Regulation of Oligodendrocyte Functions: Targeting Lipid Metabolism and Extracellular Matrix for Myelin Repair



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Lipids as Main Components of Myelin Membranes
2.1. Cholesterol Biosynthesis, Transport and Catabolism
2.2. Biosynthesis of Fatty Acids and Sphingolipids
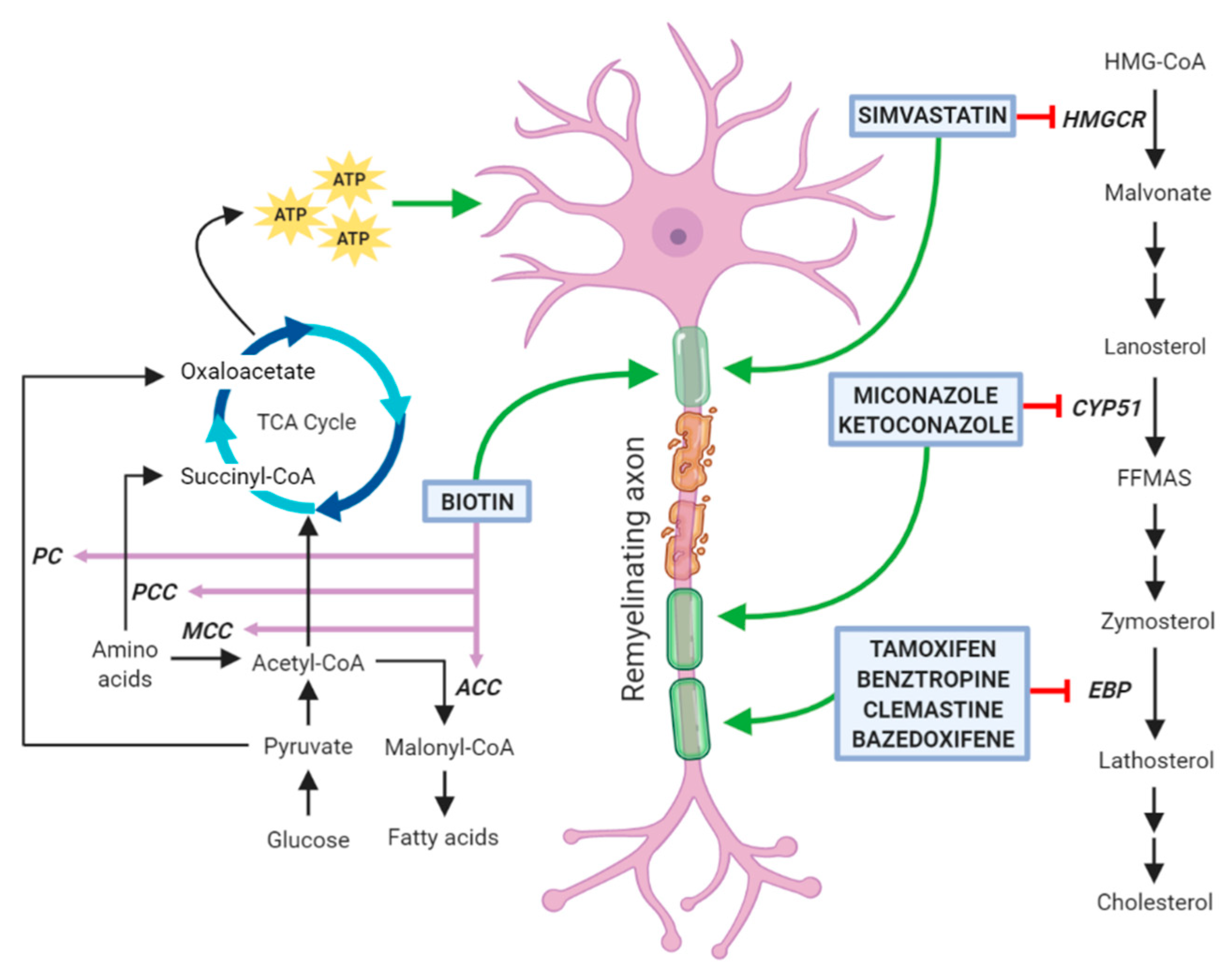
3. Drugs Promoting Remyelination through a Potential Action on Oligodendrocyte Lipid Metabolism
3.1. Clemastine
3.2. Benztropine
3.3. Selective Estrogen Receptor Modulators
3.4. Clobetasol and Miconazole
3.5. Simvastatin
3.6. Biotin
4. Essential Roles of ECM for Timely Development of OPCs into Myelinating Oligodendrocytes
4.1. Chondroitin Sulfate Proteoglycans
4.2. Hyaluronan
4.3. Fibronectin
4.4. Laminin
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saab, A.S.; Nave, K.A. Myelin dynamics: Protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Boil. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.S.; Sampson, E.L. Lipid composition of the normal human brain: Gray matter, white matter, and myelin. J. Lipid Res. 1965, 6, 537–544. [Google Scholar]
- Nave, K.A.; Werner, H.B. Myelination of the nervous system: Mechanisms and functions. Ann. Rev. Cell Dev. Boil. 2014, 30, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Micu, I.; Plemel, J.R.; Caprariello, A.V.; Nave, K.A.; Stys, P.K. Axo-myelinic neurotransmission: A novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 2018, 19, 49–58. [Google Scholar] [CrossRef]
- Morrison, B.M.; Lee, Y.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol. 2013, 23, 644–651. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—From mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Miron, V.E. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Boil. 2017, 101, 1103–1108. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Dimas, P.; Montani, L.; Pereira, J.A.; Moreno, D.; Trotzmuller, M.; Gerber, J.; Semenkovich, C.F.; Kofeler, H.C.; Suter, U. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. eLife 2019, 8. [Google Scholar] [CrossRef]
- Colognato, H.; Tzvetanova, I.D. Glia unglued: How signals from the extracellular matrix regulate the development of myelinating glia. Dev. Neurobiol. 2011, 71, 924–955. [Google Scholar] [CrossRef] [PubMed]
- Stoffels, J.M.; de Jonge, J.C.; Stancic, M.; Nomden, A.; van Strien, M.E.; Ma, D.; Siskova, Z.; Maier, O.; Ffrench-Constant, C.; Franklin, R.J.; et al. Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Br. J. Neurol. 2013, 136, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, V.R.; Benbenishty, A.; Blinder, P.; Sagi, I. Demystifying the extracellular matrix and its proteolytic remodeling in the brain: Structural and functional insights. Cell. Mol. Life Sci. CMLS 2019, 76, 3229–3248. [Google Scholar] [CrossRef] [PubMed]
- Monnerie, H.; Romer, M.; Jensen, B.K.; Millar, J.S.; Jordan-Sciutto, K.L.; Kim, S.F.; Grinspan, J.B. Reduced sterol regulatory element-binding protein (SREBP) processing through site-1 protease (S1P) inhibition alters oligodendrocyte differentiation in vitro. J. Neurochem. 2017, 140, 53–67. [Google Scholar] [CrossRef]
- Nelissen, K.; Mulder, M.; Smets, I.; Timmermans, S.; Smeets, K.; Ameloot, M.; Hendriks, J.J. Liver X receptors regulate cholesterol homeostasis in oligodendrocytes. J. Neurosci. Res. 2012, 90, 60–71. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Boil. 2019. [Google Scholar] [CrossRef]
- Moutinho, M.; Nunes, M.J.; Rodrigues, E. Cholesterol 24-hydroxylase: Brain cholesterol metabolism and beyond. Biochim. Biophys. Acta 2016, 1861, 1911–1920. [Google Scholar] [CrossRef]
- Lavrnja, I.; Smiljanic, K.; Savic, D.; Mladenovic-Djordjevic, A.; Tesovic, K.; Kanazir, S.; Pekovic, S. Expression profiles of cholesterol metabolism-related genes are altered during development of experimental autoimmune encephalomyelitis in the rat spinal cord. Sci. Rep. 2017, 7, 2702. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lutjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Voskuhl, R.R.; Itoh, N.; Tassoni, A.; Matsukawa, M.A.; Ren, E.; Tse, V.; Jang, E.; Suen, T.T.; Itoh, Y. Gene expression in oligodendrocytes during remyelination reveals cholesterol homeostasis as a therapeutic target in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 10130–10139. [Google Scholar] [CrossRef]
- Tettey, P.; Simpson, S., Jr.; Taylor, B.; Blizzard, L.; Ponsonby, A.L.; Dwyer, T.; Kostner, K.; van der Mei, I. An adverse lipid profile is associated with disability and progression in disability, in people with MS. Mult. Scler. 2014, 20, 1737–1744. [Google Scholar] [CrossRef] [PubMed]
- Camargo, N.; Goudriaan, A.; van Deijk, A.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.A.; Dijkhuizen, R.M.; Mansvelder, H.D.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Boil. 2017, 15, e1002605. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C. Long-chain omega-3 fatty acids and the brain: A review of the independent and shared effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Castelvetri, L.C.; Simons, M. Metabolism and functions of lipids in myelin. Biochim. Biophys. Acta 2015, 1851, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Lewkowicz, N.; Piatek, P.; Namiecinska, M.; Domowicz, M.; Bonikowski, R.; Szemraj, J.; Przygodzka, P.; Stasiolek, M.; Lewkowicz, P. Naturally Occurring Nervonic Acid Ester Improves Myelin Synthesis by Human Oligodendrocytes. Cells 2019, 8, 786. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.G., Jr.; Young, J.A.; Ray, S.K.; Wang, G.; Purohit, S.; Banik, N.L.; Dasgupta, S. Sphingosine Toxicity in EAE and MS: Evidence for Ceramide Generation via Serine-Palmitoyltransferase Activation. Neurochem. Res. 2017, 42, 2755–2768. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Ray, S.K. Insights into abnormal sphingolipid metabolism in multiple sclerosis: Targeting ceramide biosynthesis as a unique therapeutic strategy. Ther. Targets Neurol. Dis. 2017, 4, e1598. [Google Scholar]
- Imgrund, S.; Hartmann, D.; Farwanah, H.; Eckhardt, M.; Sandhoff, R.; Degen, J.; Gieselmann, V.; Sandhoff, K.; Willecke, K. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J. Boil. Chem. 2009, 284, 33549–33560. [Google Scholar] [CrossRef]
- Lahiri, S.; Park, H.; Laviad, E.L.; Lu, X.; Bittman, R.; Futerman, A.H. Ceramide synthesis is modulated by the sphingosine analog FTY720 via a mixture of uncompetitive and noncompetitive inhibition in an Acyl-CoA chain length-dependent manner. J. Boil. Chem. 2009, 284, 16090–16098. [Google Scholar] [CrossRef]
- Yazdi, A.; Ghasemi-Kasman, M.; Javan, M. Possible regenerative effects of fingolimod (FTY720) in multiple sclerosis disease: An overview on remyelination process. J. Neurosci. Res. 2020, 98, 524–536. [Google Scholar] [CrossRef]
- Nystad, A.E.; Lereim, R.R.; Wergeland, S.; Oveland, E.; Myhr, K.M.; Bo, L.; Torkildsen, O. Fingolimod downregulates brain sphingosine-1-phosphate receptor 1 levels but does not promote remyelination or neuroprotection in the cuprizone model. J. Neuroimmunol. 2019, 339, 577091. [Google Scholar] [CrossRef] [PubMed]
- Grassi, S.; Prioni, S.; Cabitta, L.; Aureli, M.; Sonnino, S.; Prinetti, A. The Role of 3-O-Sulfogalactosylceramide, Sulfatide, in the Lateral Organization of Myelin Membrane. Neurochem. Res. 2016, 41, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Polito, A.; Reynolds, R. NG2-expressing cells as oligodendrocyte progenitors in the normal and demyelinated adult central nervous system. J. Anat. 2005, 207, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, T.; Fujita, N.; Dupree, J.; Shi, R.; Blight, A.; Suzuki, K.; Suzuki, K.; Popko, B. Myelination in the absence of galactocerebroside and sulfatide: Normal structure with abnormal function and regional instability. Cell 1996, 86, 209–219. [Google Scholar] [CrossRef]
- Coetzee, T.; Li, X.; Fujita, N.; Marcus, J.; Suzuki, K.; Francke, U.; Popko, B. Molecular cloning, chromosomal mapping, and characterization of the mouse UDP-galactose:ceramide galactosyltransferase gene. Genomics 1996, 35, 215–222. [Google Scholar] [CrossRef]
- Jennemann, R.; Sandhoff, R.; Wang, S.; Kiss, E.; Gretz, N.; Zuliani, C.; Martin-Villalba, A.; Jager, R.; Schorle, H.; Kenzelmann, M.; et al. Cell-specific deletion of glucosylceramide synthase in brain leads to severe neural defects after birth. Proc. Natl. Acad. Sci. USA 2005, 102, 12459–12464. [Google Scholar] [CrossRef]
- Boggs, J.M.; Gao, W.; Zhao, J.; Park, H.J.; Liu, Y.; Basu, A. Participation of galactosylceramide and sulfatide in glycosynapses between oligodendrocyte or myelin membranes. FEBS Lett. 2010, 584, 1771–1778. [Google Scholar] [CrossRef]
- Aureli, M.; Grassi, S.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid membrane domains in the brain. Biochim. Biophys. Acta 2015, 1851, 1006–1016. [Google Scholar] [CrossRef]
- Ozgen, H.; Schrimpf, W.; Hendrix, J.; de Jonge, J.C.; Lamb, D.C.; Hoekstra, D.; Kahya, N.; Baron, W. The lateral membrane organization and dynamics of myelin proteins PLP and MBP are dictated by distinct galactolipids and the extracellular matrix. PLoS ONE 2014, 9, e101834. [Google Scholar] [CrossRef]
- Popescu, D.C.; Huang, H.; Singhal, N.K.; Shriver, L.; McDonough, J.; Clements, R.J.; Freeman, E.J. Vitamin K enhances the production of brain sulfatides during remyelination. PLoS ONE 2018, 13, e0203057. [Google Scholar] [CrossRef]
- Wheeler, D.; Bandaru, V.V.; Calabresi, P.A.; Nath, A.; Haughey, N.J. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Br. J. Neurol. 2008, 131, 3092–3102. [Google Scholar] [CrossRef] [PubMed]
- Vidaurre, O.G.; Haines, J.D.; Katz Sand, I.; Adula, K.P.; Huynh, J.L.; McGraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Br. J. Neurol. 2014, 137, 2271–2286. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, S.; Lekman, A.; Nilsson, S.; Blomqvist, M.; Andersen, O. Myelin glycosphingolipid immunoreactivity and CSF levels in multiple sclerosis. Acta Neurol. Scand. 2012, 125, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, A.A.; Chen, Z.W.; Cook, S.D. Antibodies to sulfatide in cerebrospinal fluid of patients with multiple sclerosis. J. Neuroimmunol. 2003, 139, 76–80. [Google Scholar] [CrossRef]
- Kanhai, K.; Goulooze, S.C.; Stevens, J.; Hay, J.L.; Dent, G.; Verma, A.; Hankemeier, T.; de Boer, T.; Meijering, H.; Chavez, J.C.; et al. Quantifying Beta-Galactosylceramide Kinetics in Cerebrospinal Fluid of Healthy Subjects Using Deuterium Labeling. Clin. Transl. Sci. 2016, 9, 321–327. [Google Scholar] [CrossRef]
- Qin, J.; Sikkema, A.H.; van der Bij, K.; de Jonge, J.C.; Klappe, K.; Nies, V.; Jonker, J.W.; Kok, J.W.; Hoekstra, D.; Baron, W. GD1a Overcomes Inhibition of Myelination by Fibronectin via Activation of Protein Kinase A: Implications for Multiple Sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 9925–9938. [Google Scholar] [CrossRef]
- Schnaar, R.L. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 2010, 584, 1741–1747. [Google Scholar] [CrossRef]
- Yang, L.J.; Lorenzini, I.; Vajn, K.; Mountney, A.; Schramm, L.P.; Schnaar, R.L. Sialidase enhances spinal axon outgrowth in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 11057–11062. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Wang, J.; Wang, Y.; Xie, X.; Meyenhofer, M.F.; Ledeen, R.W. Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: Protection with LIGA 20, a membrane-permeant analog of GM1. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 11014–11022. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Mei, F.; Fancy, S.P.J.; Shen, Y.A.; Niu, J.; Zhao, C.; Presley, B.; Miao, E.; Lee, S.; Mayoral, S.R.; Redmond, S.A.; et al. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 2014, 20, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Najm, F.J.; Madhavan, M.; Zaremba, A.; Shick, E.; Karl, R.T.; Factor, D.C.; Miller, T.E.; Nevin, Z.S.; Kantor, C.; Sargent, A.; et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 2015, 522, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hubler, Z.; Allimuthu, D.; Bederman, I.; Elitt, M.S.; Madhavan, M.; Allan, K.C.; Shick, H.E.; Garrison, E.; Molly, T.K.; Factor, D.C.; et al. Accumulation of 8,9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 2018, 560, 372–376. [Google Scholar] [CrossRef]
- Rankin, K.A.; Mei, F.; Kim, K.; Shen, Y.A.; Mayoral, S.R.; Desponts, C.; Lorrain, D.S.; Green, A.J.; Baranzini, S.E.; Chan, J.R.; et al. Selective Estrogen Receptor Modulators Enhance CNS Remyelination Independent of Estrogen Receptors. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 2184–2194. [Google Scholar] [CrossRef]
- Bertacchi, M.; Gruart, A.; Kaimakis, P.; Allet, C.; Serra, L.; Giacobini, P.; Delgado-García, J.M.; Bovolenta, P.; Studer, M. Mouse Nr2f1 haploinsufficiency unveils new pathological mechanisms of a human optic atrophy syndrome. EMBO Mol. Med. 2019, 11, e10291. [Google Scholar] [CrossRef]
- Datta, A.; Kim, H.; McGee, L.; Johnson, A.E.; Talwar, S.; Marugan, J.; Southall, N.; Hu, X.; Lal, M.; Mondal, D.; et al. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci. Rep. 2018, 8, 8161. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, Y.; Huang, D.; Luo, X.; Zhang, L.; Zhu, X.; Zhang, X.; Liu, Z.; Han, J.Y.; Xiong, J.W. Miconazole protects blood vessels from MMP9-dependent rupture and hemorrhage. Dis. Models Mech. 2017, 10, 337–348. [Google Scholar] [CrossRef]
- Fyffe-Maricich, S.L.; Schott, A.; Karl, M.; Krasno, J.; Miller, R.H. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 18402–18408. [Google Scholar] [CrossRef]
- Au, W.L.; Skinner, M.F.; Benfeldt, E.; Verbeeck, R.K.; Kanfer, I. Application of dermal microdialysis for the determination of bioavailability of clobetasol propionate applied to the skin of human subjects. Skin Pharmacol. Physiol. 2012, 25, 17–24. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Bond, M.C.; Chen, M.; Ren, X.R.; Lyerly, H.K.; Barak, L.S.; Chen, W. Identification of select glucocorticoids as Smoothened agonists: Potential utility for regenerative medicine. Proc. Natl. Acad. Sci. USA 2010, 107, 9323–9328. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; He, Y.; Fan, S.; Sun, B. Clemastine rescues behavioral changes and enhances remyelination in the cuprizone mouse model of demyelination. Neurosci. Bull. 2015, 31, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Niu, J.; Hoi, K.K.; Zhao, C.; Caganap, S.D.; Henry, R.G.; Dao, D.Q.; Zollinger, D.R.; Mei, F.; Shen, Y.A.; et al. Clemastine rescues myelination defects and promotes functional recovery in hypoxic brain injury. Br. J. Neurol. 2018, 141, 85–98. [Google Scholar] [CrossRef]
- Wang, F.; Yang, Y.J.; Yang, N.; Chen, X.J.; Huang, N.X.; Zhang, J.; Wu, Y.; Liu, Z.; Gao, X.; Li, T.; et al. Enhancing Oligodendrocyte Myelination Rescues Synaptic Loss and Improves Functional Recovery after Chronic Hypoxia. Neuron 2018, 99, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dupree, J.L.; Gacias, M.; Frawley, R.; Sikder, T.; Naik, P.; Casaccia, P. Clemastine Enhances Myelination in the Prefrontal Cortex and Rescues Behavioral Changes in Socially Isolated Mice. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Green, A.J.; Gelfand, J.M.; Cree, B.A.; Bevan, C.; Boscardin, W.J.; Mei, F.; Inman, J.; Arnow, S.; Devereux, M.; Abounasr, A.; et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): A randomised, controlled, double-blind, crossover trial. Lancet 2017, 390, 2481–2489. [Google Scholar] [CrossRef]
- Clemons, M.; Danson, S.; Howell, A. Tamoxifen (“Nolvadex”): A review. Cancer Treat. Rev. 2002, 28, 165–180. [Google Scholar] [CrossRef]
- Gonzalez, G.A.; Hofer, M.P.; Syed, Y.A.; Amaral, A.I.; Rundle, J.; Rahman, S.; Zhao, C.; Kotter, M.R.N. Tamoxifen accelerates the repair of demyelinated lesions in the central nervous system. Sci. Rep. 2016, 6, 31599. [Google Scholar] [CrossRef]
- Arevalo, M.A.; Diz-Chaves, Y.; Santos-Galindo, M.; Bellini, M.J.; Garcia-Segura, L.M. Selective oestrogen receptor modulators decrease the inflammatory response of glial cells. J. Neuroendocrinol. 2012, 24, 183–190. [Google Scholar] [CrossRef]
- Goldstein, M.R.; Mascitelli, L. Regarding long-term statin therapy: Are we trading stronger hearts for weaker brains? Med. Hypotheses 2014, 83, 6. [Google Scholar] [CrossRef]
- Ciurleo, R.; Bramanti, P.; Marino, S. Role of statins in the treatment of multiple sclerosis. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2014, 87, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Ntolkeras, G.; Barba, C.; Mavropoulos, A.; Vasileiadis, G.K.; Dardiotis, E.; Sakkas, L.I.; Hadjigeorgiou, G.; Bogdanos, D.P. On the immunoregulatory role of statins in multiple sclerosis: The effects on Th17 cells. Immunol. Res. 2019, 67, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Rajasekharan, S.; Jarjour, A.A.; Zamvil, S.S.; Kennedy, T.E.; Antel, J.P. Simvastatin regulates oligodendroglial process dynamics and survival. Glia 2007, 55, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Zehntner, S.P.; Kuhlmann, T.; Ludwin, S.K.; Owens, T.; Kennedy, T.E.; Bedell, B.J.; Antel, J.P. Statin therapy inhibits remyelination in the central nervous system. Am. J. Pathol. 2009, 174, 1880–1890. [Google Scholar] [CrossRef]
- Sim, F.J.; Lang, J.K.; Ali, T.A.; Roy, N.S.; Vates, G.E.; Pilcher, W.H.; Goldman, S.A. Statin treatment of adult human glial progenitors induces PPAR gamma-mediated oligodendrocytic differentiation. Glia 2008, 56, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Youssef, S.; Stuve, O.; Patarroyo, J.C.; Ruiz, P.J.; Radosevich, J.L.; Hur, E.M.; Bravo, M.; Mitchell, D.J.; Sobel, R.A.; Steinman, L.; et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature 2002, 420, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Br. Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Lycke, J.; Eralinna, J.P.; Edland, A.; Wu, X.; Frederiksen, J.L.; Oturai, A.; Malmestrom, C.; Stenager, E.; Sellebjerg, F.; et al. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): A placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011, 10, 691–701. [Google Scholar] [CrossRef]
- Chataway, J.; Schuerer, N.; Alsanousi, A.; Chan, D.; MacManus, D.; Hunter, K.; Anderson, V.; Bangham, C.R.; Clegg, S.; Nielsen, C.; et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): A randomised, placebo-controlled, phase 2 trial. Lancet 2014, 383, 2213–2221. [Google Scholar] [CrossRef]
- Chan, D.; Binks, S.; Nicholas, J.M.; Frost, C.; Cardoso, M.J.; Ourselin, S.; Wilkie, D.; Nicholas, R.; Chataway, J. Effect of high-dose simvastatin on cognitive, neuropsychiatric, and health-related quality-of-life measures in secondary progressive multiple sclerosis: Secondary analyses from the MS-STAT randomised, placebo-controlled trial. Lancet Neurol. 2017, 16, 591–600. [Google Scholar] [CrossRef]
- Sedel, F.; Bernard, D.; Mock, D.M.; Tourbah, A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology 2016, 110, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Sedel, F.; Papeix, C.; Bellanger, A.; Touitou, V.; Lebrun-Frenay, C.; Galanaud, D.; Gout, O.; Lyon-Caen, O.; Tourbah, A. High doses of biotin in chronic progressive multiple sclerosis: A pilot study. Mult. Scler. Relat. Disord. 2015, 4, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Tourbah, A.; Gout, O.; Vighetto, A.; Deburghgraeve, V.; Pelletier, J.; Papeix, C.; Lebrun-Frenay, C.; Labauge, P.; Brassat, D.; Toosy, A.; et al. MD1003 (High-Dose Pharmaceutical-Grade Biotin) for the Treatment of Chronic Visual Loss Related to Optic Neuritis in Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Study. CNS Drugs 2018, 32, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Seze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult. Scler. 2016, 22, 1719–1731. [Google Scholar] [CrossRef]
- You, Y.; Gupta, V. The Extracellular Matrix and Remyelination Strategies in Multiple Sclerosis. eNeuro 2018, 5. [Google Scholar] [CrossRef]
- Wang, P.; Gorter, R.P.; de Jonge, J.C.; Nazmuddin, M.; Zhao, C.; Amor, S.; Hoekstra, D.; Baron, W. MMP7 cleaves remyelination-impairing fibronectin aggregates and its expression is reduced in chronic multiple sclerosis lesions. Glia 2018, 66, 1625–1643. [Google Scholar] [CrossRef]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef]
- Srivastava, T.; Sherman, L.S.; Back, S.A. Dysregulation of Hyaluronan Homeostasis During White Matter Injury. Neurochem. Res. 2019. [Google Scholar] [CrossRef]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1326. [Google Scholar] [CrossRef]
- Romani, P.; Brian, I.; Santinon, G.; Pocaterra, A.; Audano, M.; Pedretti, S.; Mathieu, S.; Forcato, M.; Bicciato, S.; Manneville, J.B.; et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Boil. 2019, 21, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Dyck, S.M.; Karimi-Abdolrezaee, S. Chondroitin sulfate proteoglycans: Key modulators in the developing and pathologic central nervous system. Exp. Neurol. 2015, 269, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Pu, A.; Stephenson, E.L.; Yong, V.W. The extracellular matrix: Focus on oligodendrocyte biology and targeting CSPGs for remyelination therapies. Glia 2018, 66, 1809–1825. [Google Scholar] [CrossRef]
- Sun, Y.; Deng, Y.; Xiao, M.; Hu, L.; Li, Z.; Chen, C. Chondroitin sulfate proteoglycans inhibit the migration and differentiation of oligodendrocyte precursor cells and its counteractive interaction with laminin. Int. J. Mol. Med. 2017, 40, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Karus, M.; Ulc, A.; Ehrlich, M.; Czopka, T.; Hennen, E.; Fischer, J.; Mizhorova, M.; Qamar, N.; Brustle, O.; Faissner, A. Regulation of oligodendrocyte precursor maintenance by chondroitin sulphate glycosaminoglycans. Glia 2016, 64, 270–286. [Google Scholar] [CrossRef]
- Pendleton, J.C.; Shamblott, M.J.; Gary, D.S.; Belegu, V.; Hurtado, A.; Malone, M.L.; McDonald, J.W. Chondroitin sulfate proteoglycans inhibit oligodendrocyte myelination through PTPsigma. Exp. Neurol. 2013, 247, 113–121. [Google Scholar] [CrossRef]
- Keough, M.B.; Rogers, J.A.; Zhang, P.; Jensen, S.K.; Stephenson, E.L.; Chen, T.; Hurlbert, M.G.; Lau, L.W.; Rawji, K.S.; Plemel, J.R.; et al. An inhibitor of chondroitin sulfate proteoglycan synthesis promotes central nervous system remyelination. Nat. Commun. 2016, 7, 11312. [Google Scholar] [CrossRef]
- Stephenson, E.L.; Zhang, P.; Ghorbani, S.; Wang, A.; Gu, J.; Keough, M.B.; Rawji, K.S.; Silva, C.; Yong, V.W.; Ling, C.C. Targeting the Chondroitin Sulfate Proteoglycans: Evaluating Fluorinated Glucosamines and Xylosides in Screens Pertinent to Multiple Sclerosis. ACS Cent. Sci. 2019, 5, 1223–1234. [Google Scholar] [CrossRef]
- Kuboyama, K.; Tanga, N.; Suzuki, R.; Fujikawa, A.; Noda, M. Protamine neutralizes chondroitin sulfate proteoglycan-mediated inhibition of oligodendrocyte differentiation. PLoS ONE 2017, 12, e0189164. [Google Scholar] [CrossRef]
- Warford, J.R.; Lamport, A.C.; Clements, D.R.; Malone, A.; Kennedy, B.E.; Kim, Y.; Gujar, S.A.; Hoskin, D.W.; Easton, A.S. Surfen, a proteoglycan binding agent, reduces inflammation but inhibits remyelination in murine models of Multiple Sclerosis. Acta Neuropathol. Commun. 2018, 6, 4. [Google Scholar] [CrossRef]
- Vinukonda, G.; Dohare, P.; Arshad, A.; Zia, M.T.; Panda, S.; Korumilli, R.; Kayton, R.; Hascall, V.C.; Lauer, M.E.; Ballabh, P. Hyaluronidase and Hyaluronan Oligosaccharides Promote Neurological Recovery after Intraventricular Hemorrhage. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 872–889. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Foster, S.C.; Xing, R.; Feistel, K.; Olsen, R.H.; Acevedo, S.F.; Raber, J.; Sherman, L.S. CD44 Transmembrane Receptor and Hyaluronan Regulate Adult Hippocampal Neural Stem Cell Quiescence and Differentiation. J. Boil. Chem. 2017, 292, 4434–4445. [Google Scholar] [CrossRef] [PubMed]
- Tuohy, T.M.; Wallingford, N.; Liu, Y.; Chan, F.H.; Rizvi, T.; Xing, R.; Bebo, B.; Rao, M.S.; Sherman, L.S. CD44 overexpression by oligodendrocytes: A novel mouse model of inflammation-independent demyelination and dysmyelination. Glia 2004, 47, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Tuohy, T.M.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.L.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Preston, M.; Gong, X.; Su, W.; Matsumoto, S.G.; Banine, F.; Winkler, C.; Foster, S.; Xing, R.; Struve, J.; Dean, J.; et al. Digestion products of the PH20 hyaluronidase inhibit remyelination. Ann. Neurol. 2013, 73, 266–280. [Google Scholar] [CrossRef]
- Su, W.; Matsumoto, S.; Banine, F.; Srivastava, T.; Dean, J.; Foster, S.; Pham, P.; Hammond, B.; Peters, A.; Girish, K.S.; et al. A modified flavonoid accelerates oligodendrocyte maturation and functional remyelination. Glia 2020, 68, 263–279. [Google Scholar] [CrossRef]
- Ciftci, O.; Ozcan, C.; Kamisli, O.; Cetin, A.; Basak, N.; Aytac, B. Hesperidin, a Citrus Flavonoid, Has the Ameliorative Effects Against Experimental Autoimmune Encephalomyelitis (EAE) in a C57BL/J6 Mouse Model. Neurochem. Res. 2015, 40, 1111–1120. [Google Scholar] [CrossRef]
- Ginwala, R.; McTish, E.; Raman, C.; Singh, N.; Nagarkatti, M.; Nagarkatti, P.; Sagar, D.; Jain, P.; Khan, Z.K. Apigenin, a Natural Flavonoid, Attenuates EAE Severity Through the Modulation of Dendritic Cell and Other Immune Cell Functions. J. Neuroimmune Pharmacol. Off. J. Soc. Neuroimmune Pharmacol. 2016, 11, 36–47. [Google Scholar] [CrossRef]
- Stoffels, J.M.; Hoekstra, D.; Franklin, R.J.; Baron, W.; Zhao, C. The EIIIA domain from astrocyte-derived fibronectin mediates proliferation of oligodendrocyte progenitor cells following CNS demyelination. Glia 2015, 63, 242–256. [Google Scholar] [CrossRef]
- Willis, C.M.; Crocker, S.J. The Mosaic of Extracellular Matrix in the Central Nervous System as a Determinant of Glial Heterogeneity; IntechOpen: London, UK, 2016. [Google Scholar] [CrossRef]
- Yahn, S.L.; Li, J.; Goo, I.; Gao, H.; Brambilla, R.; Lee, J.K. Fibrotic scar after experimental autoimmune encephalomyelitis inhibits oligodendrocyte differentiation. Neurobiol. Dis. 2019, 134, 104674. [Google Scholar] [CrossRef]
- Barros, C.S.; Franco, S.J.; Muller, U. Extracellular matrix: Functions in the nervous system. Cold Spring Harb. Perspect. Boil. 2011, 3, a005108. [Google Scholar] [CrossRef] [PubMed]
- Buttery, P.C.; ffrench-Constant, C. Laminin-2/integrin interactions enhance myelin membrane formation by oligodendrocytes. Mol. Cell. Neurosci. 1999, 14, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.T.T.; Stewart, C.; Pallen, C.J. PTPalpha is required for laminin-2-induced Fyn-Akt signaling to drive oligodendrocyte differentiation. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, T.; Paes de Faria, J.; Bippes, C.A.; Maia, J.; Lopes-da-Silva, J.A.; Relvas, J.B.; Graos, M. Modulation of oligodendrocyte differentiation and maturation by combined biochemical and mechanical cues. Sci. Rep. 2016, 6, 21563. [Google Scholar] [CrossRef] [PubMed]
- Relucio, J.; Tzvetanova, I.D.; Ao, W.; Lindquist, S.; Colognato, H. Laminin alters fyn regulatory mechanisms and promotes oligodendrocyte development. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 11794–11806. [Google Scholar] [CrossRef]
- Aranmolate, A.; Tse, N.; Colognato, H. Myelination is delayed during postnatal brain development in the mdx mouse model of Duchenne muscular dystrophy. BMC Neurosci. 2017, 18, 63. [Google Scholar] [CrossRef]
- Baron, W.; Bijlard, M.; Nomden, A.; de Jonge, J.C.; Teunissen, C.E.; Hoekstra, D. Sulfatide-mediated control of extracellular matrix-dependent oligodendrocyte maturation. Glia 2014, 62, 927–942. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marangon, D.; Boccazzi, M.; Lecca, D.; Fumagalli, M. Regulation of Oligodendrocyte Functions: Targeting Lipid Metabolism and Extracellular Matrix for Myelin Repair. J. Clin. Med. 2020, 9, 470. https://doi.org/10.3390/jcm9020470
Marangon D, Boccazzi M, Lecca D, Fumagalli M. Regulation of Oligodendrocyte Functions: Targeting Lipid Metabolism and Extracellular Matrix for Myelin Repair. Journal of Clinical Medicine. 2020; 9(2):470. https://doi.org/10.3390/jcm9020470
Chicago/Turabian StyleMarangon, Davide, Marta Boccazzi, Davide Lecca, and Marta Fumagalli. 2020. "Regulation of Oligodendrocyte Functions: Targeting Lipid Metabolism and Extracellular Matrix for Myelin Repair" Journal of Clinical Medicine 9, no. 2: 470. https://doi.org/10.3390/jcm9020470
APA StyleMarangon, D., Boccazzi, M., Lecca, D., & Fumagalli, M. (2020). Regulation of Oligodendrocyte Functions: Targeting Lipid Metabolism and Extracellular Matrix for Myelin Repair. Journal of Clinical Medicine, 9(2), 470. https://doi.org/10.3390/jcm9020470