Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective

Abstract
1. Introduction
2. Inflammation and Immunity in PAH and CTEPH
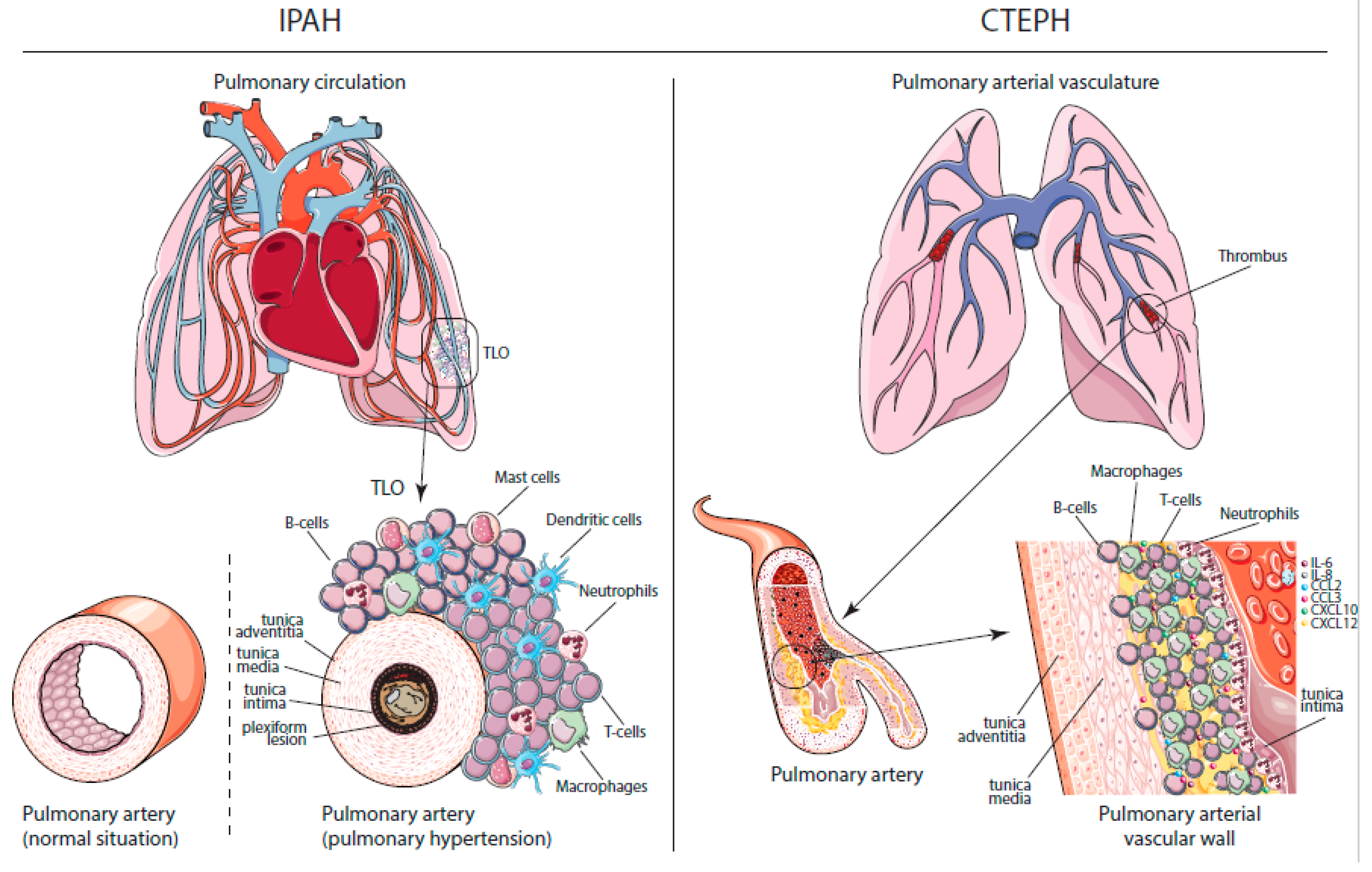
2.1. Histopathology in PAH and CTEPH
2.2. Dysregulated Immune Responses in PAH and CTEPH
2.2.1. Innate Immunity
Macrophages
Neutrophils
Mast Cells
Natural Killer Cells
2.2.2. Linking Innate and Adaptive Immunity
Dendritic Cells
2.2.3. Adaptive Immunity
T Cells
B Cells and Humoral Immune Responses
3. Inflammatory Diagnostic and Prognostic Biomarkers in PAH and CTEPH
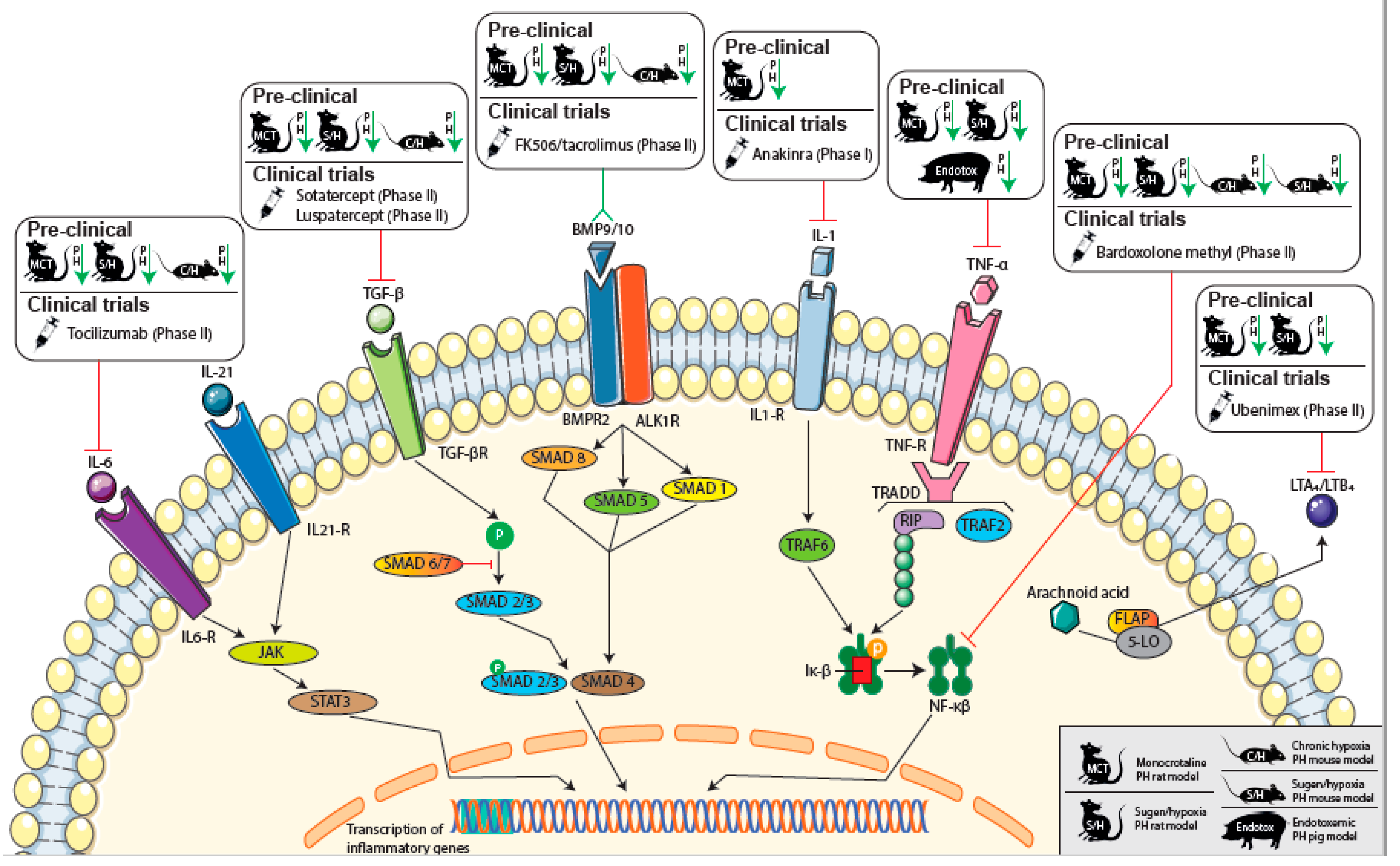
4. Immunomodulatory Therapy in PAH and CTEPH
5. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ang-1 | Angiopoietin 1 |
| APAH | associated PAH |
| BPA | Balloon pulmonary angioplasty |
| BMPR2 | Bone morphogenetic protein receptor type II |
| BMPs | Bone morphogenetic proteins |
| tacrolimus | Calcineurin inhibitor FK506 |
| CCL | Chemokine C-C motif ligand |
| CXCL | Chemokine C-X3-C motif ligand |
| CTEPH | Chronic thromboembolic pulmonary hypertension |
| CHD | Congenital heart disease |
| CTD-PAH | Connective tissue disease associated PAH |
| DC-SIGN | Dendritic cell- specific intercellular adhesion molecule-3 grabbing non- integrin |
| DCs | Dendritic cells |
| DMF | Dimethyl fumarate |
| Tfh | Follicular T-helper cells |
| FC | Functional class |
| γδ | Gamma delta |
| HPAH | Heritable PAH |
| TCRs | Heterodimeric T-cell receptors |
| HIF-1α | Hypoxia-inducible factor-1α |
| IPAH | Idiopathic PAH |
| IL | Interleukin |
| LTB4 | Leukotriene B4 |
| CCL3 | Macrophage inflammatory protein-1α |
| MHC | Major histocompatibility complex |
| MCs | Mast cells |
| MMP-9 | Matrix metallopeptidase 9 |
| MCTD | Mixed connective tissue diseases |
| MCT | Monocrotaline |
| mo-DCs | Monocyte-derived DCs |
| MPO | Myeloperoxidase |
| NIAD | National Institute of Allergy and Infectious Diseases |
| NK | Natural killer cells |
| NYHA | New York Heart Association |
| NF-κβ | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFAT | Nuclear factor of activated T cell |
| Porto-pulmonary PAH | PAH due to liver disease |
| PPHN | Persistent pulmonary hypertension of the newborn |
| PlGF | Placental growth factor |
| pDCs | Plasmacytoid DCs |
| PAH | Pulmonary arterial hypertension |
| PAP | Pulmonary arterial pressures |
| PASMCs | Pulmonary arterial smooth muscle cells |
| PEA | Pulmonary endarterectomy |
| PH | Pulmonary hypertension |
| PVR | Pulmonary vascular resistance |
| PVOD | Pulmonary veno-occlusive disease |
| ROS | Reactive oxygen species |
| RELMα | Resistin-like molecule-α |
| RV | Right ventricular |
| RVH | Right ventricular hypertrophy |
| SU/Hx | Sugen/hypoxia |
| SLE | Systemic lupus erythematosus |
| SSc | Systemic sclerosis |
| TLOs | Tertiary lymphoid organs |
| Th | T-helper cell |
| TSP-1 | Thrombospondin-1 |
| TNFAIP3 | TNF Alpha Induced Protein 3 |
| NRF2 | Transcriptional regulator nuclear factor erythroid 2-related factor 2 |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| cDC1s | Type 1 conventional DCs |
| cDC2s | Type 2 cDCs |
| VEGF | Vascular endothelial growth factor |
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar]
- Southgate, L.; Machado, R.D.; Graf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2019, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Garg, L.; Akbar, G.; Agrawal, S.; Agarwal, M.; Khaddour, L.; Handa, R.; Garg, A.; Shah, M.; Patel, B.; Dalal, B.D. Drug-induced pulmonary arterial hypertension: A Review. Heart Fail. Rev. 2017, 22, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, A.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Radegran, G.; Kjellstrom, B.; Ekmehag, B.; Larsen, F.; Rundqvist, B.; Blomquist, S.B.; Gustafsson, C.; Hesselstrand, R.; Karlsson, M.; Kornhall, B.; et al. Characteristics and survival of adult Swedish PAH and CTEPH patients 2000–2014. Scand. Cardiovasc. J. 2016, 50, 243–250. [Google Scholar] [CrossRef]
- Quadery, S.R.; Swift, A.J.; Billings, C.G.; Thompson, A.A.R.; Elliot, C.A.; Hurdman, J.; Charalampopoulos, A.; Sabroe, I.; Armstrong, I.J.; Hamilton, N.; et al. The impact of patient choice on survival in chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Boucly, A.; Weatherald, J.; Savale, L.; Jais, X.; Cottin, V.; Prevot, G.; Picard, F.; de Groote, P.; Jevnikar, M.; Bergot, E.; et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Matthews, D.T.; Hemnes, A.R. Current concepts in the pathogenesis of chronic thromboembolic pulmonary hypertension. Pulm. Circ. 2016, 6, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Bonderman, D.; Jakowitsch, J.; Adlbrecht, C.; Schemper, M.; Kyrle, P.A.; Schonauer, V.; Exner, M.; Klepetko, W.; Kneussl, M.P.; Maurer, G.; et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2005, 93, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Chazova, I.; Loyd, J.E.; Zhdanov, V.S.; Newman, J.H.; Belenkov, Y.; Meyrick, B. Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am. J. Pathol. 1995, 146, 389–397. [Google Scholar] [PubMed]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Wagenvoort, C.A. Pathology of pulmonary thromboembolism. Chest 1995, 107, 10S–17S. [Google Scholar] [CrossRef]
- Lang, I.M.; Dorfmuller, P.; Noordegraaf, A.V. The Pathobiology of Chronic Thromboembolic Pulmonary Hypertension. Ann. Am. Thorac. Soc. 2016, 13, S215–S221. [Google Scholar] [CrossRef]
- Perros, F.; Dorfmüller, P.; Montani, D.; Hammad, H.; Waelput, W.; Girerd, B.; Raymond, N.; Mercier, O.; Mussot, S.; Cohen-Kaminsky, S.; et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 311–321. [Google Scholar] [CrossRef]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Quarck, R.; Wynants, M.; Verbeken, E.; Meyns, B.; Delcroix, M. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2015, 46, 431–443. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Tan, S.Y.; Krasnow, M.A. Developmental origin of lung macrophage diversity. Development 2016, 143, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv 2019. [Google Scholar] [CrossRef]
- Pugliese, S.C.; Kumar, S.; Janssen, W.J.; Graham, B.B.; Frid, M.G.; Riddle, S.R.; El Kasmi, K.C.; Stenmark, K.R. A Time- and Compartment-Specific Activation of Lung Macrophages in Hypoxic Pulmonary Hypertension. J. Immunol. 2017, 198, 4802–4812. [Google Scholar] [CrossRef]
- Franke-Ullmann, G.; Pförtner, C.; Walter, P.; Steinmüller, C.; Lohmann-Matthes, M.L.; Kobzik, L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J. Immunol. 1996, 157, 3097–3104. [Google Scholar]
- Bedoret, D.; Wallemacq, H.; Marichal, T.; Desmet, C.; Quesada Calvo, F.; Henry, E.; Closset, R.; Dewals, B.; Thielen, C.; Gustin, P.; et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Investig. 2009, 119, 3723–3738. [Google Scholar] [CrossRef]
- Laskin, D.L.; Weinberger, B.; Laskin, J.D. Functional heterogeneity in liver and lung macrophages. J. Leukoc. Biol. 2001, 70, 163–170. [Google Scholar]
- Florentin, J.; Coppin, E.; Vasamsetti, S.B.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Zhang, Y.; Watson, A.; Sembrat, J.; Rojas, M.; et al. Inflammatory Macrophage Expansion in Pulmonary Hypertension Depends upon Mobilization of Blood-Borne Monocytes. J. Immunol. 2018, 200, 3612–3625. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, O.; Marcos, E.; Perros, F.; Fadel, E.; Tu, L.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Adnot, S.; Eddahibi, S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2007, 176, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.; Marcos, E.; Parpaleix, A.; Amsellem, V.; Breau, M.; Houssaini, A.; Vienney, N.; Lefevre, M.; Derumeaux, G.; Evans, S.; et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur. Respir. J. 2019, 54, 1802308. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Kassa, B.; Sanders, L.; Hernandez-Saavedra, D.; E Koyanagi, D.; Kumar, S.; Pugliese, S.C.; Thomas, S.; McClendon, J.; et al. Interstitial Macrophage-Derived Thrombospondin-1 Contributes to Hypoxia-Induced Pulmonary Hypertension. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.D.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, A.; Kourembanas, S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef]
- Frid, M.G.; Brunetti, J.A.; Burke, D.L.; Carpenter, T.C.; Davie, N.J.; Reeves, J.T.; Roedersheimer, M.T.; Van Rooijen, N.; Stenmark, K.R. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006, 168, 659–669. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Tamosiuniene, R.; Sung, Y.K.; Qian, J.; Dhillon, G.; Gera, L.; Farkas, L.; Rabinovitch, M.; Zamanian, R.T.; et al. Bocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci. Transl. Med. 2013, 5, 200ra117. [Google Scholar] [CrossRef]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef]
- Kojima, H.; Tokunou, T.; Takahara, Y.; Sunagawa, K.; Hirooka, Y.; Ichiki, T.; Tsutsui, H. Hypoxia-inducible factor-1 alpha deletion in myeloid lineage attenuates hypoxia-induced pulmonary hypertension. Physiol. Rep. 2019, 7, e14025. [Google Scholar] [CrossRef]
- Marsh, L.M.; Jandl, K.; Grunig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1701214. [Google Scholar] [CrossRef]
- Lin, Q.; Fan, C.; Skinner, J.T.; Hunter, E.N.; Macdonald, A.A.; Illei, P.B.; Yamaji-Kegan, K.; Johns, R.A. RELMalpha Licenses Macrophages for Damage-Associated Molecular Pattern Activation to Instigate Pulmonary Vascular Remodeling. J. Immunol. 2019, 203, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Heinemann, A.; Foris, V.; Nagaraj, C.; Nierlich, P.; Balint, Z.; Kwapiszewska, G.; Lang, I.M.; Klepetko, W.; Olschewski, H.; et al. Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur. Respir. J. 2014, 44, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Florentin, J.; Dutta, P. Origin and production of inflammatory perivascular macrophages in pulmonary hypertension. Cytokine 2017, 100, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Savai, R. Macrophage Regulation during Vascular Remodeling: Implications for Pulmonary Hypertension Therapy. Am. J. Respir. Cell Mol. Biol. 2017, 56, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Ozpelit, E.; Akdeniz, B.; Özpelit, M.E.; Tas, S.; Bozkurt, S.; Tertemiz, K.C.; Sevinç, C.; Badak, Ö. Prognostic value of neutrophil-to-lymphocyte ratio in pulmonary arterial hypertension. J. Int. Med. Res. 2015, 43, 661–671. [Google Scholar] [CrossRef]
- Harbaum, L.; Baaske, K.M.; Simon, M.; Oqueka, T.; Sinning, C.; Glatzel, A.; Lüneburg, N.; Sydow, K.; Bokemeyer, C.; Klose, H. Exploratory analysis of the neutrophil to lymphocyte ratio in patients with pulmonary arterial hypertension. BMC Pulm. Med. 2017, 17, 72. [Google Scholar] [CrossRef]
- Yanartaş, M.; Kalkan, M.E.; Arslan, A.; Tas, S.G.; Koksal, C.; Bekiroğlu, N.; Yildizeli, B. Neutrophil/Lymphocyte Ratio Can Predict Postoperative Mortality in Patients with Chronic Thromboembolic Pulmonary Hypertension. Ann. Thorac. Cardiovasc. Surg. 2015, 21, 229–235. [Google Scholar] [CrossRef][Green Version]
- Schultze, A.E.; Wagner, J.G.; White, S.M.; Roth, R.A. Early indications of monocrotaline pyrrole-induced lung injury in rats. Toxicol. Appl. Pharmacol. 1991, 109, 41–50. [Google Scholar] [CrossRef]
- Klinke, A.; Berghausen, E.; Friedrichs, K.; Molz, S.; Lau, D.; Remane, L.; Berlin, M.; Kaltwasser, C.; Adam, M.; Mehrkens, D.; et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight 2018, 3, e97530. [Google Scholar] [CrossRef]
- Theoharides, T.C. Mast cells and pancreatic cancer. N. Engl. J. Med. 2008, 358, 1860–1861. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Ueda, M.; Maruyama, K.; Shimpo, H.; Kojima, A.; Matsumura, M.; Aoki, K.; Sakurai, M. Mast cell chymase in pulmonary hypertension. Thorax 1999, 54, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Heath, D.; Yacoub, M. Lung mast cells in plexogenic pulmonary arteriopathy. J. Clin. Pathol. 1991, 44, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, A.M.; Rivera, J. The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 2009, 228, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Yin, J.; Kukucka, M.; Yin, N.; Saarikko, I.; Sterner-Kock, A.; Fujii, H.; Leong-Poi, H.; Kuppe, H.; Schermuly, R.T.; et al. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur. Respir. J. 2011, 37, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Bartelds, B.; Van Loon, R.L.E.; Mohaupt, S.; Wijnberg, H.; Dickinson, M.G.; Boersma, B.; Takens, J.; Van Albada, M.; Berger, R.M. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest 2012, 141, 651–660. [Google Scholar] [CrossRef]
- Dahal, B.K.; Kosanovic, D.; Kaulen, C.; Cornitescu, T.; Savai, R.; Hoffmann, J.; Reiss, I.K.M.; Ghofrani, A.; Weissmann, N.; Kuebler, W.M.; et al. Involvement of mast cells in monocrotaline-induced pulmonary hypertension in rats. Respir. Res. 2011, 12, 60. [Google Scholar] [CrossRef]
- Banasova, A.; Maxova, H.; Hampl, V.; Vizek, M.; Povysilova, V.; Novotna, J.; Vajnerova, O.; Hnilickova, O.; Herget, J. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration 2008, 76, 102–107. [Google Scholar]
- Farha, S.; Sharp, J.; Asosingh, K.; Park, M.; Comhair, S.A.; Tang, W.H.; Thomas, J.; Farver, C.; Hsieh, F.; Loyd, J.E.; et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm. Circ. 2012, 2, 220–228. [Google Scholar] [CrossRef]
- Hamada, H.; Terai, M.; Kimura, H.; Hirano, K.; Oana, S.; Niimi, H. Increased expression of mast cell chymase in the lungs of patients with congenital heart disease associated with early pulmonary vascular disease. Am. J. Respir. Crit. Care Med. 1999, 160, 1303–1308. [Google Scholar] [CrossRef]
- Doggrell, S.A.; Wanstall, J.C. Vascular chymase: Pathophysiological role and therapeutic potential of inhibition. Cardiovasc. Res. 2004, 61, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Kosanovic, D.; Luitel, H.; Dahal, B.K.; Cornitescu, T.; Janssen, W.; Danser, A.H.; Garrelds, I.M.; De Mey, J.G.; Fazzi, G.; Schiffers, P.; et al. Chymase: A multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur. Respir. J. 2015, 46, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Riley, D.J.; Thakker-Varia, S.; Wilson, F.J.; Poiani, G.J.; Tozzi, C.A. Role of proteolysis and apoptosis in regression of pulmonary vascular remodeling. Physiol. Res. 2000, 49, 577–585. [Google Scholar] [PubMed]
- Xu, J.; Wang, J.; Shao, C.; Zeng, X.; Sun, L.; Kong, H.; Xie, W.; Wang, H. New dynamic viewing of mast cells in pulmonary arterial hypertension (PAH): Contributors or outsiders to cardiovascular remodeling. J. Thorac. Dis. 2018, 10, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Ormiston, M.L.; Chang, C.; Long, L.L.; Soon, E.; Jones, D.; Machado, R.; Treacy, C.; Toshner, M.R.; Campbell, K.; Riding, A.; et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 2012, 126, 1099–1109. [Google Scholar] [CrossRef]
- Ratsep, M.T.; Moore, S.D.; Jafri, S.; Mitchell, M.; Brady, H.J.M.; Mandelboim, O.; Southwood, M.; Morrell, N.W.; Colucci, F.; Ormiston, M.L. Spontaneous pulmonary hypertension in genetic mouse models of natural killer cell deficiency. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L977–L990. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.L.; Reizis, B. Dendritic cells: Arbiters of immunity and immunological tolerance. Cold Spring Harb. Perspect. Biol. 2012, 4, a007401. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 13, 566–577. [Google Scholar] [CrossRef]
- Blanco, P.; Palucka, A.K.; Pascual, V.; Banchereau, J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008, 19, 41–52. [Google Scholar] [CrossRef]
- Kool, M.; van Loo, G.; Waelput, W.; De Prijck, S.; Muskens, F.; Sze, M.; van Praet, J.; Branco-Madeira, F.; Janssens, S.; Reizis, B.; et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 2011, 35, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Van Uden, D.; Boomars, K.; Kool, M. Dendritic Cell Subsets and Effector Function in Idiopathic and Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Front. Immunol. 2019, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Dorfmüller, P.; Souza, R.; Durand-Gasselin, I.; Mussot, S.; Mazmanian, M.; Hervé, P.; Emilie, D.; Simonneau, G.; Humbert, M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 462–468. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yan, H.; Zhu, W.; Cui, Y.; Chen, J.; Wang, X.; Li, S.; Zhu, J. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J. Clin. Immunol. 2009, 29, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Mauad, T.; Pozzan, G.; Lancas, T.; Overbeek, M.J.; Souza, R.; Jardim, C.; Dolhnikoff, M.; Mello, G.; Pires-Neto, R.C.; Bernardi Fdel, C.; et al. Immunopathological aspects of schistosomiasis-associated pulmonary arterial hypertension. J. Infect. 2014, 68, 90–98. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Hashimoto-Kataoka, T.; Hosen, N.; Sonobe, T.; Arita, Y.; Yasui, T.; Masaki, T.; Minami, M.; Inagaki, T.; Miyagawa, S.; Sawa, Y.; et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2015, 112, E2677–E2686. [Google Scholar] [CrossRef]
- Van Hamburg, J.P.; Tas, S.W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 2018, 87, 69–81. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef]
- Gaowa, S.; Zhou, W.; Yu, L.; Zhou, X.; Liao, K.; Yang, K.; Lu, Z.; Jiang, H.; Chen, X. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediat. Inflamm. 2014, 2014, 247372. [Google Scholar] [CrossRef] [PubMed]
- Vroman, H.; Bergen, I.M.; Van Hulst, J.A.; Van Nimwegen, M.; Van Uden, D.; Schuijs, M.J.; Pillai, S.Y.; Van Loo, G.; Hammad, H.; Lambrecht, B.N.; et al. TNF-alpha-induced protein 3 levels in lung dendritic cells instruct TH2 or TH17 cell differentiation in eosinophilic or neutrophilic asthma. J. Allergy Clin. Immunol. 2018, 141, 1620–1633. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef] [PubMed]
- De Bourcy, C.F.A.; Dekker, C.L.; Davis, M.M.; Nicolls, M.R.; Quake, S.R. Dynamics of the human antibody repertoire after B cell depletion in systemic sclerosis. Sci. Immunol. 2017, 2, eaan8289. [Google Scholar] [CrossRef]
- Ulrich, S.; Taraseviciene-Stewart, L.; Huber, L.C.; Speich, R.; Voelkel, N.F. Peripheral blood B lymphocytes derived from patients with idiopathic pulmonary arterial hypertension express a different RNA pattern compared with healthy controls: A cross sectional study. Respir. Res. 2008, 9, 20. [Google Scholar] [CrossRef]
- Blum, L.K.; Cao, R.R.L.; Sweatt, A.J.; Bill, M.; Lahey, L.J.; Hsi, A.C.; Lee, C.S.; Kongpachith, S.; Ju, C.H.; Mao, R.; et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur. J. Immunol. 2018, 48, 874–884. [Google Scholar] [CrossRef]
- Rich, S.; Kieras, K.; Hart, K.; Groves, B.M.; Stobo, J.D.; Brundage, B.H. Antinuclear antibodies in primary pulmonary hypertension. J. Am. Coll. Cardiol. 1986, 8, 1307–1311. [Google Scholar] [CrossRef]
- Arends, S.J.; Damoiseaux, J.; Duijvestijn, A.; Debrus-Palmans, L.; Boomars, K.; Broers, B.; Tervaert, J.W.; van Paassen, P. Prevalence of anti-endothelial cell antibodies in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2010, 35, 923–925. [Google Scholar] [CrossRef]
- Dib, H.; Tamby, M.C.; Bussone, G.; Regent, A.; Berezne, A.; Lafine, C.; Broussard, C.; Simonneau, G.; Guillevin, L.; Witko-Sarsat, V.; et al. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur. Respir. J. 2012, 39, 1405–1414. [Google Scholar] [CrossRef]
- Tamby, M.C.; Chanseaud, Y.; Humbert, M.; Fermanian, J.; Guilpain, P.; Garcia-de-la-Pena-Lefebvre, P.; Brunet, S.; Servettaz, A.; Weill, B.; Simonneau, G.; et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax 2005, 60, 765–772. [Google Scholar] [CrossRef]
- Arends, S.J.; Damoiseaux, J.G.M.C.; Duijvestijn, A.M.; Debrus-Palmans, L.; Vroomen, M.; Boomars, K.A.; Rocca, H.-P.B.-L.; Reutelingsperger, C.P.M.; Tervaert, J.W.C.; Van Paassen, P. Immunoglobulin G anti-endothelial cell antibodies: Inducers of endothelial cell apoptosis in pulmonary arterial hypertension? Clin. Exp. Immunol. 2013, 174, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kühl, A.A.; Heidecke, H.; Ghofrani, A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Colvin, K.L.; Cripe, P.J.; Ivy, D.D.; Stenmark, K.R.; Yeager, M.E. Bronchus-associated lymphoid tissue in pulmonary hypertension produces pathologic autoantibodies. Am. J. Respir. Crit. Care Med. 2013, 188, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Cracowski, J.L.; Chabot, F.; Labarere, J.; Faure, P.; Degano, B.; Schwebel, C.; Chaouat, A.; Reynaud-Gaubert, M.; Cracowski, C.; Sitbon, O.; et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur. Respir. J. 2014, 43, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Säleby, J.; Bouzina, H.; Lundgren, J.; Rådegran, G. Angiogenic and inflammatory biomarkers in the differentiation of pulmonary hypertension. Scand. Cardiovasc. J. 2017, 51, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Okada, O.; Tanabe, N.; Tanaka, Y.; Terai, M.; Takiguchi, Y.; Masuda, M.; Nakajima, N.; Hiroshima, K.; Inadera, H.; et al. Plasma monocyte chemoattractant protein-1 and pulmonary vascular resistance in chronic thromboembolic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2001, 164, 319–324. [Google Scholar] [CrossRef]
- Langer, F.; Schramm, R.; Bauer, M.; Tscholl, D.; Kunihara, T.; Schäfers, H.-J. Cytokine response to pulmonary thromboendarterectomy. Chest 2004, 126, 135–141. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.; Treacy, C.; Barker, L.; Suntharalingam, J.; Southwood, M.; Nicklin, P.; Walker, C.; Budd, D.; Jenkins, D.; et al. Inflammatory cytokines are elevated in patients with operable chronic thromboembolic pulmonary hypertension and predict outcome post-endarterectomy. Thorax 2010, 65, A45. [Google Scholar] [CrossRef]
- Kylhammar, D.; Hesselstrand, R.; Nielsen, S.; Scheele, C.; Rådegran, G. Angiogenic and inflammatory biomarkers for screening and follow-up in patients with pulmonary arterial hypertension. Scand. J. Rheumatol. 2018, 47, 319–324. [Google Scholar] [CrossRef]
- Low, A.; George, S.; Howard, L.; Bell, N.; Millar, A.; Tulloh, R. Lung Function, Inflammation, and Endothelin-1 in Congenital Heart Disease-Associated Pulmonary Arterial Hypertension. J. Am. Heart Assoc. 2018, 7, e007249. [Google Scholar] [CrossRef]
- Joshi, A.A.; Davey, R.; Rao, Y.; Shen, K.; Benza, R.L.; Raina, A. Association between cytokines and functional, hemodynamic parameters, and clinical outcomes in pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045894018794051. [Google Scholar] [CrossRef] [PubMed]
- Dorfmüller, P.; Zarka, V.; Durand-Gasselin, I.; Monti, G.; Balabanian, K.; Garcia, G.; Capron, F.; Coulomb-Lhermine, A.; Marfaing-Koka, A.; Simonneau, G.; et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2002, 165, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef]
- Golembeski, S.M.; West, J.; Tada, Y.; Fagan, K.A. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest 2005, 128, 572S–573S. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Sakuma, F.; Yoshimura, A.; Ishikawa, H.; Nishimaki, T.; Kasukawa, R. Pulmonary hypertension in rats. 2. Role of interleukin-6. Int. Arch. Allergy Immunol. 1995, 108, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Tu, L.; Rideau, D.; Izziki, M.; Maitre, B.; Adnot, S.; Eddahibi, S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 2009, 10, 6. [Google Scholar] [CrossRef]
- Matura, L.A.; Ventetuolo, C.E.; Palevsky, H.I.; Lederer, D.J.; Horn, E.M.; Mathai, S.C.; Pinder, D.; Archer-Chicko, C.; Bagiella, E.; Roberts, K.E.; et al. Interleukin-6 and tumor necrosis factor-alpha are associated with quality of life-related symptoms in pulmonary arterial hypertension. Ann. Am. Thorac. Soc. 2015, 12, 370–375. [Google Scholar] [CrossRef]
- Sweatt, A.J.; Hedlin, H.K.; Balasubramanian, V.; Hsi, A.; Blum, L.K.; Robinson, W.H.; Haddad, F.; Hickey, P.M.; Condliffe, R.; Lawrie, A.; et al. Discovery of Distinct Immune Phenotypes Using Machine Learning in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 904–919. [Google Scholar] [CrossRef]
- Fujita, M.; Mason, R.J.; Cool, C.; Shannon, J.M.; Hara, N.; Fagan, K.A. Pulmonary hypertension in TNF-alpha-overexpressing mice is associated with decreased VEGF gene expression. J. Appl. Physiol. 2002, 93, 2162–2170. [Google Scholar] [CrossRef]
- Price, L.C.; Montani, D.; Tcherakian, C.; Dorfmuller, P.; Souza, R.; Gambaryan, N.; Chaumais, M.C.; Shao, D.M.; Simonneau, G.; Howard, L.S.; et al. Dexamethasone reverses monocrotaline-induced pulmonary arterial hypertension in rats. Eur. Respir. J. 2011, 37, 813–822. [Google Scholar] [CrossRef]
- Suzuki, C.; Takahashi, M.; Morimoto, H.; Izawa, A.; Ise, H.; Hongo, M.; Hoshikawa, Y.; Ito, T.; Miyashita, H.; Kobayashi, E.; et al. Mycophenolate mofetil attenuates pulmonary arterial hypertension in rats. Biochem. Biophys. Res. Commun. 2006, 349, 781–788. [Google Scholar] [CrossRef]
- Bonnet, S.; Rochefort, G.Y.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Bonnet, S.N.; Michelakis, E.D. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423. [Google Scholar] [CrossRef] [PubMed]
- Jais, X.; Launay, D.; Yaici, A.; Le Pavec, J.; Tcherakian, C.; Sitbon, O.; Simonneau, G.; Humbert, M. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: A retrospective analysis of twenty-three cases. Arthritis Rheum. 2008, 58, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Tuder, R.M.; Bridges, J.; Arend, W.P. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 1994, 11, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Phan, C.; Tu, L.; Le Hiress, M.; Thuillet, R.; Jutant, E.M.; Fadel, E.; Savale, L.; Huertas, A.; Humbert, M.; et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J. Clin. Investig. 2018, 128, 1956–1970. [Google Scholar] [CrossRef]
- Hernández-Sánchez, J.; Harlow, L.; Church, C.; Gaine, S.; Knightbridge, E.; Bunclark, K.; Gor, D.; Bedding, A.; Morrell, N.; A Corris, P.; et al. Clinical trial protocol for TRANSFORM-UK: A therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045893217735820. [Google Scholar] [CrossRef]
- Roeleveld, D.M.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.; van de Loo, F.A.; van Lent, P.L.; van der Kraan, P.M.; van den Berg, W.B.; Koenders, M.I. Higher efficacy of anti-IL-6/IL-21 combination therapy compared to monotherapy in the induction phase of Th17-driven experimental arthritis. PLoS ONE 2017, 12, e0171757. [Google Scholar] [CrossRef]
- Wang, Q.; Zuo, X.R.; Wang, Y.Y.; Xie, W.P.; Wang, H.; Zhang, M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-alpha antagonists via the suppression of TNF-alpha expression and NF-kappaB pathway in rats. Vascul. Pharmacol. 2013, 58, 71–77. [Google Scholar] [CrossRef]
- Zhang, L.L.; Lu, J.; Li, M.T.; Wang, Q.; Zeng, X.F. Preventive and remedial application of etanercept attenuate monocrotaline-induced pulmonary arterial hypertension. Int. J. Rheum. Dis. 2016, 19, 192–198. [Google Scholar] [CrossRef]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFalpha drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef]
- Mutschler, D.; Wikström, G.; Lind, L.; Larsson, A.; Lagrange, A.; Eriksson, M. Etanercept reduces late endotoxin-induced pulmonary hypertension in the pig. J. Interferon Cytokine Res. 2006, 26, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Pousada, G.; Lupo, V.; Castro-Sánchez, S.; Álvarez-Satta, M.; Sánchez-Monteagudo, A.; Baloira, A.; Espinós, C.; Valverde, D. Molecular and functional characterization of the BMPR2 gene in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 1923. [Google Scholar] [CrossRef] [PubMed]
- Rol, N.; Kurakula, K.B.; Happe, C.; Bogaard, H.J.; Goumans, M.J. TGF-beta and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585. [Google Scholar] [CrossRef]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Nikolic, I.; Yung, L.M.; Yang, P.; Malhotra, R.; Paskin-Flerlage, S.D.; Dinter, T.; Bocobo, G.A.; Tumelty, K.E.; Faugno, A.J.; Troncone, L.; et al. Bone Morphogenetic Protein 9 Is a Mechanistic Biomarker of Portopulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef]
- Hensley, M.K.; Levine, A.; Gladwin, M.T.; Lai, Y.C. Emerging therapeutics in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L769–L781. [Google Scholar] [CrossRef]
- Reynolds, A.M.; Xia, W.; Holmes, M.D.; Hodge, S.; Danilov, S.; Curiel, D.T.; Morrell, N.W.; Reynolds, P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1182–L1192. [Google Scholar] [CrossRef]
- Long, L.; Ormiston, M.; Yang, X.; Southwood, M.; Gräf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef]
- Morrell, N.W.; Yang, X.; Upton, P.D.; Jourdan, K.B.; Morgan, N.; Sheares, K.K.; Trembath, R.C. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation 2001, 104, 790–795. [Google Scholar] [CrossRef]
- Long, L.; Crosby, A.; Yang, X.; Southwood, M.; Upton, P.D.; Kim, D.K.; Morrell, N.W. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009, 119, 566–576. [Google Scholar] [CrossRef]
- Yung, L.M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective Transforming Growth Factor-beta Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Egashira, K.; Chen, L.; Nakano, K.; Iwata, E.; Miyagawa, M.; Tsujimoto, H.; Hara, K.; Morishita, R.; Sueishi, K.; et al. Nanoparticle-mediated delivery of nuclear factor kappaB decoy into lungs ameliorates monocrotaline-induced pulmonary arterial hypertension. Hypertension 2009, 53, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kaminski, P.M.; Edwards, J.G.; Yeh, A.; Wolin, M.S.; Frishman, W.H.; Gewitz, M.H.; Mathew, R. Pyrrolidine dithiocarbamate restores endothelial cell membrane integrity and attenuates monocrotaline-induced pulmonary artery hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1250–L1259. [Google Scholar] [CrossRef]
- Li, L.; Wei, C.; Kim, I.K.; Janssen-Heininger, Y.; Gupta, S. Inhibition of nuclear factor-kappaB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 2014, 63, 1260–1269. [Google Scholar] [CrossRef]
- Farkas, D.; Alhussaini, A.A.; Kraskauskas, D.; Kraskauskiene, V.; Cool, C.D.; Nicolls, M.R.; Natarajan, R.; Farkas, L. Nuclear factor kappaB inhibition reduces lung vascular lumen obliteration in severe pulmonary hypertension in rats. Am. J. Respir. Cell Mol. Biol. 2014, 51, 413–425. [Google Scholar] [CrossRef]
- Kumar, S.; Wei, C.; Thomas, C.M.; Kim, I.K.; Seqqat, R.; Kumar, R.; Baker, K.M.; Jones, W.K.; Gupta, S. Cardiac-specific genetic inhibition of nuclear factor-kappaB prevents right ventricular hypertrophy induced by monocrotaline. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1655–H1666. [Google Scholar] [CrossRef]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear factor kappa-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS ONE 2013, 8, e75415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [PubMed]
- Heiss, E.H.; Schachner, D.; Werner, E.R.; Dirsch, V.M. Active NF-E2-related factor (Nrf2) contributes to keep endothelial NO synthase (eNOS) in the coupled state: Role of reactive oxygen species (ROS), eNOS, and heme oxygenase (HO-1) levels. J. Biol. Chem. 2009, 284, 31579–31586. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.; Hayes, J. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.-S.; Yu, S.; Kong, A.-N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef]
- Grzegorzewska, A.P.; Seta, F.; Han, R.; Czajka, C.A.; Makino, K.; Stawski, L.; Isenberg, J.S.; Browning, J.L.; Trojanowska, M. Dimethyl Fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci. Rep. 2017, 7, 41605. [Google Scholar] [CrossRef]
- Hennigan, S.; Channick, R.N.; Silverman, G.J. Rituximab treatment of pulmonary arterial hypertension associated with systemic lupus erythematosus: A case report. Lupus 2008, 17, 754–756. [Google Scholar] [CrossRef]
- Samuelsson, B.; Dahlen, S.E.; Lindgren, J.A.; Rouzer, C.A.; Serhan, C.N. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science 1987, 237, 1171–1176. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Sung, Y.K.; Qian, J.; Yuan, K.; Nicolls, M.R. Leukotrienes in pulmonary arterial hypertension. Immunol. Res. 2014, 58, 387–393. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PAH | CTEPH | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Biomarker | Incident Patients | Prevalent Patients | Hemodynamic Correlation | Prognosis | Etiology | Ref. | Prevalent Patients | Hemodynamic Correlation | Prognosis | Ref. |
| IL-1α | ↑[94] | ↑ | N/A | + | IPAH, HPAH | [94] | ||||
| IL-1β | ↑[94] | ↑ | − | + [94] | IPAH, HPAH, CHD-PAH | [80,94] | ↑[98] | − | − | [21,96,98] |
| IL-2 | = | ↑ | − | − | PAH, HPAH | [80,94] | ↑ | − | − | [98] |
| IL-4 | = | ↑ | − | − | PAH, HPAH | [80,94] | ↑ | − | − | [98] |
| IL-5 | N/A | = | − | − | PAH, HPAH | [80] | = | − | − | [98] |
| IL-6 | ↑[94,95,99] | ↑ | + | + | IPAH, HPAH, CTD-PAH, CHD-PAH | [80,94,95,99,100] | ↑[42,97] | + | + [97,98] | [21,42,95,97,98] |
| IL-8 | = | ↑ | − | + [80] | IPAH, HPAH, CTD-PAH, CHD-PAH | [80,94,95,99,100] | ↑[42,97,98] | − | + [97,98] | [42,95,97,98] |
| IL-10 | = | ↑ | − | + [80] | IPAH, HPAH | [80,94] | ↑ | − | + [97,98] | [21,97,98] |
| IL-12 | = | ↑ | − | + [80] | IPAH, HPAH | [80,94] | = | − | − | [98] |
| IL-13 | = | = | − | + [94] | IPAH, HPAH | [80,94] | = | − | − | [98] |
| IFN-γ | = | = | − | − | IPAH, HPAH | [79,93] | = | − | − | [98] |
| TNF-α | ↑[94,95,99] | ↑ | N/A | + [94] | IPAH, HPAH, CTD-PAH, CHD-PAH | [80,94,95,99,100] | = | − | + [96,97] | [95,96,97] |
| MMP-9 | ↑ | ↑ | + | + | PAH | [101] | ↑ | − | N/A | [21] |
| VEGF | = | ↑ | N/A | + [101] | IPAH, HPAH, CHD-PAH | [80,94,100,101] | = | − | N/A | [21] |
| CCL-2 | = | ↑[33] | N/A | − | IPAH | [33,94] | ↑ | − | N/A | [21,42] |
| MIG | N/A | ↑ | − | N/A | IPAH | [42] | ↑ | − | N/A | [42] |
| CCL-3 | = | = | − | N/A | IPAH | [42] | ↑ | + | N/A | [21,42] |
| CXCL-10 | N/A | ↑ | − | N/A | IPAH | [42] | ↑ | + | N/A | [42] |
| CCL-5 | N/A | ↑ | − | N/A | IPAH, PAH | [42,102] | ↓ | − | N/A | [42] |
| CX3CL-1 | = | = | − | N/A | IPAH | [42] | = | − | N/A | [42] |
| CXCL-12 | = | = | − | N/A | IPAH | [42] | = | − | N/A | [42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koudstaal, T.; Boomars, K.A.; Kool, M. Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. J. Clin. Med. 2020, 9, 561. https://doi.org/10.3390/jcm9020561
Koudstaal T, Boomars KA, Kool M. Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. Journal of Clinical Medicine. 2020; 9(2):561. https://doi.org/10.3390/jcm9020561
Chicago/Turabian StyleKoudstaal, Thomas, Karin A. Boomars, and Mirjam Kool. 2020. "Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective" Journal of Clinical Medicine 9, no. 2: 561. https://doi.org/10.3390/jcm9020561
APA StyleKoudstaal, T., Boomars, K. A., & Kool, M. (2020). Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. Journal of Clinical Medicine, 9(2), 561. https://doi.org/10.3390/jcm9020561