myeloid leukemia patients resistant to or intolerant of tyrosine kinase inhibitors. Am. J. Hematol. 2013, 88, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Ibrahim, A.R.; Soverini, S.; Martinelli, G.; Müller, M.C.; Hochhaus, A.; Dufva, I.H.; Kim, N.-W.; Cortes, J.; Mauro, M.J.; et al. The BCR-ABLT315I mutation compromises survival in chronic phase chronic myelogenous leukemia patients resistant to tyrosine kinase inhibitors, in a matched pair analysis. Haematologica 2013, 98, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Hadzijusufovic, E.; Albrecht-Schgoer, K.; Huber, K.V.M.; Hoermann, G.; Grebien, F.; Eisenwort, G.; Schgoer, W.; Herndlhofer, S.; Kaun, C.; Theurl, M.; et al. Nilotinib-induced vasculopathy: Identification of vascular endothelial cells as a primary target site. Leukemia 2017, 31, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; Raj, K. The role of Notch in tumorigenesis: Oncogene or tumour suppressor? Nat. Rev. Cancer 2003, 3, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Guentchev, M.; McKay, R.D. Notch controls proliferation and differentiation of stem cells in a dose-dependent manner. Eur. J. Neurosci. 2006, 23, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Pandolfi, A.; Massaro, M.; Consoli, A.; De Caterina, R. Insulin enhances vascular cell adhesion molecule-1 expression in human cultured endothelial cells through a pro-atherogenic pathway mediated by p38 mitogen-activated protein-kinase. Diabetologia 2004, 47, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.; Falk, A.; Dyson, M.; Melidoni, A.; Parthiban, K.; Young, J.L.; Roake, W.; McCafferty, J. Generation of anti-Notch antibodies and their application in blocking Notch signaling in neural stem cells. Methods 2012, 58, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; D’Alessandro, M.; Di Ioia, M.; Rossi, C.; Zucchelli, M.; Urbani, A.; Di Ilio, C.; Lugaresi, A.; Sacchetta, P.; Del Boccio, P. An integrated metabolomics approach for the research of new cerebrospinal fluid biomarkers of multiple sclerosis. Mol. Biosyst. 2015, 11, 1563–1572. [Google Scholar] [CrossRef]
- Pieragostino, D.; Agnifili, L.; Fasanella, V.; D’Aguanno, S.; Mastropasqua, R.; Di Ilio, C.; Sacchetta, P.; Urbani, A.; Del Boccio, P. Shotgun proteomics reveals specific modulated protein patterns in tears of patients with primary open angle glaucoma naive to therapy. Mol. Biosyst. 2013, 9, 1108–1116. [Google Scholar] [CrossRef]
- Pieragostino, D.; Lanuti, P.; Cicalini, I.; Cufaro, M.C.; Ciccocioppo, F.; Ronci, M.; Simeone, P.; Onofrj, M.; Van Der Pol, E.; Fontana, A.; et al. Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J. Proteom. 2019, 204, 103403. [Google Scholar] [CrossRef]
- Madonna, R.; De Caterina, R. Circulating endothelial progenitor cells: Do they live up to their name? Vasc. Pharmacol. 2015, 67, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Van der Loo, B.; Fenton, M.J.; Erusalimsky, J.D. Cytochemical detection of a senescence-associated beta-galactosidase in endothelial and smooth muscle cells from human and rabbit blood vessels. Exp. Cell Res. 1998, 241, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Rea, D.; Schwarz, M.; Grille, P.; Nicolini, F.E.; Rosti, G.; Levato, L.; Giles, F.J.; Dombret, H.; Mirault, T.; et al. Peripheral artery occlusive disease in chronic phase chronic myeloid leukemia patients treated with nilotinib or imatinib. Leukemia 2013, 27, 1316–1321. [Google Scholar] [CrossRef]
- Alivon, M.; Giroux, J.; Briet, M.; Goldwasser, F.; Laurent, S.; Boutouyrie, P. Large artery stiffness and hypertension after antiangiogenic drugs: Influence on cancer progression. J. Hypertens. 2015, 33, 1310–1317. [Google Scholar] [CrossRef]
- Zangari, M.; Fink, L.M.; Elice, F.; Zhan, F.; Adcock, D.M.; Tricot, G.J. Thrombotic events in patients with cancer receiving antiangiogenesis agents. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4865–4873. [Google Scholar] [CrossRef]
- Ghatalia, P.; Morgan, C.J.; Je, Y.; Nguyen, P.L.; Trinh, Q.-D.; Choueiri, T.K.; Sonpavde, G. Congestive heart failure with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit. Rev. Oncol. /Hematol. 2015, 94, 228–237. [Google Scholar] [CrossRef]
- Gover-Proaktor, A.; Granot, G.; Pasmanik-Chor, M.; Pasvolsky, O.; Shapira, S.; Raz, O.; Raanani, P.; Leader, A. Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib differentially affect the vascular molecular pathways and functionality of human endothelial cells. Leuk. Lymphoma 2019, 60, 189–199. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, J.; Ji, C. The emerging roles of Notch signaling in leukemia and stem cells. Biomark. Res. 2013, 1, 23. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Madonna, R.; Melino, G.; Caruso, C. The emerging role of Notch pathway in ageing: Focus on the related mechanisms in age-related diseases. Ageing Res. Rev. 2016, 29, 50–65. [Google Scholar] [CrossRef]
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madonna, R.; Pieragostino, D.; Cufaro, M.C.; Doria, V.; Del Boccio, P.; Deidda, M.; Pierdomenico, S.D.; Dessalvi, C.C.; De Caterina, R.; Mercuro, G. Ponatinib Induces Vascular Toxicity through the Notch-1 Signaling Pathway. J. Clin. Med. 2020, 9, 820. https://doi.org/10.3390/jcm9030820
Madonna R, Pieragostino D, Cufaro MC, Doria V, Del Boccio P, Deidda M, Pierdomenico SD, Dessalvi CC, De Caterina R, Mercuro G. Ponatinib Induces Vascular Toxicity through the Notch-1 Signaling Pathway. Journal of Clinical Medicine. 2020; 9(3):820. https://doi.org/10.3390/jcm9030820
Chicago/Turabian StyleMadonna, Rosalinda, Damiana Pieragostino, Maria Concetta Cufaro, Vanessa Doria, Piero Del Boccio, Martino Deidda, Sante Donato Pierdomenico, Christian Cadeddu Dessalvi, Raffaele De Caterina, and Giuseppe Mercuro. 2020. "Ponatinib Induces Vascular Toxicity through the Notch-1 Signaling Pathway" Journal of Clinical Medicine 9, no. 3: 820. https://doi.org/10.3390/jcm9030820
APA StyleMadonna, R., Pieragostino, D., Cufaro, M. C., Doria, V., Del Boccio, P., Deidda, M., Pierdomenico, S. D., Dessalvi, C. C., De Caterina, R., & Mercuro, G. (2020). Ponatinib Induces Vascular Toxicity through the Notch-1 Signaling Pathway. Journal of Clinical Medicine, 9(3), 820. https://doi.org/10.3390/jcm9030820