You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS

 ,
,  , , ,
, , ,  , and
, and
Abstract
1. The Enteric Nervous System and the Gut–Brain Axis
Circuitry of the Enteric Nervous System
2. Commensal Bacteria and the Enteric Nervous System
2.1. Microbiota and Social Behavior
2.2. Microbiota, Sleep Cycle, and Mood Disorders
2.3. Microbiota and Alzheimer’s Disease
2.4. Microbiota and Parkinson’s Disease
2.5. Microbiota and Other Neurodegenerative Disease
3. Pathogenic Bacteria and the Enteric Nervous System
3.1. Toxins Promoting Secretion
3.2. Toxins Promoting Emesis
4. Viral Influence on the Enteric Nervous System
5. Parasitic Influence on the Enteric Nervous System
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Furness, J.B. Types of neurons in the enteric nervous system. J. Auton. Nerv. Syst. 2000, 81, 87–96. [Google Scholar] [CrossRef]
- Yang, N.J.; Chiu, I.M. Bacterial Signaling to the Nervous System through Toxins and Metabolites. J. Mol. Biol. 2017, 429, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Fekete, E.; Timmermans, J.P.; Resch, B.A.; Scheuermann, D.W. Different distribution of S-100 protein and glial fibrillary acidic protein (GFAP) immunoreactive cells and their relations with nitrergic neurons in the human fetal small intestine. Histol. Histopathol. 1999, 14, 785–790. [Google Scholar] [PubMed]
- Christofi, F.L. TRPV1 Sensory Neurons and Enteric Glia in ENS Link Tachykinins to Neuroinflammation and Nociception. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 354–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-X.; Wang, Y.-P. Gut Microbiota-brain Axis. Chin. Med. J. 2016, 129, 2373–2380. [Google Scholar] [CrossRef]
- Foster, J.A.; Neufeld, K.-A.M. Gut–brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J. Clin. Investig. 2015, 125, 926–938. [Google Scholar] [CrossRef]
- Schemann, M.; Neunlist, M. The human enteric nervous system. Neurogastroenterol. Motil. 2004, 16, 55–59. [Google Scholar] [CrossRef]
- Anlauf, M.; Schäfer, M.K.-H.; Eiden, L.E.; Weihe, E. Chemical coding of the human gastrointestinal nervous system: Cholinergic, VIPergic, and catecholaminergic phenotypes. J. Comp. Neurol. 2003, 459, 90–111. [Google Scholar] [CrossRef]
- Szurszewski, J.H. Physiology of Mammalian Prevertebral Ganglia. Annu. Rev. Physiol. 1981, 43, 53–68. [Google Scholar] [CrossRef]
- Chang, H.Y.; Mashimo, H.; Goyal, R.K. Musings on the wanderer: What’s new in our understanding of vago-vagal reflex? IV. Current concepts of vagal efferent projections to the gut. Am. J. Physiol. Liver Physiol. 2003, 284, G357–G366. [Google Scholar] [CrossRef]
- Mulak, A. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.-W.; Gao, X.-B.; et al. A Neural Circuit for Gut-Induced Reward. Cell 2018, 175, 665–678.e23. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, E.; Bordenstein, S.R.; Quinn, J.L.; Dinan, T.G.; Cryan, J.F. Microbiota and the social brain. Science 2019, 366, eaar2016. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.L.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Gonkowski, S.; Rytel, L. Somatostatin as an Active Substance in the Mammalian Enteric Nervous System. Int. J. Mol. Sci. 2019, 20, 4461. [Google Scholar] [CrossRef] [PubMed]
- Mourad, F.H.; Barada, K.; Rached, N.A.B.; Khoury, C.I.; Saadé, N.E.; Nassar, C.F. Inhibitory effect of experimental colitis on fluid absorption in rat jejunum: Role of the enteric nervous system, VIP, and nitric oxide. Am. J. Physiol. Liver Physiol. 2006, 290, G262–G268. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, J.A.; Freitas, M.A.R.; De Oliveira, E.C.; Jabari, S.; Brehmer, A.; Da Silveira, A.B.M. 5-HT3A serotonin receptor in the gastrointestinal tract: The link between immune system and enteric nervous system in the digestive form of Chagas disease. Parasitol. Res. 2019, 118, 1325–1329. [Google Scholar] [CrossRef]
- Natale, G.; Ryskalin, L.; Busceti, C.L.; Biagioni, F.; Fornai, F. The nature of catecholamine-containing neurons in the enteric nervous system in relationship with organogenesis, normal human anatomy and neurodegeneration. Arch. Ital. Biol 2017, 155, 118–130. [Google Scholar]
- Lundgren, O. Enteric Nerves and Diarrhoea. Pharmacol. Toxicol. 2002, 90, 109–120. [Google Scholar] [CrossRef]
- Hagbom, M.; Istrate, C.; Engblom, D.; Karlsson, T.; Rodriguez-Diaz, J.; Buesa, J.; Taylor, J.A.; Loitto, V.-M.; Magnusson, K.-E.; Ahlman, H.; et al. Rotavirus Stimulates Release of Serotonin (5-HT) from Human Enterochromaffin Cells and Activates Brain Structures Involved in Nausea and Vomiting. PLOS Pathog. 2011, 7, e1002115. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, M.; Campigotto, M.; Campisciano, G.; Comar, M.; Crocè, L.S. A story of liver and gut microbes: How does the intestinal flora affect liver disease? A review of the literature. Am. J. Physiol. Liver Physiol. 2020, 318, G889–G906. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.R.; Mizrahi-Man, O.; Michelini, K.; Barreiro, L.B.; Ober, C.; Gilad, Y. Seasonal Variation in Human Gut Microbiome Composition. PLoS ONE 2014, 9, e90731. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Rigatto, K.; Gazzana, M.B.; Knorst, M.M.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. Altered Gut Microbiome Profile in Patients With Pulmonary Arterial Hypertension. Hypertension 2020, 75, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- Bercik, P.; Park, A.J.; Sinclair, D.; Khoshdel, A.; Lu, J.; Huang, X.; Deng, Y.; Blennerhassett, P.A.; Fahnestock, M.; Moine, D.; et al. The anxiolytic effect of Bifidobacterium longum NCC3001 involves vagal pathways for gut-brain communication. Neurogastroenterol. Motil. 2011, 23, 1132–1139. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef]
- Forsythe, P.; Bienenstock, J.; Kunze, W.A. Vagal Pathways for Microbiome-Brain-Gut Axis Communication. Adv. Exp. Medi. Biol. 2014, 817, 115–133. [Google Scholar] [CrossRef]
- Bercik, P.; Verdu, E.F.; Foster, J.A.; Macri, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A.; et al. Chronic Gastrointestinal Inflammation Induces Anxiety-Like Behavior and Alters Central Nervous System Biochemistry in Mice. Gastroenterology 2010, 139, 2102–2112.e1. [Google Scholar] [CrossRef]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.J.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; De Angelis, A.L.H.; Jaitin, D.A.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Van De Wouw, M.; Boehme, M.; Lyte, J.M.; Wiley, N.; Strain, C.; O’Sullivan, O.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 2018, 596, 4923–4944. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The Role of Short-Chain Fatty Acids in Health and Disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar]
- Heng, B.C.; Aubel, D.; Fussenegger, M. An overview of the diverse roles of G-protein coupled receptors (GPCRs) in the pathophysiology of various human diseases. Biotechnol. Adv. 2013, 31, 1676–1694. [Google Scholar] [CrossRef]
- Tazoe, H.; Otomo, Y.; Kaji, I.; Tanaka, R.; Karaki, S.-I.; Kuwahara, A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J. Physiol. Pharmacol. 2008, 59, 251–262. [Google Scholar]
- Galland, L. The Gut Microbiome and the Brain. J. Med. Food 2014, 17, 1261–1272. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef]
- Nankova, B.B.; Agarwal, R.; Macfabe, D.F.; La Gamma, E.F. Enteric Bacterial Metabolites Propionic and Butyric Acid Modulate Gene Expression, Including CREB-Dependent Catecholaminergic Neurotransmission, in PC12 Cells - Possible Relevance to Autism Spectrum Disorders. PLoS ONE 2014, 9, e103740. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. The gut microbiota of insects – diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef]
- Martinson, V.G.; Danforth, B.N.; Minckley, R.L.; Rueppell, O.; Tingek, S.; Moran, N.A. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 2011, 20, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Alberoni, D.; Baffoni, L.; Gaggìa, F.; Ryan, P.; Murphy, K.; Ross, P.; Stanton, C.; Di Gioia, D. Impact of beneficial bacteria supplementation on the gut microbiota, colony development and productivity of Apis mellifera L. Benef. Microbes 2018, 9, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Grieneisen, L.E.; Livermore, J.; Alberts, S.; Tung, J.; Archie, E.A. Group Living and Male Dispersal Predict the Core Gut Microbiome in Wild Baboons. Integr. Comp. Biol. 2017, 57, 770–785. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Varian, B.J.; Poutahidis, T.; DiBenedictis, B.T.; Levkovich, T.; Ibrahim, Y.; Didyk, E.; Shikhman, L.; Cheung, H.K.; Hardas, A.; Ricciardi, C.E.; et al. Microbial lysate upregulates host oxytocin. Brain Behav. Immun. 2017, 61, 36–49. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef]
- Gosselin, D.; Rivest, S. MyD88 signaling in brain endothelial cells is essential for the neuronal activity and glucocorticoid release during systemic inflammation. Mol. Psychiatry 2008, 13, 480–497. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011, 23, 255-e119. [Google Scholar] [CrossRef]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Shropshire, J.D.; Bordenstein, S.R. Speciation by Symbiosis: The Microbiome and Behavior. mBio 2016, 7, e01785-15. [Google Scholar] [CrossRef] [PubMed]
- Bordenstein, S.R.; Theis, K.R. Host Biology in Light of the Microbiome: Ten Principles of Holobionts and Hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef] [PubMed]
- Theis, K.R.; Venkataraman, A.; Dycus, J.A.; Koonter, K.D.; Schmitt-Matzen, E.N.; Wagner, A.P.; Holekamp, K.E.; Schmidt, T.M. Symbiotic bacteria appear to mediate hyena social odors. Proc. Natl. Acad. Sci. USA 2013, 110, 19832–19837. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Korzan, W.J.; Ferrero, D.M.; Chang, R.B.; Roy, D.S.; Buchi, M.; Lemon, J.K.; Kaur, A.W.; Stowers, L.; Fendt, M.; et al. Synchronous Evolution of an Odor Biosynthesis Pathway and Behavioral Response. Curr. Biol. 2013, 23, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chafee, M.E.; Zecher, C.N.; Gourley, M.L.; Schmidt, V.T.; Chen, J.H.; Bordenstein, S.R.; Clark, M.E.; Bordenstein, S.R. Decoupling of Host–Symbiont–Phage Coadaptations Following Transfer Between Insect Species. Genetics 2010, 187, 203–215. [Google Scholar] [CrossRef]
- Tung, J.; Barreiro, L.B.; Burns, M.B.; Grenier, J.-C.; Lynch, J.; Grieneisen, L.E.; Altmann, J.; Alberts, S.C.; Blekhman, R.; Archie, E.A. Social networks predict gut microbiome composition in wild baboons. eLife 2015, 4, e05224. [Google Scholar] [CrossRef]
- Phillips, J.G.P. The Treatment of Melancholia by the Lactic Acid Bacillus. J. Ment. Sci. 1910, 56, 422–430. [Google Scholar] [CrossRef]
- Duerkop, B.A.; Vaishnava, S.; Hooper, L.V. Immune Responses to the Microbiota at the Intestinal Mucosal Surface. Immunology 2009, 31, 368–376. [Google Scholar] [CrossRef]
- Heumann, D.; Barras, C.; Severin, A.; Glauser, M.P.; Tomasz, A. Gram-positive cell walls stimulate synthesis of tumor necrosis factor alpha and interleukin-6 by human monocytes. Infect. Immun. 1994, 62, 2715–2721. [Google Scholar] [CrossRef]
- Alam, N.; McGinty, D.; Bashir, T.; Kumar, S.; Imeri, L.; Opp, M.R.; Szymusiak, R. Interleukin-1beta modulates state-dependent discharge activity of preoptic area and basal forebrain neurons: Role in sleep regulation. Eur. J. Neurosci. 2004, 20, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Schuld, A.; Haack, M.; Hinze-Selch, D.; Mullington, J.; Pollmacher, T. Experimentelle Untersuchungen der Interaktion zwischen Schlaf und Immunsystem beim Menschen. PPmP-Psychother. Psychosom. Med. Psychol. 2005, 55, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Fang, J.; Brown, R.A.; Krueger, J.M. Interleukin-18 promotes sleep in rabbits and rats. Am. J. Physiol. Integr. Comp. Physiol. 2001, 281, R828–R838. [Google Scholar] [CrossRef] [PubMed]
- Cermakian, N.; Lange, T.; Golombek, D.; Sarkar, D.; Nakao, A.; Shibata, S.; Mazzoccoli, G. Crosstalk between the circadian clock circuitry and the immune system. Chrono- Int. 2013, 30, 870–888. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Huang, J.-W.; Chiang, C.-K.; Pan, C.-C.; Wu, K.-D.; Tsai, T.-J.; Chen, W.-Y. Higher plasma interleukin-18 levels associated with poor quality of sleep in peritoneal dialysis patients. Nephrol. Dial. Transplant. 2007, 22, 3606–3609. [Google Scholar] [CrossRef] [PubMed]
- Grigoleit, J.-S.; Kullmann, J.S.; Wolf, O.T.; Hammes, F.; Wegner, A.; Jablonowski, S.; Engler, H.; Gizewski, E.R.; Oberbeck, R.; Schedlowski, M. Dose-Dependent Effects of Endotoxin on Neurobehavioral Functions in Humans. PLoS ONE 2011, 6, e28330. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ozawa, N.; Shinozaki, T.; Wakabayashi, K.-I.; Suzuki, K.; Kawano, Y.; Ohtsu, I.; Tatebayashi, Y. Ergothioneine, a metabolite of the gut bacterium Lactobacillus reuteri, protects against stress-induced sleep disturbances. Transl. Psychiatry 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ozawa, N.; Shinozaki, T.; Aoki, K.; Nihonmatsu-Kikuchi, N.; Shinba, T.; Tatebayashi, Y. Chronic antidepressant treatments rescue reduced REM sleep theta power in a rat social defeat stress model of depression. bioRxiv 2020. [Google Scholar] [CrossRef]
- Leclercq, S.; Matamoros, S.; Cani, P.D.; Neyrinck, A.M.; Jamar, F.; Stärkel, P.; Windey, K.; Tremaroli, V.; Bäckhed, F.; Verbeke, K.; et al. Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. USA 2014, 111, E4485–E4493. [Google Scholar] [CrossRef]
- Alzheimer’s Association 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Tiraboschi, P.; Hansen, L.A.; Thal, L.J.; Corey-Bloom, J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 2004, 62, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Higgins, G.A.; Mayford, M.; Barzilai, A.; Keller, F.; Schacher, S.; Kandel, E. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Yankner, B.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and neurotoxic effects of amyloid beta protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Gavrilova, S.I.; Samsonova, A.; Barreto, G.E.; Aliev, G. Mild cognitive impairment due to Alzheimer disease: Contemporary approaches to diagnostics and pharmacological intervention. Pharmacol. Res. 2018, 129, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease--A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Carecchio, M.; Fleetwood, T.; Magistrelli, L.; Cantello, R.; Dianzani, U.; Comi, C.A.M. Immunity and inflammation in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 89–107. [Google Scholar]
- Ma, Q.; Xing, C.; Long, W.; Wang, H.Y.; Liu, Q.; Wang, R.-F. Impact of microbiota on central nervous system and neurological diseases: The gut-brain axis. J. Neuroinflammation 2019, 16, 1–14. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Lambert, J.-C.; Vrièze, F.W.-D.; Wollmer, M.; Von Der Kammer, H.; Streffer, J.R.; Maddalena, A.; Huynh, K.-D.; Wolleb, S.; Lütjohann, D.; et al. Cholesterol 25-Hydroxylase on Chromosome 10q Is a Susceptibility Gene for Sporadic Alzheimer’s Disease. Neurodegener. Dis. 2005, 2, 233–241. [Google Scholar] [CrossRef]
- Wozniak, M.; Frost, A.L.; Itzhaki, R.F. Alzheimer’s Disease-Specific Tau Phosphorylation is Induced by Herpes Simplex Virus Type 1. J. Alzheimer’s Dis. 2009, 16, 341–350. [Google Scholar] [CrossRef]
- Stojković, D.; Kostić, M.; Smiljković, M.; Aleksić, M.; Vasiljević, P.; Nikolić, M.; Soković, M. Linking Antimicrobial Potential of Natural Products Derived from Aquatic Organisms and Microbes Involved in Alzheimer’s Disease - A Review. Curr. Med. Chem. 2020, 27, 4372–4391. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Hammond, C.J.; Hingley, S.T.; Balin, B.J. Chlamydia pneumoniae infection of monocytes in vitro stimulates innate and adaptive immune responses relevant to those in Alzheimer’s disease. J. Neuroinflammation 2014, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Romano, K.A.; Darst, B.F.; Engelman, C.D.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B.; et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.B.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, srep41802. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Xiayu, X.; Shi, C.; Chen, W.; Song, N.; Fu, X.; Zhou, R.; Xu, Y.-F.; Huang, L.; et al. Altered Gut Microbiota in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1241–1257. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.-J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2013, 124, 307–321. [Google Scholar] [CrossRef]
- Gatz, M.; Mortimer, J.A.; Fratiglioni, L.; Johansson, B.; Berg, S.; Reynolds, C.A.; Pedersen, N.L. Potentially modifiable risk factors for dementia in identical twins. Alzheimer’s Dement. 2006, 2, 110–117. [Google Scholar] [CrossRef]
- Stein, P.S.; Desrosiers, M.; Donegan, S.J.; Yepes, J.F.; Kryscio, R.J. Tooth loss, dementia and neuropathology in the Nun Study. J. Am. Dent. Assoc. 2007, 138, 1314–1322. [Google Scholar] [CrossRef]
- Noble, J.M.; Borrell, L.N.; Papapanou, P.N.; Elkind, M.S.V.; Scarmeas, N.; Wright, C.B. Periodontitis is associated with cognitive impairment among older adults: Analysis of NHANES-III. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1206–1211. [Google Scholar] [CrossRef]
- Stewart, R.; Sabbah, W.; Tsakos, G.; D’aiuto, F.; Watt, R.G. Oral Health and Cognitive Function in the Third National Health and Nutrition Examination Survey (NHANES III). Psychosom. Med. 2008, 70, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Paganini-Hill, A.; White, S.C.; Atchison, K.A. Dentition, Dental Health Habits, and Dementia: The Leisure World Cohort Study. J. Am. Geriatr. Soc. 2012, 60, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Pirraglia, E.; Tsui, W.; Rusinek, H.; Vallabhajosula, S.; Mosconi, L.; Yi, L.; McHugh, P.; Craig, R.G.; Svetcov, S.; et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging 2015, 36, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, A.F.; Dehlin, J.M.; Ngan, T.; Crout, R.J.; Boskovic, G.; Denvir, J.; Primerano, D.A.; Plassman, B.; Wu, B.; Cuff, C.F. High throughput DNA sequencing to detect differences in the subgingival plaque microbiome in elderly subjects with and without dementia. Investig. Genet. 2012, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Craig, R.G.; Pirraglia, E.; Dasanayake, A.P.; Norman, R.G.; Boylan, R.J.; Nehorayoff, A.; Glodzik, L.; Brys, M.; De Leon, M.J. TNF-α and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J. Neuroimmunol. 2009, 216, 92–97. [Google Scholar] [CrossRef]
- Stein, P.S.; Steffen, M.J.; Smith, C.; Jicha, G.; Ebersole, J.L.; Abner, E.; Dawson, D. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 196–203. [Google Scholar] [CrossRef]
- Noble, J.M.; Scarmeas, N.; Celenti, R.S.; Elkind, M.S.V.; Wright, C.B.; Schupf, N.; Papapanou, P.N. Serum IgG Antibody Levels to Periodontal Microbiota Are Associated with Incident Alzheimer Disease. PLoS ONE 2014, 9, e114959. [Google Scholar] [CrossRef]
- Hayashi, C.; Gudino, C.; Iii, F.G.; Genco, C. REVIEW: Pathogen-induced inflammation at sites distant from oral infection: Bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol. Oral Microbiol. 2010, 25, 305–316. [Google Scholar] [CrossRef]
- Fasano, A.; Visanji, N.P.; Liu, L.W.C.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015, 14, 625–639. [Google Scholar] [CrossRef]
- Braak, H.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Ruffmann, C.; Parkkinen, L. Gut Feelings About α-Synuclein in Gastrointestinal Biopsies: Biomarker in the Making? Mov. Disord. 2016, 31, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Arizona Parkinson’s Disease Consortium; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; Iii, C.L.W.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Goto, S.; Tsuji, H.; Okuno, T.; Asahara, T.; Nomoto, K.; Shibata, A.; Fujisawa, Y.; Minato, T.; Okamoto, A.; et al. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson’s Disease. PLoS ONE 2015, 10, e0142164. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Msc, P.A.B.P.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schã¤Fer, K.-H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Park. Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Krishnamoorthy, G. Microbial view of central nervous system autoimmunity. FEBS Lett. 2014, 588, 4207–4213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kasper, L.H. The role of microbiome in central nervous system disorders. Brain Behav. Immun. 2014, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Goverman, J.; Woods, A.; Larson, L.; Weiner, L.P.; Hood, L.; Zaller, D.M. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell 1993, 72, 551–560. [Google Scholar] [CrossRef]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108, 4615–4622. [Google Scholar] [CrossRef]
- Berer, K.; Mues, M.; Koutrolos, M.; Al Rasbi, Z.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nat. Cell Biol. 2011, 479, 538–541. [Google Scholar] [CrossRef]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking Cessation Induces Profound Changes in the Composition of the Intestinal Microbiota in Humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nat. Cell Biol. 2014, 516, 94–98. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.M.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; Von Bergen, M.; McCoy, K.; MacPherson, A.J.; Danska, J.S. Sex Differences in the Gut Microbiome Drive Hormone-Dependent Regulation of Autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Zhu, F.; Hart, J.; Roalstad, S.; Graves, J.; Lynch, S.; Waubant, E.; Centers, T.U.N.O.P.M. Gut microbiota in early pediatric multiple sclerosis: A case-control study. Eur. J. Neurol. 2016, 23, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Soldan, M.M.P.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; Von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.-W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Waubant, E.; Chehoud, C.; Kuczynski, J.; DeSantis, T.Z.; Warrington, J.; Venkatesan, A.; Fraser, C.M.; Mowry, E.M. Gut Microbiota in Multiple Sclerosis: Possible influence of immunomodulators. J. Investig. Med. 2015, 63, 729–734. [Google Scholar] [CrossRef]
- Rumah, K.R.; Linden, J.; Fischetti, V.A.; Vartanian, T. Isolation of Clostridium perfringens Type B in an Individual at First Clinical Presentation of Multiple Sclerosis Provides Clues for Environmental Triggers of the Disease. PLoS ONE 2013, 8, e76359. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Farrokhi, V.; Nemati, R.; Nichols, F.C.; Yao, X.; Anstadt, E.; Fujiwara, M.; Grady, J.J.; Wakefield, D.; Castro, W.; Donaldson, J.; et al. Bacterial lipodipeptide, Lipid 654, is a microbiome-associated biomarker for multiple sclerosis. Clin. Transl. Immunol. 2013, 2, e8. [Google Scholar] [CrossRef] [PubMed]
- Branton, W.; Lu, J.; Surette, M.; Holt, R.; Lind, J.; Laman, J.; Power, C. Multiple sclerosis lesions show perturbations in cerebral microbiota. Neurology 2016. Available online: https://n.neurology.org/content/86/16_Supplement/S37.005.short (accessed on 25 October 2020).
- Varrin-Doyer, M.; Bs, C.M.S.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Cree, B.A.C.; Zamvil, S.S. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann. Neurol. 2012, 72, 53–64. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Spencer, C.M.; Varrin-Doyer, M.; Baranzini, S.E.; Zamvil, S.S. Gut microbiome analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens. Ann. Neurol. 2016, 80, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Banati, M.; Csecsei, P.; Kőszegi, É.; Nielsen, H.H.; Sütõ, G.; Bors, L.; Trauninger, A.; Csépány, T.; Rozsa, C.; Jakab, G.; et al. Antibody response against gastrointestinal antigens in demyelinating diseases of the central nervous system. Eur. J. Neurol. 2013, 20, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Goehler, L.E.; Gaykema, R.P.; Opitz, N.; Reddaway, R.; Badr, N.; Lyte, M. Activation in vagal afferents and central autonomic pathways: Early responses to intestinal infection with Campylobacter jejuni. Brain Behav. Immun. 2005, 19, 334–344. [Google Scholar] [CrossRef]
- Cassuto, J.; Siewert, A.; Jodal, M.; Lundgren, O. The involvement of intramural nerves in cholera toxin induced intestinal secretion. Acta Physiol. Scand. 1983, 117, 195–202. [Google Scholar] [CrossRef]
- Farthing, M. Enterotoxins and the enteric nervous system — a fatal attraction. Int. J. Med. Microbiol. 2000, 290, 491–496. [Google Scholar] [CrossRef]
- Koussoulas, K.; Gwynne, R.M.; Foong, J.P.P.; Bornstein, J.C. Cholera Toxin Induces Sustained Hyperexcitability in Myenteric, but Not Submucosal, AH Neurons in Guinea Pig Jejunum. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Popoff, M.R.; Poulain, B. Bacterial Toxins and the Nervous System: Neurotoxins and Multipotential Toxins Interacting with Neuronal Cells. Toxins 2010, 2, 683–737. [Google Scholar] [CrossRef]
- Rolfe, V.E.; Levin, R.J. Vagotomy inhibits the jejunal fluid secretion activated by luminal ileal Escherichia coli STa in the rat in vivo. Gut 1999, 44, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, V.; Levin, R.J. Enterotoxin Escherichia coli STa activates a nitric oxide-dependent myenteric plexus secretory reflex in the rat ileum. J. Physiol. 1994, 475, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Eklund, S.; Jodal, M.; Lundgren, O. The enteric nervous system participates in the secretory response to the heat stable enterotoxins of Escherichia coli in rats and cats. Neurosci. 1985, 14, 673–681. [Google Scholar] [CrossRef]
- Pothoulakis, C.; Castagliuolo, I.; Lamont, J.T. Nerves and Intestinal Mast Cells Modulate Responses to Enterotoxins. News Physiol. Sci. Int. J. Physiol. Prod. Jointly Int. Union Physiol. Sci. Am. Physiol. Soc. 1998, 13, 58–63. [Google Scholar] [CrossRef]
- Kelly, C.P.; Becker, S.; Linevsky, J.K.; Joshi, M.A.; O’Keane, J.C.; Dickey, B.F.; Lamont, J.T.; Pothoulakis, C. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J. Clin. Investig. 1994, 93, 1257–1265. [Google Scholar] [CrossRef]
- Castagliuolo, I.; Lamont, J.T.; Létourneau, R.; Kelly, C.; O’Keane, J.; Jaffer, A.; Theoharides, T.C.; Pothoulakis, C. Neuronal involvement in the intestinal effects of Clostridium difficile toxin A and Vibrio cholerae enterotoxin in rat ileum. Gastroenterol. 1994, 107, 657–665. [Google Scholar] [CrossRef]
- Xia, Y.; Hu, H.Z.; Liu, S.; Pothoulakis, C.; Wood, J.D. Clostridium difficile toxin A excites enteric neurones and suppresses sympathetic neurotransmission in the guinea pig. Gut 2000, 46, 481–486. [Google Scholar] [CrossRef]
- Fettucciari, K.; Ponsini, P.; Gioè, D.; Macchioni, L.; Palumbo, C.; Antonelli, E.; Coaccioli, S.; Villanacci, V.; Corazzi, L.; Marconi, P.; et al. Enteric glial cells are susceptible to Clostridium difficile toxin B. Cell. Mol. Life Sci. 2016, 74, 1527–1551. [Google Scholar] [CrossRef]
- Fettucciari, K.; Macchioni, L.; Davidescu, M.; Scarpelli, P.; Palumbo, C.; Corazzi, L.; Marchegiani, A.; Cerquetella, M.; Spaterna, A.; Marconi, P.; et al. Clostridium difficile toxin B induces senescence in enteric glial cells: A potential new mechanism of Clostridium difficile pathogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1945–1958. [Google Scholar] [CrossRef]
- Hu, D.-L.; Zhu, G.; Mori, F.; Omoe, K.; Okada, M.; Wakabayashi, K.; Kaneko, S.; Shinagawa, K.; Nakane, A. Staphylococcal enterotoxin induces emesis through increasing serotonin release in intestine and it is downregulated by cannabinoid receptor 1. Cell. Microbiol. 2007, 9, 2267–2277. [Google Scholar] [CrossRef]
- Toh, M.; Moffitt, M.C.; Henrichsen, L.; Raftery, M.; Barrow, K.; Cox, J.M.; Marquis, C.P.; Neilan, B.A. Cereulide, the emetic toxin of Bacillus cereus, is putatively a product of nonribosomal peptide synthesis. J. Appl. Microbiol. 2004, 97, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Klem, F.; Wadhwa, A.; Prokop, L.J.; Sundt, W.J.; Farrugia, G.; Camilleri, M.; Singh, S.; Grover, M. Prevalence, Risk Factors, and Outcomes of Irritable Bowel Syndrome After Infectious Enteritis: A Systematic Review and Meta-analysis. Gastroenterol. 2017, 152, 1042–1054.e1. [Google Scholar] [CrossRef]
- Zanini, B.; Ricci, C.; Bandera, F.; Caselani, F.; Magni, A.; Laronga, A.M.; Lanzini, A. Incidence of Post-Infectious Irritable Bowel Syndrome and Functional Intestinal Disorders Following a Water-Borne Viral Gastroenteritis Outbreak. Am. J. Gastroenterol. 2012, 107, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.K.; Thabane, M.; Borgaonkar, M.R.; James, C. Postinfectious Irritable Bowel Syndrome After a Food-Borne Outbreak of Acute Gastroenteritis Attributed to a Viral Pathogen. Clin. Gastroenterol. Hepatol. 2007, 5, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.; Nguyen, L.; Gill, J.; Kulkarni, S.; Pasricha, P.J.; Habtezion, A. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut 2018, 67, 827–836. [Google Scholar] [CrossRef]
- Gershon, A.A.; Chen, J.; Gershon, M.D. A Model of Lytic, Latent, and Reactivating Varicella-Zoster Virus Infections in Isolated Enteric Neurons. J. Infect. Dis. 2008, 197, S61–S65. [Google Scholar] [CrossRef]
- Benini, L.; Sembenini, C.; Bulighin, G.M.; Polo, A.; Ederle, A.; Zambito, A.; Vantini, I. Achalasia: A Possible Late Cause of Postpolio Dysphagia. Dig. Dis. Sci. 1996, 41, 516–518. [Google Scholar] [CrossRef]
- Debinski, H.S.; Kamm, M.A.; Talbot, I.C.; Khan, G.; Kangro, H.O.; Jeffries, D.J. DNA viruses in the pathogenesis of sporadic chronic idiopathic intestinal pseudo-obstruction. Gut 1997, 41, 100–106. [Google Scholar] [CrossRef]
- Jucglà, A.; Badell, A.; Ballesta, C.; Arbizu, T. Colonic pseudo-obstruction: A complication of herpes zoster. Br. J. Dermatol. 1996, 134, 788–790. [Google Scholar] [CrossRef]
- Gershon, A.A.; Chen, J.; Davis, L.; Krinsky, C.; Cowles, R.; Reichard, R.; Gershon, M. Latency of Varicella Zoster Virus in Dorsal Root, Cranial, and Enteric Ganglia in Vaccinated Children. Trans. Am. Clin. Clim. Assoc. 2012, 123, 17–35. [Google Scholar]
- Gulbransen, B.D.; Sharkey, K.A. Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-B. Enteric glial cells and their role in the intestinal epithelial barrier. World J. Gastroenterol. 2014, 20, 11273–11280. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Giron, M.C.; Qesari, M.; Porzionato, A.; Caputi, V.; Zoppellaro, C.; Banzato, S.; Grillo, A.R.; Spagnol, L.; De Caro, R.; et al. Toll-Like Receptor 2 Regulates Intestinal Inflammation by Controlling Integrity of the Enteric Nervous System. Gastroenterol. 2013, 145, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Barajon, I.; Serrao, G.; Arnaboldi, F.; Opizzi, E.; Ripamonti, G.; Balsari, A.; Rumio, C. Toll-like Receptors 3, 4, and 7 Are Expressed in the Enteric Nervous System and Dorsal Root Ganglia. J. Histochem. Cytochem. 2009, 57, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Capoccia, E.; Gigli, S.; Pesce, M.; Bruzzese, E.; D’Alessandro, A.; Cirillo, C.; Di Cerbo, A.; Cuomo, R.; Seguella, L.; et al. HIV-1 Tat-induced diarrhea evokes an enteric glia-dependent neuroinflammatory response in the central nervous system. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut 2013, 63, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Turco, F.; Sarnelli, G.; Cirillo, C.; Palumbo, I.; De Giorgi, F.; D’Alessandro, A.; Cammarota, M.; Giuliano, M.; Cuomo, R. Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut 2013, 63, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Liñán-Rico, A.; Turco, F.; Ochoa-Cortes, F.; Harzman, A.; Needleman, B.J.; Arsenescu, R.; Abdel-Rasoul, M.; Fadda, P.; Grants, I.; Whitaker, E.; et al. Molecular Signaling and Dysfunction of the Human Reactive Enteric Glial Cell Phenotype: Implications for GI Infection, IBD, POI, Neurological, Motility, and GI Disorders. Inflamm. Bowel Dis. 2016, 22, 1812–1834. [Google Scholar] [CrossRef]
- Brun, P.; Giron, M.C.; Zoppellaro, C.; Bin, A.; Porzionato, A.; De Caro, R.; Barbara, G.; Stanghellini, V.; Corinaldesi, R.; Zaninotto, G.; et al. Herpes Simplex Virus Type 1 Infection of the Rat Enteric Nervous System Evokes Small-Bowel Neuromuscular Abnormalities. Gastroenterol. 2010, 138, 1790–1801. [Google Scholar] [CrossRef]
- Facco, M.; Brun, P.; Baesso, I.; Costantini, M.; Rizzetto, C.; Berto, A.; Baldan, N.; Pal, G.; Semenzato, G.; Castagliuolo, I.; et al. T Cells in the Myenteric Plexus of Achalasia Patients Show a Skewed TCR Repertoire and React to HSV-1 Antigens. Am. J. Gastroenterol. 2008, 103, 1598–1609. [Google Scholar] [CrossRef]
- Khoury-Hanold, W.; Yordy, B.; Kong, P.; Kong, Y.; Ge, W.; Szigeti-Buck, K.; Ralevski, A.; Horvath, T.L.; Iwasaki, A. Viral Spread to Enteric Neurons Links Genital HSV-1 Infection to Toxic Megacolon and Lethality. Cell Host Microbe 2016, 19, 788–799. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Xiong, S.; Malvin, N.P.; Khoury-Hanold, W.; Heuckeroth, R.O.; Stappenbeck, T.S.; Diamond, M.S. Intestinal Dysmotility Syndromes following Systemic Infection by Flaviviruses. Cell 2018, 175, 1198–1212.e12. [Google Scholar] [CrossRef] [PubMed]
- Mori, I. Viremic attack explains the dual-hit theory of Parkinson’s disease. Med. Hypotheses 2017, 101, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed]
- Gesser, R.M.; Koo, S.C. Oral inoculation with herpes simplex virus type 1 infects enteric neuron and mucosal nerve fibers within the gastrointestinal tract in mice. J. Virol. 1996, 70, 4097–4102. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution ofα-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, A.; Fronzoni, L.; Cogliandro, L.; Cogliandro, R.F.; Caputo, C.; De Giorgio, R.; Pallotti, F.; Barbara, G.; Corinaldesi, R.; Stanghellini, V. Chronic intestinal pseudo-obstruction. World J. Gastroenterol. 2008, 14, 2953–2961. [Google Scholar] [CrossRef]
- Déchelotte, P.J.; Mulliez, N.M.; Bouvier, R.J.; Vanlieféringhen, P.C.; Lemery, D.J. Pseudo-Meconium Ileus Due to Cytomegalovirus Infection: A Report of Three Cases. Pediatr. Pathol. 1992, 12, 73–82. [Google Scholar] [CrossRef]
- Besnard, M.; Faure, C.; Fromont-Hankard, G.; Ansart-Pirenne, H.; Peuchmaur, M.; Cezard, J.; Navarro, J. Intestinal pseudo-obstruction and acute pandysautonomia associated with epstein-barr virus infection. Am. J. Gastroenterol. 2000, 95, 280–284. [Google Scholar] [CrossRef]
- Chang, A.E.; Young, N.A.; Reddick, R.L.; Orenstein, J.M.; Hosea, S.W.; Katz, P.; Brennan, M.F. Small bowel obstruction as a complication of disseminated varicella-zoster infection. Surgery 1978, 83, 371–374. [Google Scholar]
- Vassallo, M.; Camilleri, M.; Caron, B.; Low, P.A. Gastrointestinal motor dysfunction in acquired selective cholinergic dysautonomia associated with infectious mononucleosis. Gastroenterology. 1991, 100, 252–258. [Google Scholar] [CrossRef]
- Selgrad, M.; De Giorgio, R.; Fini, L.; Cogliandro, R.F.; Williams, S.; Stanghellini, V.; Barbara, G.; Tonini, M.; Corinaldesi, R.; Genta, R.M.; et al. JC virus infects the enteric glia of patients with chronic idiopathic intestinal pseudo-obstruction. Gut 2009, 58, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sarnelli, G.; Seguella, L.; Pesce, M.; Lu, J.; Gigli, S.; Bruzzese, E.; Lattanzi, R.; D’Alessandro, A.; Cuomo, R.; Steardo, L.; et al. HIV-1 Tat-induced diarrhea is improved by the PPARalpha agonist, palmitoylethanolamide, by suppressing the activation of enteric glia. J. Neuroinflammation 2018, 15, 94. [Google Scholar] [CrossRef] [PubMed]
- Galligan, J.J. HIV, opiates, and enteric neuron dysfunction. Neurogastroenterol. Motil. 2015, 27, 449–454. [Google Scholar] [CrossRef]
- Fitting, S.; Ngwainmbi, J.; Kang, M.; Khan, F.A.; Stevens, D.L.; Dewey, W.L.; Knapp, P.E.; Hauser, K.F.; Akbarali, H.I. Sensitization of enteric neurons to morphine by HIV-1 Tat protein. Neurogastroenterol. Motil. 2015, 27, 468–480. [Google Scholar] [CrossRef]
- Guedia, J.; Brun, P.; Bhave, S.; Fitting, S.; Kang, M.; Dewey, W.L.; Hauser, K.F.; Akbarali, H.I. HIV-1 Tat exacerbates lipopolysaccharide-induced cytokine release via TLR4 signaling in the enteric nervous system. Sci. Rep. 2016, 6, 31203. [Google Scholar] [CrossRef]
- Hansen, M.B.; Witte, A.-B. The role of serotonin in intestinal luminal sensing and secretion. Acta Physiol. 2008, 193, 311–323. [Google Scholar] [CrossRef]
- Manocha, M.; Khan, W.I. Serotonin and GI Disorders: An Update on Clinical and Experimental Studies. Clin. Transl. Gastroenterol. 2012, 3, e13. [Google Scholar] [CrossRef]
- Gershon, M. Review article: Roles played by 5-hydroxytryptamine in the physiology of the bowel. Aliment. Pharmacol. Ther. 1999, 13, 15–30. [Google Scholar] [CrossRef]
- Bialowas, S.; Hagbom, M.; Nordgren, J.; Karlsson, T.; Sharma, S.; Magnusson, K.-E.; Svensson, L. Rotavirus and Serotonin Cross-Talk in Diarrhoea. PLoS ONE 2016, 11, e0159660. [Google Scholar] [CrossRef]
- Hagbom, M.; De Faria, F.M.; Winberg, M.E.; Westerberg, S.; Nordgren, J.; Sharma, S.; Keita, Å.V.; Loitto, V.; Magnusson, K.-E.; Svensson, L. Neurotrophic Factors Protect the Intestinal Barrier from Rotavirus Insult in Mice. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, S.; Hagbom, M.; Rajan, A.; Loitto, V.; Persson, B.D.; Allard, A.; Nordgren, J.; Sharma, S.; Magnusson, K.-E.; Arnberg, N.; et al. Interaction of Human Enterochromaffin Cells with Human Enteric Adenovirus 41 Leads to Serotonin Release and Subsequent Activation of Enteric Glia Cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Pesce, M.; Seguella, L.; Sanseverino, W.; Lu, J.; Sarnelli, G. Can the enteric nervous system be an alternative entrance door in SARS-CoV2 neuroinvasion? Brain, Behav. Immun. 2020, 87, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, G.; Giuffrè, M.; Sambataro, D.; Palermo, A.; Vignigni, G.; Cesareo, R.; Crimi, N.; Torrisi, S.E.; Vancheri, C.; Malatino, L.; et al. The Model for Early COvid-19 Recognition (MECOR) Score: A Proof-of-Concept for a Simple and Low-Cost Tool to Recognize a Possible Viral Etiology in Community-Acquired Pneumonia Patients during COVID-19 Outbreak. Diagnostics 2020, 10, 619. [Google Scholar] [CrossRef]
- Giuffrè, M.; Di Bella, S.; Sambataro, G.; Zerbato, V.; Cavallaro, M.; Occhipinti, A.A.; Palermo, A.; Crescenzi, A.; Monica, F.; Luzzati, R.; et al. COVID-19-Induced Thrombosis in Patients without Gastrointestinal Symptoms and Elevated Fecal Calprotectin: Hypothesis Regarding Mechanism of Intestinal Damage Associated with COVID-19. Trop. Med. Infect. Dis. 2020, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, M.; Bozzato, A.M.; Di Bella, S.; Occhipinti, A.A.; Martingano, P.; Cavallaro, M.F.M.; Luzzati, R.; Monica, F.; Cova, M.A.; Crocé, L.S. Spontaneous Rectal Perforation in a Patient with SARS–CoV-2 Infection. J. Pers. Med. 2020, 10, 157. [Google Scholar] [CrossRef]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costapinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef]
- Robinson, C.M.; Pfeiffer, J.K. Viruses and the Microbiota. Annu. Rev. Virol. 2014, 1, 55–69. [Google Scholar] [CrossRef]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Segal, J.P.; Carding, S.R.; Hart, A.L.; Hold, G.L. The gut virome: The ‘missing link’ between gut bacteria and host immunity? Ther. Adv. Gastroenterol. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Toledo, A.; Osorio, R.; Matus, C.; Lopez, Y.M.; Cruz, N.R.; Sciutto, E.; Fragoso, G.; Arauz, A.; Carrillo-Mezo, R.; Fleury, A. Human Extraparenchymal Neurocysticercosis: The Control of Inflammation Favors the Host…but Also the Parasite. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Worthington, J.J.; Samuelson, L.C.; Grencis, R.K.; McLaughlin, J. Adaptive Immunity Alters Distinct Host Feeding Pathways during Nematode Induced Inflammation, a Novel Mechanism in Parasite Expulsion. PLOS Pathog. 2013, 9, e1003122. [Google Scholar] [CrossRef]
- Whelan, R.; Rausch, S.; Ebner, F.; Günzel, D.; Richter, J.F.; Hering, N.A.; Schulzke, J.-D.; Kühl, A.A.; Keles, A.; Janczyk, P.; et al. A Transgenic Probiotic Secreting a Parasite Immunomodulator for Site-Directed Treatment of Gut Inflammation. Mol. Ther. 2014, 22, 1730–1740. [Google Scholar] [CrossRef]
- Podolska, M.; Nadolna, K. Acetylcholinesterase secreted by Anisakis simplex larvae (Nematoda: Anisakidae) parasitizing herring, Clupea harengus: An inverse relationship of enzyme activity in the host-parasite system. Parasitol. Res. 2014, 113, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Liu, C.; Du, X.; Nawaratna, S.S.; Rivera, V.; Harvie, M.; Jones, M.K.; McManus, D.P. Suppression of Schistosoma japonicum Acetylcholinesterase Affects Parasite Growth and Development. Int. J. Mol. Sci. 2018, 19, 2426. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.; Lee, D.L. A vasoactive intestinal polypeptide-like protein excreted/secreted byNippostrongylus brasiliensisand its effect on contraction of uninfected rat intestine. Parasitology 1996, 112, 97–104. [Google Scholar] [CrossRef]
- Argenzio, R.A.; Armstrong, M.; Rhoads, J.M. Role of the enteric nervous system in piglet cryptosporidiosis. J. Pharmacol. Exp. Ther. 1996, 279, 1109–1115. [Google Scholar]
- Laurent, F.; Kagnoff, M.F.; Savidge, T.C.; Naciri, M.; Eckmann, L. Human Intestinal Epithelial Cells Respond toCryptosporidium parvum Infection with Increased Prostaglandin H Synthase 2 Expression and Prostaglandin E2and F2α Production. Infect. Immun. 1998, 66, 1787–1790. [Google Scholar] [CrossRef]
- Dizdar, V.; Spiller, R.C.; Singh, G.; Hanevik, K.; Gilja, O.; El-Salhy, M.; Hausken, T. Relative importance of abnormalities of CCK (cholecystokinin) and 5-HT (serotonin) inGiardia-induced post-infectious irritable bowel syndrome and functional dyspepsia. Aliment. Pharmacol. Ther. 2010, 31, 883–891. [Google Scholar] [CrossRef]
- Leslie, F.; Thompson, D.; McLaughlin, J.; Varro, A.; Dockray, G.; Mandal, B. Plasma cholecystokinin concentrations are elevated in acute upper gastrointestinal infections. Qjm: Int. J. Med. 2003, 96, 870–871. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Zhao, A.; Shea-Donohue, T.; Singer, S.M. Mast Cell-Mediated Changes in Smooth Muscle Contractility during Mouse Giardiasis. Infect. Immun. 2007, 75, 4514–4518. [Google Scholar] [CrossRef] [PubMed]
- Swain, M.G.; Agro, A.; Blennerhassett, P.; Stanisz, A.; Collins, S.M. Increased levels of substance P in the myenteric plexus of Trichinella-infected rats. Gastroenterology 1992, 102, 1913–1919. [Google Scholar] [CrossRef]
- Kataeva, G.; Agro, A.; Stanisz, A.M. Substance-P-Mediated Intestinal Inflammation: Inhibitory Effects of CP 96,345 and SMS 201-995. Neuroimmunomodulation 1994, 1, 350–356. [Google Scholar] [CrossRef]
- Palmer, J.; Greenwood, B. Regional content of enteric substance P and vasoactive intestinal peptide during intestinal inflammation in the parasitized ferret. Neuropeptides 1993, 25, 95–103. [Google Scholar] [CrossRef]
- Arnhold, S.; When, M.; Andressen, C.; Addicks, K. Transient expression of NOS-II during development of the murine enteric nervous system. J. Mol. Histol. 2004, 35, 741–748. [Google Scholar] [CrossRef]
- Scheschowitsch, K.; De Moraes, J.A.; Sordi, R.; Barja-Fidalgo, C.; Assreuy, J. Rapid NOS-1-derived nitric oxide and peroxynitrite formation act as signaling agents for inducible NOS-2 expression in vascular smooth muscle cells. Pharmacol. Res. 2015, 100, 73–84. [Google Scholar] [CrossRef]
- Lourenssen, S.; Houpt, E.R.; Chadee, K.; Blennerhassett, M.G. Entamoeba histolytica Infection and Secreted Proteins Proteolytically Damage Enteric Neurons. Infect. Immun. 2010, 78, 5332–5340. [Google Scholar] [CrossRef]
- Suzuki, Y.; Orellana, M.A.; Schreiber, R.D.; Remington, J.S. Interferon-gamma: The major mediator of resistance against Toxoplasma gondii. Science 1988, 240, 516–518. [Google Scholar] [CrossRef]
- Araújo, E.J.D.A.; Zaniolo, L.M.; Vicentino, S.L.; Góis, M.B.; Zanoni, J.N.; Da Silva, A.V.; Sant’Ana, D.D.M.G. Toxoplasma gondiicauses death and plastic alteration in the jejunal myenteric plexus. World J. Gastroenterol. 2015, 21, 4829–4839. [Google Scholar] [CrossRef]
- Pentreath, V. Trypanosomiasis and the nervous system: Pathology and immunology. Trans. R. Soc. Trop. Med. Hyg. 1995, 89, 9–15. [Google Scholar] [CrossRef]
- Petry, K.; Van Voorhis, W. Antigens of Trypanosoma cruzi that mimic mammalian nervous tissues: Investigations of their role in the autoimmune pathophysiology of chronic Chagas’ disease. Res. Immunol. 1991, 142, 151–156. [Google Scholar] [CrossRef]
- Desalegn, G.; Pabst, O. Inflammation triggers immediate rather than progressive changes in monocyte differentiation in the small intestine. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Blennerhassett, M.G.; Vignjevic, P.; Vermillion, D.L.; Collins, S.M. Inflammation causes hyperplasia and hypertrophy in smooth muscle of rat small intestine. Am. J. Physiol. Liver Physiol. 1992, 262, G1041–G1046. [Google Scholar] [CrossRef]
- Beatty, J.K.; Bhargava, A.; Buret, A.G. Post-infectious irritable bowel syndrome: Mechanistic insights into chronic disturbances following enteric infection. World J. Gastroenterol. 2014, 20, 3976–3985. [Google Scholar] [CrossRef]
- Chaudhury, A.; Dendi, V.S.R.; Chaudhury, M.; Jain, A.; Kasarla, M.R.; Panuganti, K.; Jain, G.; Ramanujam, A.; Rena, B.; Koyagura, S.R.; et al. HSV1/2 Genital Infection in Mice Cause Reversible Delayed Gastrointestinal Transit: A Model for Enteric Myopathy. Front. Med. 2018, 5. [Google Scholar] [CrossRef]
- Notari, L.; Riera, D.C.; Sun, R.; Bohl, J.A.; McLean, L.P.; Madden, K.B.; Van Rooijen, N.; Vanuytsel, T.; Urban, J.F., Jr.; Zhao, A.; et al. Role of Macrophages in the Altered Epithelial Function during a Type 2 Immune Response Induced by Enteric Nematode Infection. PLoS ONE 2014, 9, e84763. [Google Scholar] [CrossRef]
- Wang, H.; Kim, J.J.; Denou, E.; Gallagher, A.; Thornton, D.J.; Shajib, M.S.; Xia, L.; Schertzer, J.D.; Grencis, R.K.; Philpott, D.J.; et al. New Role of Nod Proteins in Regulation of Intestinal Goblet Cell Response in the Context of Innate Host Defense in an Enteric Parasite Infection. Infect. Immun. 2015, 84, 275–285. [Google Scholar] [CrossRef]



{kind=link}
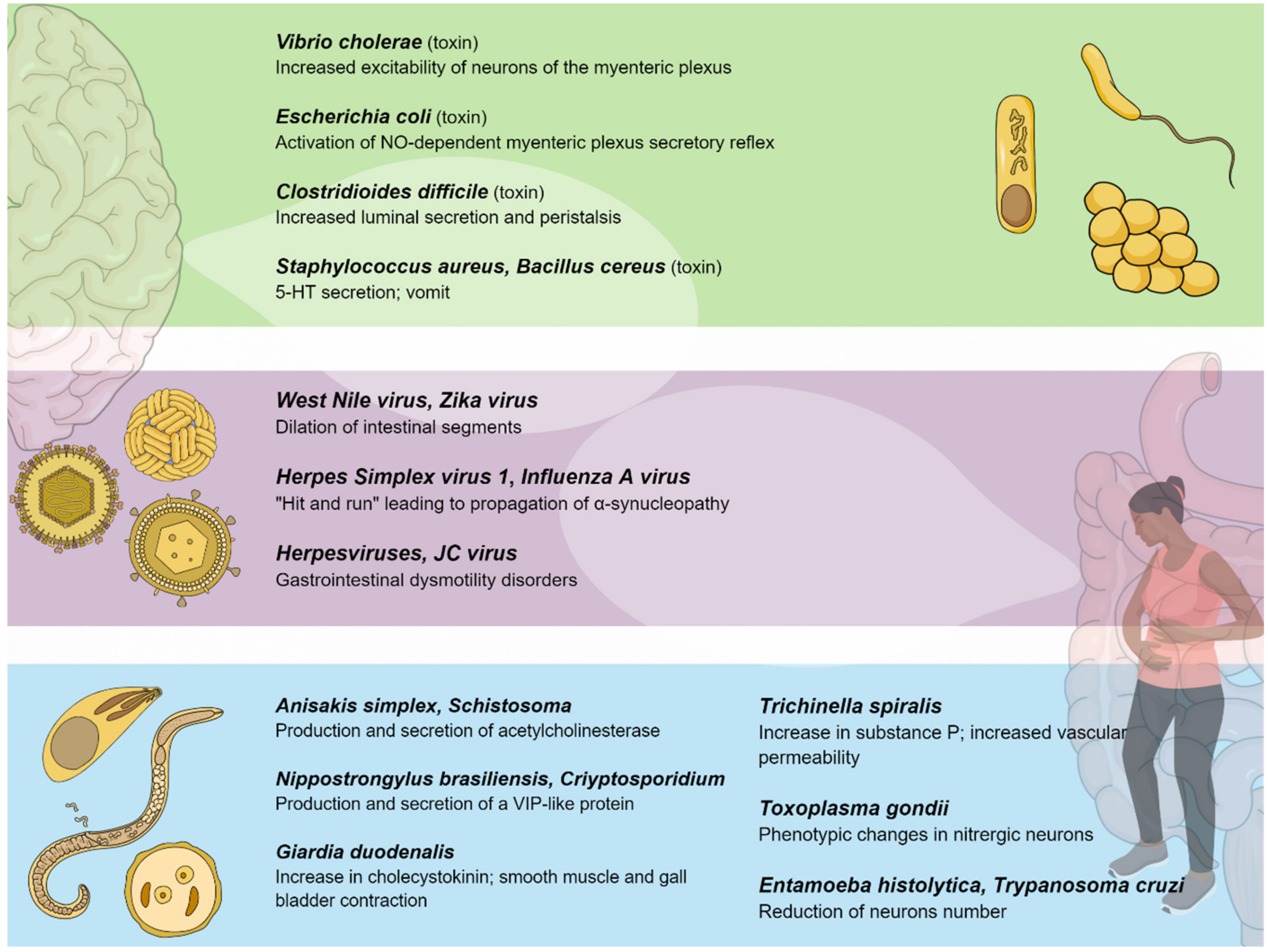{kind=link}
| Field of Interest | Key Findings |
|---|---|
| Gut Microbiota | |
| Social behavior | Social events allow horizontal transmission of microbes between individuals of the same species (as observed in Blattodea or baboons). Rodent models with high-fat diets and reduction of Lactobacillus spp. give birth to offspring with reduced ability to discriminate between familiar and unknown individuals of the same species. Dysbiosis promotes drastic changes in social behavior in rodents and supplementation with Bifidobacteria and Lactobacilli leads to improvement in early life and adulthood. |
| Sleep cycle and mood dysorders | Gut microbiota can alter sleep cycles through the systemic production of inflammatory cytokines, which have been proven to alter non-REM sleep and alter cortisol and norepinephrine production. These phenomena are related to gut permeability and systemic translocation of gut bacteria. |
| Alzheimer’s disease (AD) | Several bacteria promote neuro-inflammatory response typical of AD. Increased phosphorylated tau in patients with microbiota metabolites in cerebrospinal fluid. |
| Parkinson’s disease (PD) | High microbial density in the olfactory bulbs of patients with PD. Postural instability and gait symptoms can be associated with abundance of particular species. |
| Pathogenic bacteria | |
| Toxin-producing bacteria | Toxin-induced diarrhea is favored by the promotion of serotonin (5-HT) from the mucosa, resulting in activation of the secretomotor reflex pathways through local 5-HT receptors. In cases of emesis, 5-HT receptors are located in vagus nerve (VN) sensory terminals that project up to the emetic center in the brainstem. |
| Viral Agent(s) | Pathogenetic Mechanism(s) | Disease(s) |
|---|---|---|
| TBEV | Myenteric plexus infection | Irreversible ileus |
| WNV, ZIKV | Viral replication within enteric neurons causing cell death | Intestinal dysmotility |
| Influenza A virus/HSV-1 | Influenza A virus alterations in the ENS structures, followed by HSV-1 life-long persistency | Parkinson’s disease |
| Herpesviruses (EBV, VZV) | VZV latency in ganglia of the ENS; EBV induction of inflammatory infiltrates within the myenteric plexuses | Ogilvie’s syndrome, CIIPO, myenteric ganglionitis |
| JCV | Infection of the EGCs of the myenteric plexus | CIIPO |
| HIV | HIV-1 Tat protein activation of EGCs causing a neuroinflammatory response and synergistic action with morphine | Diarrhea and neurotoxic effects |
| Rotaviruses | Rotavirus infection of the EC cells and stimulation of serotonin secretion | Rotavirus-related diarrhea |
| HAdV-41 | Serotonin release from EC cells leading to activation of EGCs | Diarrhea |
| HSV-1 | Destruction of the enteric neurons by the massive recruitment of neutrophils | Loss of peristalsis and toxic megacolon |
| SARS-CoV-2 | Activation of EGCs with massive release of IL-6 and other inflammatory mediators (cytokine storm) | SARS-CoV-2 related-diarrhea |
| Parasite | Pathophysiological Modifications | Involved Factors |
|---|---|---|
| Cryptosporidium parvum | Altered transmembrane ionic transport/hypersecretion | Cholinergic and VIPergic response through prostacyclins Increased levels of substance P |
| Giardia duodenalis | Altered intestinal contractility Promotion of malabsorption and hypersecretion | Depletion in NO synthesis Reduction of 5-HT secretion Increase of CCK secretion |
| Entamoeba histolytica | Neuron and axon depletion | Process dependent on cysteine-proteases |
| Nippostrongylus brasiliensis | Motility dysfunction | Production of a VIP-similar peptide |
| Trichinella spiralis | Increased intestinal contractility | Altered neurotransmitter releases with 5-HT receptor disfunction |
| Trypanosoma cruzi | Decrease of enteric glial cells | Cross reaction between parasitic antigens and the human hosts Reduced contractility due to loss of Ach receptor function |
| Toxoplasma gondii | Phenotypic changes in enteric neurons | Increased NO response Reduction of VIPergic neurons |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuffrè, M.; Moretti, R.; Campisciano, G.; da Silveira, A.B.M.; Monda, V.M.; Comar, M.; Di Bella, S.; Antonello, R.M.; Luzzati, R.; Crocè, L.S. You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS. J. Clin. Med. 2020, 9, 3705. https://doi.org/10.3390/jcm9113705
Giuffrè M, Moretti R, Campisciano G, da Silveira ABM, Monda VM, Comar M, Di Bella S, Antonello RM, Luzzati R, Crocè LS. You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS. Journal of Clinical Medicine. 2020; 9(11):3705. https://doi.org/10.3390/jcm9113705
Chicago/Turabian StyleGiuffrè, Mauro, Rita Moretti, Giuseppina Campisciano, Alexandre Barcelos Morais da Silveira, Vincenzo Maria Monda, Manola Comar, Stefano Di Bella, Roberta Maria Antonello, Roberto Luzzati, and Lory Saveria Crocè. 2020. "You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS" Journal of Clinical Medicine 9, no. 11: 3705. https://doi.org/10.3390/jcm9113705
APA StyleGiuffrè, M., Moretti, R., Campisciano, G., da Silveira, A. B. M., Monda, V. M., Comar, M., Di Bella, S., Antonello, R. M., Luzzati, R., & Crocè, L. S. (2020). You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS. Journal of Clinical Medicine, 9(11), 3705. https://doi.org/10.3390/jcm9113705