Plasma Krebs Cycle Intermediates in Nonalcoholic Fatty Liver Disease

 ,
,
Abstract
1. Introduction
2. Experimental Section
2.1. Subjects
2.2. Sample Preparation for Krebs Cycle Intermediates
2.3. Gas Chromatography-mass Spectrometry (GCMS) Analysis
Aconitate and Isocitrate Quantification
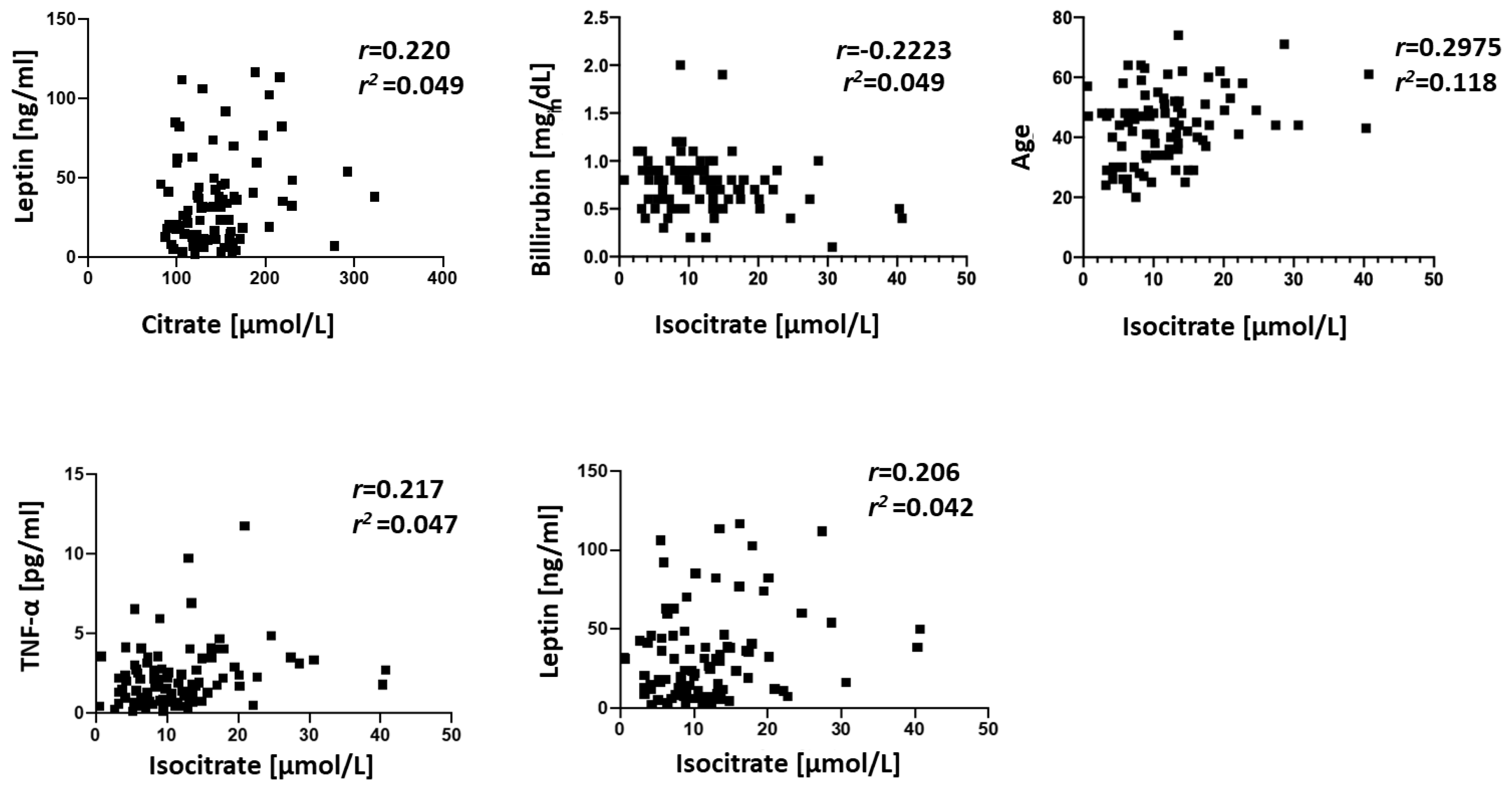
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexander, M.; Katrina Loomis, A.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019. [Google Scholar] [CrossRef]
- Wong, S.-W.; Ting, Y.-W.; Chan, W.-K. Epidemiology of non-alcoholic fatty liver disease-related hepatocellular carcinoma and its implications. JGH Open 2018, 2, 235–241. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. 2008, 14, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-L.; Chen, H.; Wang, C.-L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef] [PubMed]
- Kovalic, A.J.; Cholankeril, G.; Satapathy, S.K. Nonalcoholic fatty liver disease and alcoholic liver disease: Metabolic diseases with systemic manifestations. Transl. Gastroenterol. Hepatol. 2019, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Kugelmas, M.; Hill, D.B.; Vivian, B.; Marsano, L.; McClain, C.J. Cytokines and NASH: A pilot study of the effects of lifestyle modification and vitamin E. Hepatology 2003, 38, 413–419. [Google Scholar] [CrossRef]
- Eccleston, H.B.; Andringa, K.K.; Betancourt, A.M.; King, A.L.; Mantena, S.K.; Swain, T.M.; Tinsley, H.N.; Nolte, R.N.; Nagy, T.R.; Abrams, G.A.; et al. Chronic exposure to a high-fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid. Redox Signal. 2011, 15, 447–459. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Dasarathy, S.; Dasarathy, J.; Khiyami, A.; Joseph, R.; Lopez, R.; McCullough, A.J. Validity of real time ultrasound in the diagnosis of hepatic steatosis: A prospective study. J. Hepatol. 2009, 51, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Anstee, Q.M.; McPherson, S. Non-alcoholic fatty liver disease: A practical approach to diagnosis and staging. Frontline Gastroenterol. 2014, 5, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.; Caldwell, S.H. Nonalcoholic steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003, 37, 1202–1219. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; McCullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.S.; Kim, D.; Chung, G.E.; Kang, S.J.; Park, M.J.; Kim, Y.J.; Yoon, J.H.; Lee, H.S. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2012, 18, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Peng, L.; Xia, Z.; Zhang, L.; Song, Z.; Wang, Y.; Peng, Y. Triglycerides to high-density lipoprotein cholesterol ratio as a surrogate for nonalcoholic fatty liver disease: A cross-sectional study. Lipids Health Dis. 2019, 18, 39. [Google Scholar] [CrossRef]
- Fan, R.; Wang, J.; Du, J. Association between body mass index and fatty liver risk: A dose-response analysis. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, D.; Kim, H.J.; Lee, C.H.; Yang, J.I.; Kim, W.; Kim, Y.J.; Yoon, J.H.; Cho, S.H.; Sung, M.W.; et al. Hepatic steatosis index: A simple screening tool reflecting nonalcoholic fatty liver disease. Dig. Liver Dis. 2010, 42, 503–508. [Google Scholar] [CrossRef]
- Naseri, N.N.; Bonica, J.; Xu, H.; Park, L.C.; Arjomand, J.; Chen, Z.; Gibson, G.E. Novel Metabolic Abnormalities in the Tricarboxylic Acid Cycle in Peripheral Cells from Huntington’s Disease Patients. PLoS ONE 2016, 11, e0160384. [Google Scholar] [CrossRef]
- Legault, J.T.; Strittmatter, L.; Tardif, J.; Rioux, J.D.; Mootha, V.K.; Des, C.; Sharma, R.; Tremblay-Vaillancourt, V.; Aubut, C.; Boucher, G.; et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome in Brief A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep. 2015, 13, 981–989. [Google Scholar]
- Rustin, P.; Bourgeron, T.; Parfait, B.; Chretien, D.; Munnich, A.; Rötig, A. Inborn errors of the Krebs cycle: A group of unusual mitochondrial diseases in human. Biochim. Biophys. Acta Mol. Basis Dis. 1997, 1361, 185–197. [Google Scholar] [CrossRef]
- Delaney, N.F.; Sharma, R.; Tadvalkar, L.; Clish, C.B.; Haller, R.G.; Mootha, V.K. Metabolic profiles of exercise in patients with McArdle disease or mitochondrial myopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 8402–8407. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Talamanca, A.A.; Castello, G.; Cordero, M.D.; D’ischia, M.; Gadaleta, M.N.; Pallardó, F.V.; Petrovit, S.; Tiano, L.; Zatterale, A. Oxidative Stress and Mitochondrial Dysfunction across Broad-Ranging Pathologies: Toward Mitochondria-Targeted Clinical Strategies. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef]
- Lee, K.; Haddad, A.; Osme, A.; Kim, C.; Borzou, A.; Ilchenko, S.; Allende, D.; Dasarathy, S.; McCullough, A.; Sadygov, R.G.; et al. Hepatic mitochondrial defects in a nonalcoholic fatty liver disease mouse model are associated with increased degradation of oxidative phosphorylation subunits. Mol. Cell. Proteom. 2018, 17, 2371–2386. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Schlensak, M.; Roden Correspondence, M.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis Cell Metabolism Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Liang, J. Blood urea nitrogen is elevated in patients with non-alcoholic fatty liver disease. Hepatogastroenterology 2013, 60, 343–345. [Google Scholar]
- Bahcecioglu, I.H.; Yalniz, M.; Ataseven, H.; Ilhan, N.; Ozercan, I.H.; Seckin, D.; Sahin, K. Levels of serum hyaluronic acid, TNF-α and IL-8 in patients with nonalcoholic steatohepatitis. Hepatogastroenterology 2005, 52, 1549–1553. [Google Scholar]
- Siebler, J.; Galle, P.R.; Weber, M.M. The gut-liver-axis: Endotoxemia, inflammation, insulin resistance and NASH. J. Hepatol. 2008, 48, 1032–1034. [Google Scholar] [CrossRef]
- Uygun, A.; Kadayifci, A.; Yesilova, Z.; Erdil, A.; Yaman, H.; Saka, M.; Deveci, M.S.; Bagci, S.; Gulsen, M.; Karaeren, N.; et al. Serum leptin levels in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2000, 95, 3584–3589. [Google Scholar] [CrossRef]
- Chitturi, S.; Farrell, G.; Frost, L.; Kriketos, A.; Lin, R.; Liddle, C.; Samarasinghe, D.; George, J. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: A manifestation of lipotoxicity? Hepatology 2002, 36, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Serin, E.; Özer, B.; Gümürdülü, Y.; Kayaselçuk, F.; Kul, K.; Boyacioǧlu, S. Serum leptin level can be a negative marker of hepatocyte damage in nonalcoholic fatty liver. J. Gastroenterol. 2003, 38, 471–476. [Google Scholar] [CrossRef] [PubMed]
- van de Wier, B.; Balk, J.M.; Haenen, G.R.M.M.; Giamouridis, D.; Bakker, J.A.; Bast, B.C.; den Hartog, G.J.M.; Koek, G.H.; Bast, A. Elevated citrate levels in non-alcoholic fatty liver disease: The potential of citrate to promote radical production. FEBS Lett. 2013, 587, 2461–2466. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Li, Z.; Erion, D.; Maurer, T. Model-Based Assessment of Plasma Citrate Flux into the Liver: Implications for NaCT as a Therapeutic Target. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 132–139. [Google Scholar] [CrossRef]
- Gong, F.; Gao, L.; Ding, T. IDH2 protects against nonalcoholic steatohepatitis by alleviating dyslipidemia regulated by oxidative stress. Biochem. Biophys. Res. Commun. 2019, 514, 593–600. [Google Scholar] [CrossRef]
- De Chiara, F.; Thomsen, K.L.; Habtesion, A.; Jones, H.; Davies, N.; Gracia-Sancho, J.; Manicardi, N.; Hall, A.; Andreola, F.; Paish, H.L.; et al. Ammonia Scavenging Prevents Progression of Fibrosis in Experimental Nonalcoholic Fatty Liver Disease. Hepatology 2019. [Google Scholar] [CrossRef]
- Davuluri, G.; Allawy, A.; Thapaliya, S.; Rennison, J.H.; Singh, D.; Kumar, A.; Sandlers, Y.; Van Wagoner, D.R.; Flask, C.A.; Hoppel, C.; et al. Hyperammonaemia-induced skeletal muscle mitochondrial dysfunction results in cataplerosis and oxidative stress. J. Physiol. 2016, 594, 7341–7360. [Google Scholar] [CrossRef]
- Batshaw, M.L.; Walser, M.; Brusilow, S.W. Plasma α-ketoglutarate in urea cycle enzymopathies and its role as a harbinger of hyperammonemic coma. Pediatr. Res. 1980, 14, 1316–1319. [Google Scholar] [CrossRef]
- Shigiyama, F.; Kumashiro, N.; Furukawa, Y.; Funayama, T.; Takeno, K.; Wakui, N.; Ikehara, T.; Nagai, H.; Taka, H.; Fujimura, T.; et al. Characteristics of hepatic insulin-sensitive nonalcoholic fatty liver disease. Hepatol. Commun. 2017, 1, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.L.; Plaut, G.W. Specific site inhibitors of NAD-specific isocitrate dehydrogenase from bovine heart. J. Biol. Chem. 1982, 257, 8021–8029. [Google Scholar] [PubMed]
- Ogasawara, N.; Watanabe, T.; Goto, H. Bilirubin: A potent inhibitor of NAD+-linked isocitrate dehydrogenase. Biochim. Biophys. Acta 1973, 327, 233–237. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 67) | NAFLD (n = 22) | p-Value | |
|---|---|---|---|
| Age (years) | 42.25 | 47.3 | |
| Male/Female | 21/67 | 1/22 | |
| BMI (kg/m2) | 29.7 ± 7.5 | 40 ± 7.5 | <0.001 |
| Albumin (g/dL) | 4.01 ± 0.22 | 3.84 ± 0.43 | 0.02 |
| Bilirubin (mg/dL) | 0.77 ± 0.24 | 0.72 ± 0.40 | 0.50 |
| ALP(U/L) | 93.6 ± 16.8 | 95.4 ± 16.37 | 0.70 |
| ALT (IU/L) | 22.12 ± 6.5 | 30.19 ± 11.53 | 0.001 |
| AST (IU/L) | 21.4 ± 4.54 | 28.42 ± 11.12 | <0.001 |
| AST/ALT | 1.00 ± 0.21 | 0.94 ± 0.23 | 0.3 |
| BUN (mg/dL) | 14.2 ± 3.8 | 15 ± 6.7 | 0.49 |
| Creatinine (mg/dL) | 0.82 ± 0.22 | 0.8 ± 0.2 | 0.70 |
| Glucose (mg/dL) | 90.7 ± 17.45 | 127.8 ± 77.1 | 0.005 |
| HbA1c (%) | 5.5 ± 0.77 | 6.5 ± 1.6 | 0.001 |
| Total chol (mg/dL) | 201 ± 47 | 192.3 ± 41.04 | 0.40 |
| HDL-C (mg/dL) | 47 ± 9.95 | 48 ± 13 | 0.39 |
| TG (mg/dL) | 171 ± 9.2 | 192.2 ± 123 | 0.89 |
| TG/HDL-C | 3.72 ± 2.22 | 4.21 ± 2.22 | 0.38 |
| TNF-α (pg/mL) | 1.86 ± 1.62 | 3.63 ± 2.8 | <0.001 |
| Leptin (ng/mL) | 23 ± 18.09 | 63.0 ± 34 | <0.001 |
| HSI | 39.19 ± 8.07 | 50.42 ± 14.9 | <0.001 |
| Control (n = 57) | NAFLD (n = 7) | p Value | |
|---|---|---|---|
| Age (years) | 42.1 | 46 | |
| Male/Female | 16/41 | 1/8 | |
| BMI (kg/m2) | 29.5 ± 7.9 | 32.4 ± 10.5 | 0.3 |
| Albumin (g/dL) | 3.98 ± 0.21 | 3.97 ± 0.32 | 0.92 |
| Bilirubin (mg/dL) | 0.77 ± 0.24 | 0.62 ± 0.16 | 0.07 |
| ALP(U/L) | 94.8 ± 16.1 | 95.8 ± 21.4 | 0.8 |
| ALT (IU/L) | 21.7 ± 5.4 | 29.6 ± 13.3 | 0.002 |
| AST (IU/L) | 21.1 ± 4.5 | 30.0 ± 13.0 | <0.001 |
| BUN (mg/dL) | 14.03 ± 3.66 | 15.0 ± 5.5 | 0.50 |
| Creatinine (mg/dL) | 0.79 ± 0.18 | 0.75 ± 0.26 | 0.55 |
| Glucose (mg/dL) | 85.35 ± 9.38 | 88.88 ± 10.7 | 0.30 |
| HbA1c (%) | 5.45 ± 0.54 | 5.9 ± 0.4 | 0.44 |
| Total chol (mg/dL) | 199.3 ± 48 | 179.6 ± 26.2 | 0.23 |
| HDL-C (mg/dL) | 47 ± 10 | 47 ± 12 | 0.76 |
| TG (mg/dL) | 159 ± 84 | 150 ± 58.1 | 0.90 |
| TG/HDL-C | 3.4 ± 1.7 | 1.9 ± 0.84 | 0.84 |
| TNF-α (pg/mL) | 1.8 ± 1.1 | 3.57 ± 3.01 | 0.01 |
| Leptin (ng/mL) | 24.18 ± 18.6 | 61.68 ± 32.2 | <0.001 |
| HSI | 38.9 ± 8.35 | 41.27 ± 11.36 | 0.45 |
| Marker | AUC | p-Value | Error |
|---|---|---|---|
| Citrate | 0.749 | p < 0.05 | 0.097 |
| Isocitrate | 0.750 | p < 0.05 | 0.102 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandlers, Y.; Shah, R.R.; Pearce, R.W.; Dasarathy, J.; McCullough, A.J.; Dasarathy, S. Plasma Krebs Cycle Intermediates in Nonalcoholic Fatty Liver Disease. J. Clin. Med. 2020, 9, 314. https://doi.org/10.3390/jcm9020314
Sandlers Y, Shah RR, Pearce RW, Dasarathy J, McCullough AJ, Dasarathy S. Plasma Krebs Cycle Intermediates in Nonalcoholic Fatty Liver Disease. Journal of Clinical Medicine. 2020; 9(2):314. https://doi.org/10.3390/jcm9020314
Chicago/Turabian StyleSandlers, Yana, Rohan R. Shah, Ryan W. Pearce, Jaividhya Dasarathy, Arthur J. McCullough, and Srinivasan Dasarathy. 2020. "Plasma Krebs Cycle Intermediates in Nonalcoholic Fatty Liver Disease" Journal of Clinical Medicine 9, no. 2: 314. https://doi.org/10.3390/jcm9020314
APA StyleSandlers, Y., Shah, R. R., Pearce, R. W., Dasarathy, J., McCullough, A. J., & Dasarathy, S. (2020). Plasma Krebs Cycle Intermediates in Nonalcoholic Fatty Liver Disease. Journal of Clinical Medicine, 9(2), 314. https://doi.org/10.3390/jcm9020314