Characteristics and Dysbiosis of the Gut Microbiome in Renal Transplant Recipients

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Experimental Section
2.1. Study Population
2.2. Patient Characteristics
2.3. Sample Collection
2.4. DNA Extraction and 16S rRNA Sequencing
2.5. Microbiome Profiling
2.6. Statistical Analyses
3. Results
3.1. Baseline Characteristics
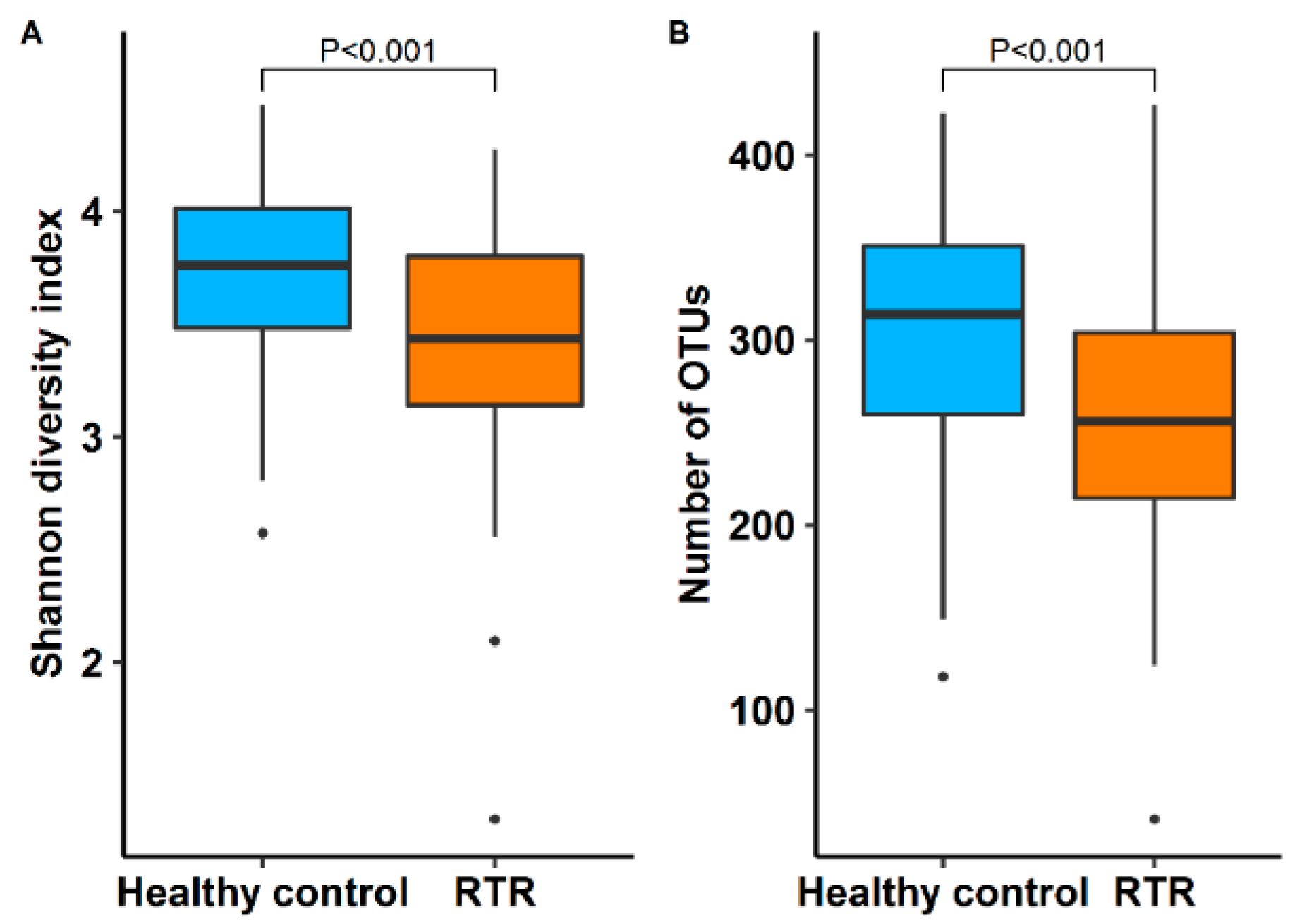
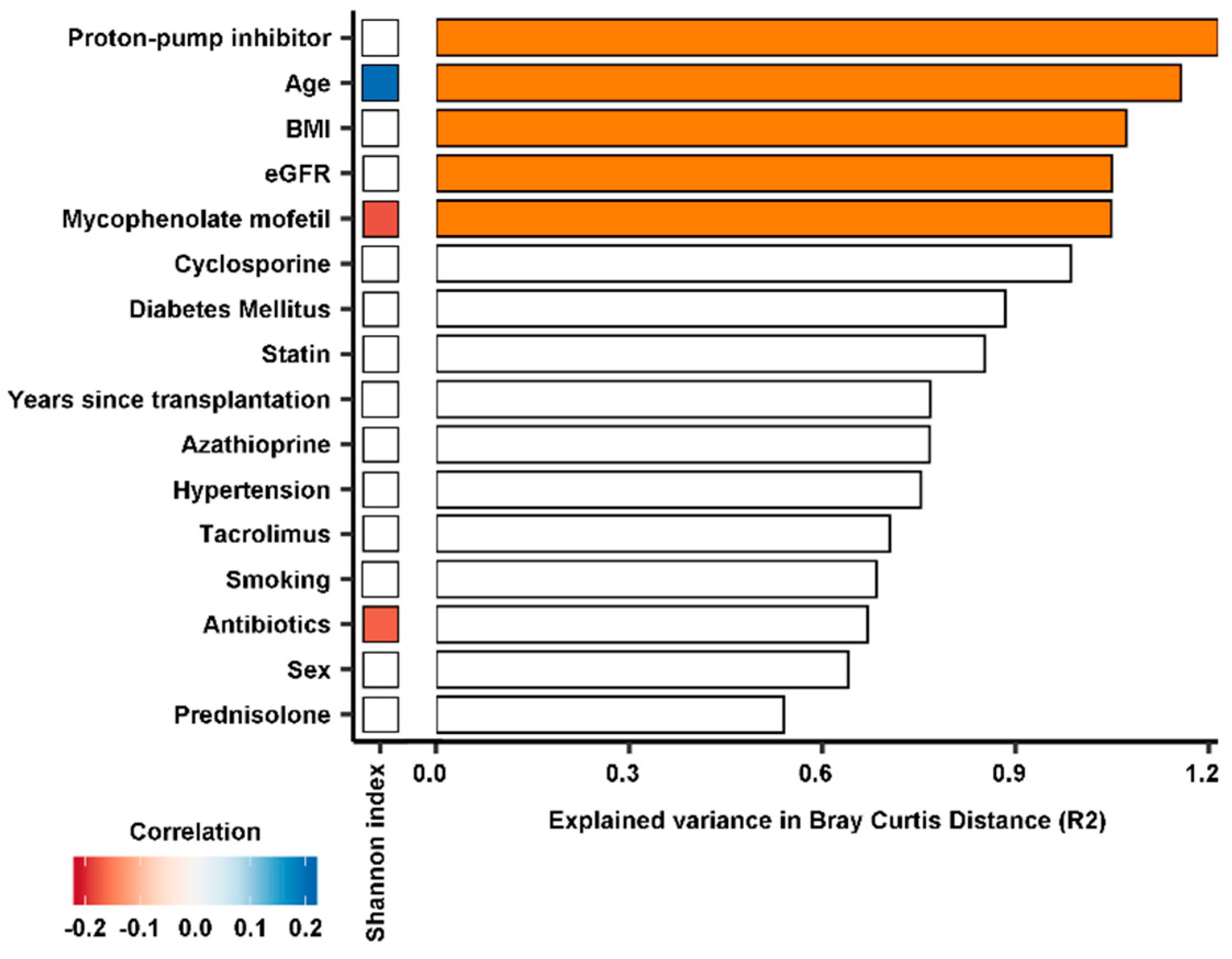
3.2. Diversity of the Gut Microbiome
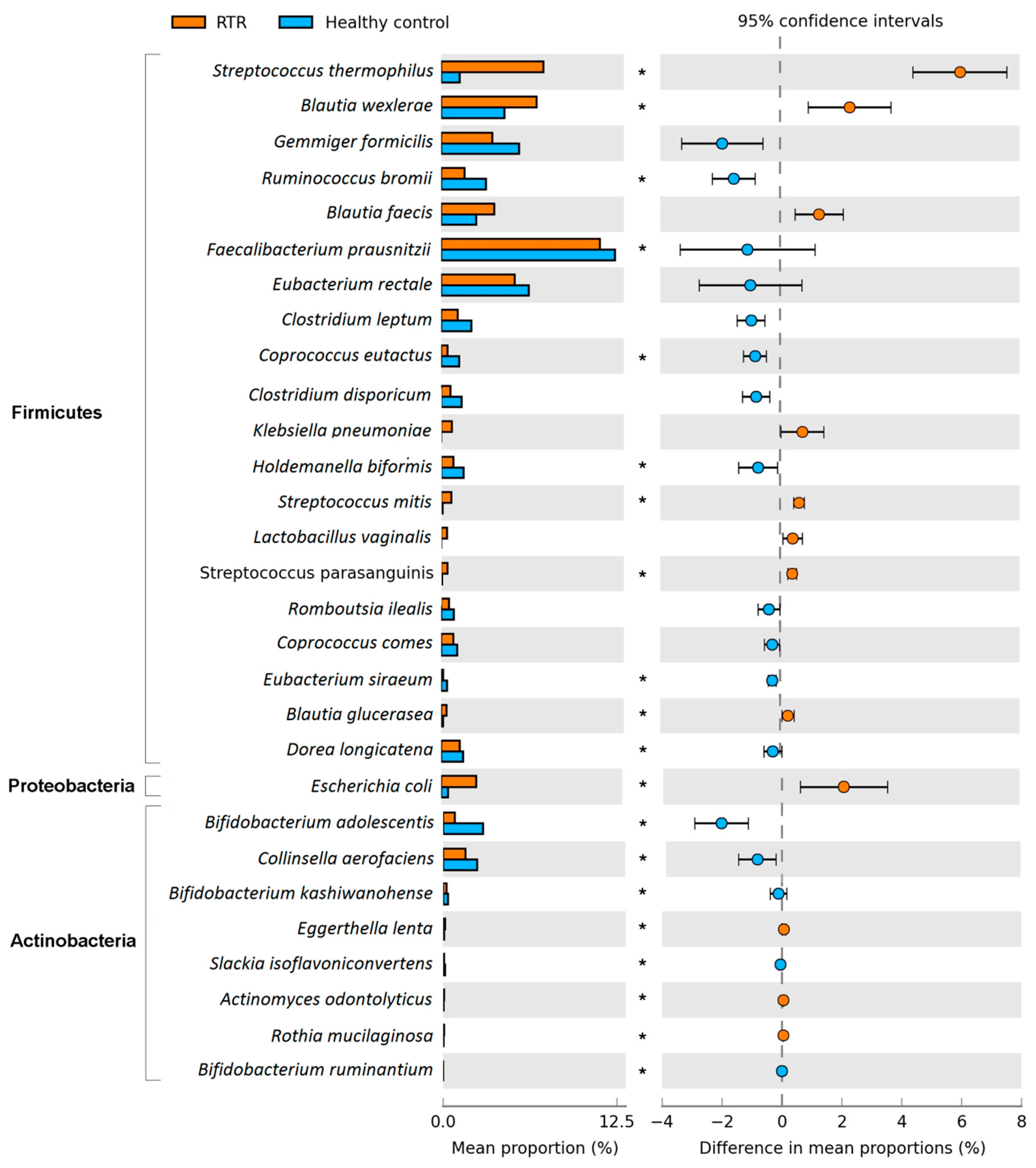
3.3. Composition of the Gut Microbiome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Ekberg, H.; Kyllönen, L.; Madsen, S.; Grave, G.; Solbu, D.; Holdaas, H. Clinicians underestimate gastrointestinal symptoms and overestimate quality of life in renal transplant recipients: A multinational survey of nephrologists. Transplantation 2007, 84, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Bunnapradist, S.; Neri, L.; Wong, W.; Lentine, K.L.; Burroughs, T.E.; Pinsky, B.W.; Takemoto, S.K.; Schnitzler, M.A. Incidence and Risk Factors for Diarrhea Following Kidney Transplantation and Association with Graft Loss and Mortality. Am. J. Kidney Dis. 2008, 51, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Magruder, M.; Zhang, L.; Westblade, L.F.; Satlin, M.J.; Robertson, A.; Edusei, E.; Crawford, C.; Ling, L.; Taur, Y.; et al. Gut microbiota dysbiosis and diarrhea in kidney transplant recipients. Am. J. Transplant. 2019, 19, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 2019, 570, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Tourret, J.; Willing, B.P.; Dion, S.; MacPherson, J.; Denamur, E.; Finlay, B.B. Immunosuppressive treatment alters secretion of ileal antimicrobial peptides and gut microbiota, and favors subsequent colonization by uropathogenic Escherichia coli. Transplantation 2017, 101, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Taur, Y.; Jenq, R.R.; Perales, M.A.; Littmann, E.R.; Morjaria, S.; Ling, L.; No, D.; Gobourne, A.; Viale, A.; Dahi, P.B.; et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 2014, 124, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Muthukumar, T.; Dadhania, D.; Toussaint, N.C.; Ling, L.; Pamer, E.; Suthanthiran, M. Gut microbial community structure and complications after kidney transplantation: A pilot study. Transplantation 2014, 98, 697–705. [Google Scholar] [CrossRef]
- Taur, Y.; Jenq, R.R.; Ubeda, C.; Van Den Brink, M.; Pamer, E.G. Role of intestinal microbiota in transplantation outcomes. Best Pract. Res. Clin. Haematol. 2015, 28, 155–161. [Google Scholar] [CrossRef]
- Annavajhala, M.K.; Gomez-Simmonds, A.; Macesic, N.; Sullivan, S.B.; Kress, A.; Khan, S.D.; Giddins, M.J.; Stump, S.; Kim, G.I.; Narain, R.; et al. Colonizing multidrug-resistant bacteria and the longitudinal evolution of the intestinal microbiome after liver transplantation. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Eisenga, M.F.; Gomes-Neto, A.W.; Van Londen, M.; Ziengs, A.L.; Douwes, R.M.; Stam, S.P.; Osté, M.C.J.; Knobbe, T.J.; Hessels, N.R.; Buunk, A.M.; et al. Rationale and design of TransplantLines: A prospective cohort study and biobank of solid organ transplant recipients. BMJ Open 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gavin, J.R.; Alberti, K.G.M.M.; Davidson, M.B.; DeFronzo, R.A.; Drash, A.; Gabbe, S.G.; Genuth, S.; Harris, M.I.; Kahn, R.; Keen, H.; et al. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 2002, 25, 5–20. [Google Scholar]
- De Goffau, M.C.; Luopajärvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Härkönen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef]
- Heida, F.H.; Van Zoonen, A.G.J.F.; Hulscher, J.B.F.; Te Kiefte, B.J.C.; Wessels, R.; Kooi, E.M.W.; Bos, A.F.; Harmsen, H.J.M.; De Goffau, M.C. A necrotizing enterocolitis-associated gut microbiota is present in the meconium: Results of a prospective study. Clin. Infect. Dis. 2016, 62, 863–870. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2011, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinformatics 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar, A.; Buchner, A.; Lai, T.; Steppi, S.; Jacob, G.; et al. ARB: A software environment for sequence data. Nucleic Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community Ecology Package. 2018. Available online: https://cran.ism.ac.jp/web/packages/vegan/vegan.pdf (accessed on 1 February 2020).
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef]
- Consortium, T.H.M.P. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2013, 486, 1049–1058. [Google Scholar]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Canani, R.B.; Costanzo, M.; Di Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2014, 760, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Sampaio-Maia, B.; Simões-Silva, L.; Pestana, M.; Araujo, R.; Soares-Silva, I.J. The Role of the Gut Microbiome on Chronic Kidney Disease. Adv. Appl. Microbiol. 2016, 96, 65–94. [Google Scholar] [PubMed]
- Mathewson, N.D.; Jenq, R.; Mathew, A.V.; Koenigsknecht, M.; Hanash, A.; Toubai, T.; Oravecz-Wilson, K.; Wu, S.R.; Sun, Y.; Rossi, C.; et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 2016, 17, 505–513. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef]
- Tang, Y.; Forsyth, C.B.; Keshavarzian, A. New molecular insights into inflammatory bowel disease-induced diarrhea. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 615–625. [Google Scholar] [CrossRef]
- Imhann, F.; Bonder, M.J.; Vila, A.V.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.M.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef]
- Spasić, A.; Catić-Đorđević, A.; Veličković-Radovanović, R.; Stefanović, N.; Džodić, P.; Cvetković, T. Adverse effects of mycophenolic acid in renal transplant recipients: Gender differences. Int. J. Clin. Pharm. 2019, 41, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.M.; Gross, A.L.; Shaffer, A.A.; Haugen, C.E.; Norman, S.P.; Xue, Q.L.; Sharrett, A.R.; Carlson, M.C.; Bandeen-Roche, K.; Segev, D.L.; et al. Frailty and changes in cognitive function after kidney transplantation. J. Am. Soc. Nephrol. 2019, 30, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Pruett, T. Spectrum of Cancer Risk Among US Solid Organ Transplant Recipients. Yearb. Surg. 2012, 2012, 105–106. [Google Scholar] [CrossRef]
- Appleton, J. The gut-brain axis: Influence of microbiota on mood and mental health. Integr. Med. 2018, 17, 28–32. [Google Scholar]
- Salem, I.; Ramser, A.; Isham, N.; Ghannoum, M.A. The gut microbiome as a major regulator of the gut-skin axis. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Eisenga, M.F.; Kieneker, L.M.; Soedamah-Muthu, S.S.; Van Den Berg, E.; Deetman, P.E.; Navis, G.J.; Gans, R.O.B.; Gaillard, C.A.J.M.; Bakker, S.J.L.; Joosten, M.M. Urinary potassium excretion, renal ammoniagenesis, and risk of graft failure and mortality in renal transplant recipients1-3. Am. J. Clin. Nutr. 2016, 104, 1703–1711. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Fouque, D. Nutritional management of chronic kidney disease. N. Engl. J. Med. 2017, 377, 1765–1776. [Google Scholar] [CrossRef]
- Novotný, M.; Klimova, B.; Valis, M. Microbiome and cognitive impairment: Can any diets influence learning processes in a positive way? Front. Aging Neurosci. 2019, 11, 1–7. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4, 1–12. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographics | Control | RTRs | p-Value |
|---|---|---|---|
| Number of Subjects, n (%) | 105 | 139 | - |
| Age (years) | 59.2 ± 10.6 | 58.3 ± 12.8 | 0.96 |
| Male, n (%) | 60 (57) | 69 (50) | 0.24 |
| BMI (kg/m²) | 27.2 ± 6.0 | 27.7 ± 5.4 | 0.60 |
| Diabetes Mellitus, n (%) | 3 (3) | 38 (27) | <0.001 |
| Hypertension, n (%) | 10 (10) | 115 (83) | <0.001 |
| Smoking, n (%) | - | 12 (9) | - |
| Years since Transplantation, Median (IQR) | - | 6.0 (1.5–12.5) | - |
| Cardiovascular Parameters | |||
| Glucose, mmol/L, Median (IQR) | 5.4 (4.0–5.9) | 5.4 (4.9–6.2) | 0.06 |
| HbA1c, mmol/L, Median (IQR) | 37.5 (36.0–40.0) | 40.0 (37.0–46.0) | <0.001 |
| Systolic Blood Pressure (mmHg) | 130.4 ± 14.2 | 136.5 ± 17.7 | 0.02 |
| Diastolic Blood Pressure (mmHg) | 75.8 ± 9.4 | 78.5 ± 9.6 | 0.03 |
| Heart Frequency (bpm) | 69.7 ± 25.8 | 72.1 ± 13.1 | 0.02 |
| Renal Function Parameters | |||
| Serum Creatinine (µmol/L) | 97.3 ± 22.1 | 133.1 ± 42.6 | <0.001 |
| eGFR (mL/min/1.73 m2) | 69.0 ± 19.2 | 48.3 ± 16.7 | <0.001 |
| Proteinuria (0.5 g/24 h), n (%) | 0 (0) | 11 (7.9) | - |
| Medication, n (%) | |||
| Antibiotics (n = 1) | 0 (0) | 7 (5) | - |
| Antihypertensive Agents (n = 8) | 10 (10) | 115 (83) | <0.001 |
| Proton-pump Inhibitors | 8 (8) | 96 (69) | <0.001 |
| Statins | 8 (8) | 66 (47) | <0.001 |
| Cyclosporine | - | 25 (18) | - |
| Tacrolimus | - | 79 (57) | - |
| Azathioprine | - | 13 (9) | - |
| Mycophenolate mofetil | - | 100 (72) | - |
| Prednisolone | - | 133 (96) | - |
| Taxonomic Level | Total Number of Taxa 1 | Number of Significant Taxa 2 |
|---|---|---|
| Phylum | 6 | 2 |
| Class | 17 | 5 |
| Order | 31 | 8 |
| Family | 60 | 18 |
| Genus | 123 | 27 |
| Species | 205 | 63 |
| Total | 442 | 123 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swarte, J.C.; Douwes, R.M.; Hu, S.; Vich Vila, A.; Eisenga, M.F.; van Londen, M.; Gomes-Neto, A.W.; Weersma, R.K.; Harmsen, H.J.M.; Bakker, S.J.L. Characteristics and Dysbiosis of the Gut Microbiome in Renal Transplant Recipients. J. Clin. Med. 2020, 9, 386. https://doi.org/10.3390/jcm9020386
Swarte JC, Douwes RM, Hu S, Vich Vila A, Eisenga MF, van Londen M, Gomes-Neto AW, Weersma RK, Harmsen HJM, Bakker SJL. Characteristics and Dysbiosis of the Gut Microbiome in Renal Transplant Recipients. Journal of Clinical Medicine. 2020; 9(2):386. https://doi.org/10.3390/jcm9020386
Chicago/Turabian StyleSwarte, J. Casper, Rianne M. Douwes, Shixian Hu, Arnau Vich Vila, Michele F. Eisenga, Marco van Londen, António W. Gomes-Neto, Rinse K. Weersma, Hermie J.M. Harmsen, and Stephan J.L. Bakker. 2020. "Characteristics and Dysbiosis of the Gut Microbiome in Renal Transplant Recipients" Journal of Clinical Medicine 9, no. 2: 386. https://doi.org/10.3390/jcm9020386
APA StyleSwarte, J. C., Douwes, R. M., Hu, S., Vich Vila, A., Eisenga, M. F., van Londen, M., Gomes-Neto, A. W., Weersma, R. K., Harmsen, H. J. M., & Bakker, S. J. L. (2020). Characteristics and Dysbiosis of the Gut Microbiome in Renal Transplant Recipients. Journal of Clinical Medicine, 9(2), 386. https://doi.org/10.3390/jcm9020386