Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure


Abstract
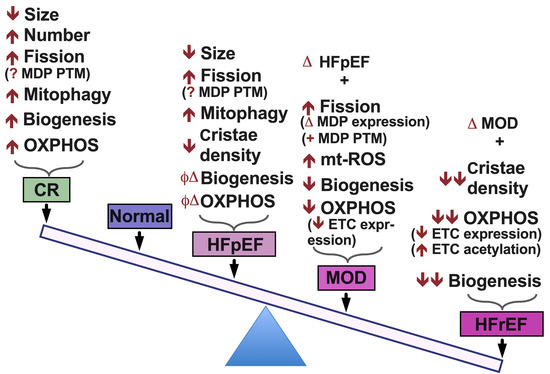
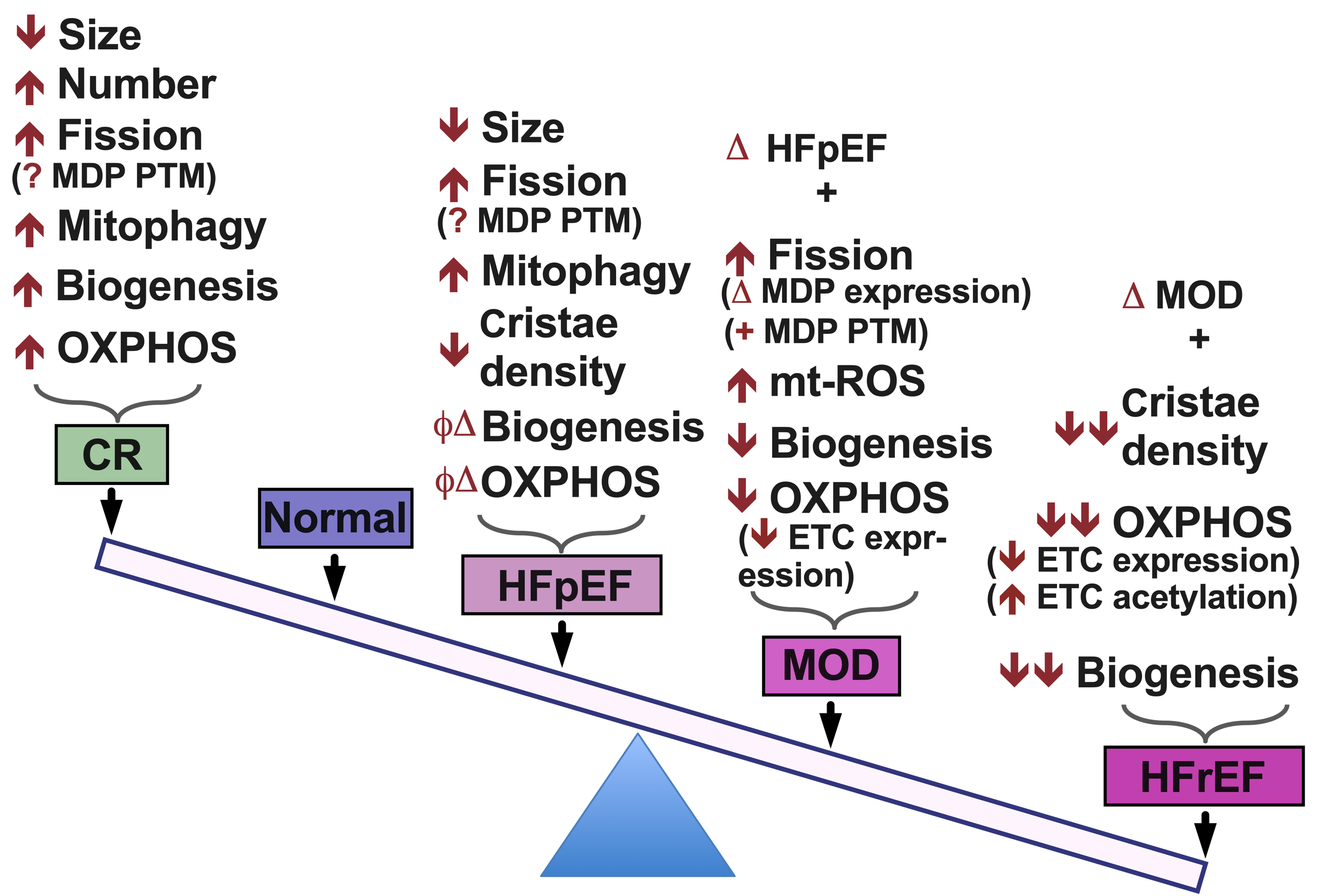{kind=link}
{kind=link}
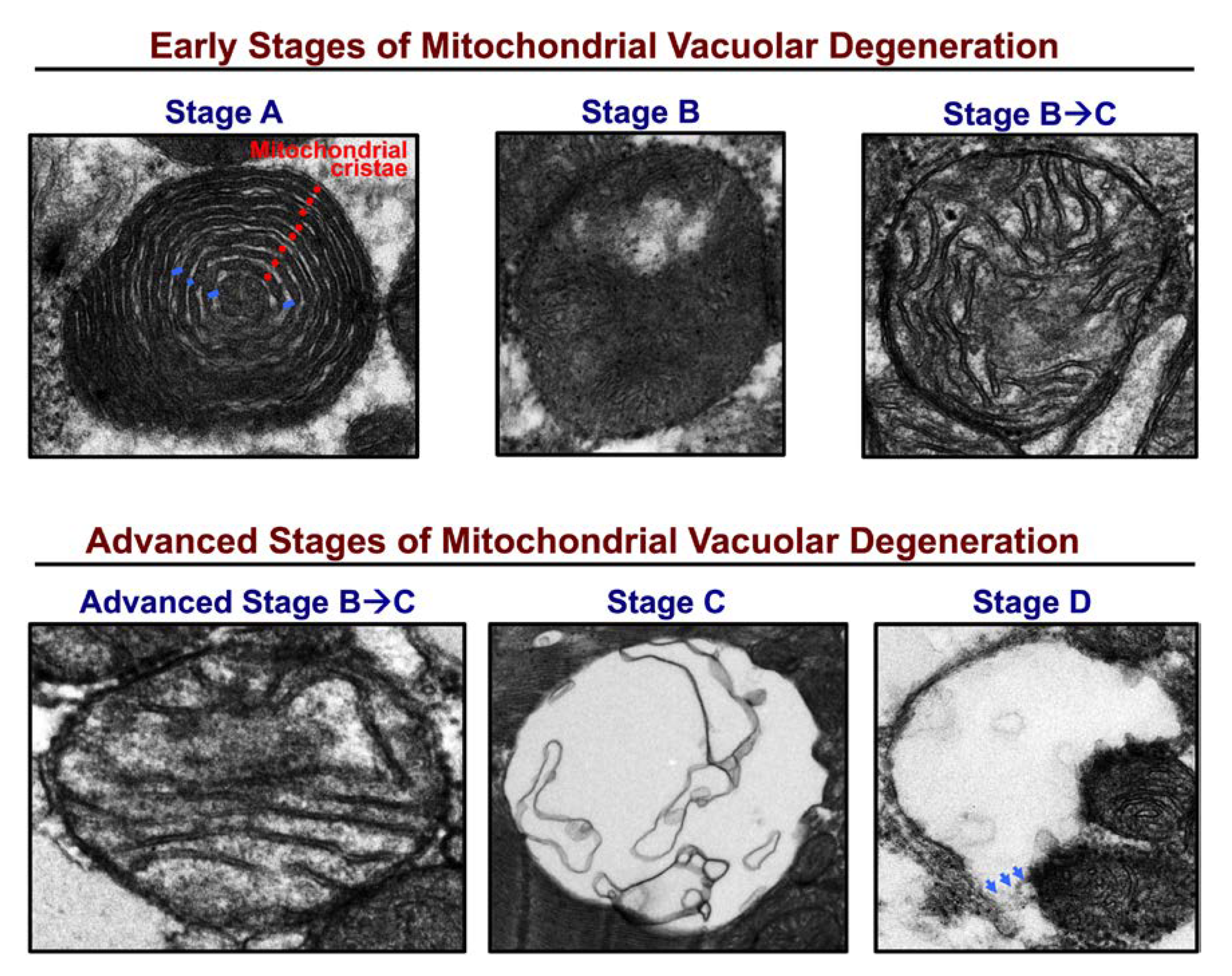{kind=link}
{kind=link}
{kind=link}
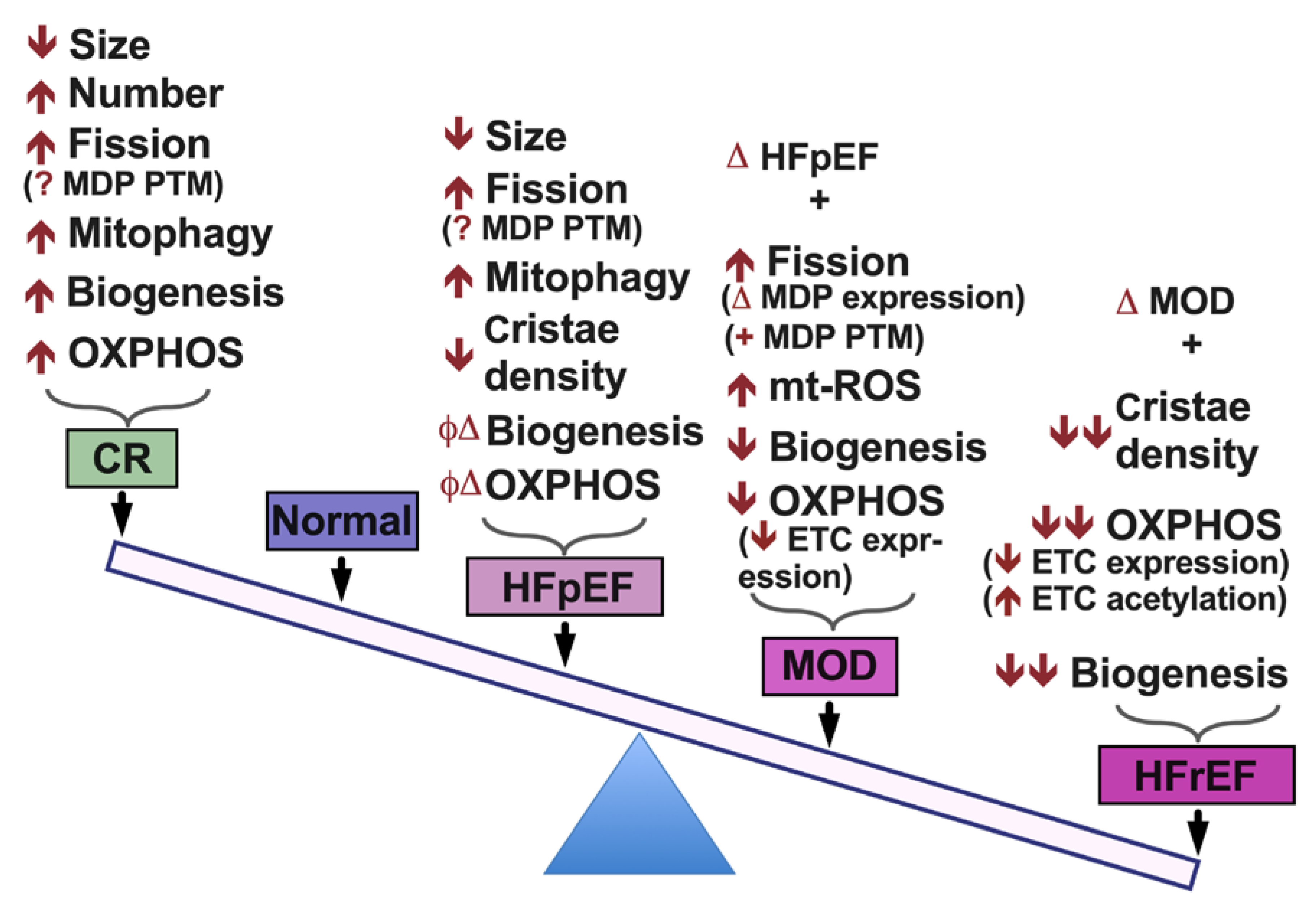{kind=link}
1. Introduction
2. Mitochondrial Morphology and Dynamics in HF
3. Mitochondrial Matrix Calcium, Redox Balance and Oxidative Capacity in HF
4. Metabolic Remodeling in HF
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hill, J.A.; Olson, E.N. Cardiac Plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Trojel-Hansen, C.; Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 2012, 111, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Youle, R.J. Mitochondrial fission and fusion and their roles in the heart. J. Mol. Med. 2010, 88, 971–979. [Google Scholar] [CrossRef]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; De Brito, O.M.; Zilio, B.D.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, e265. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, N.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef]
- Chaanine, A.H. Morphological stages of mitochondrial vacuolar degeneration in phenylephrine-stressed cardiac myocytes and in animal models and human heart failure. Medicina 2019, 55, 239. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial morphology, dynamics, and function in human pressure overload or ischemic heart disease with preserved or reduced ejection fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002, 80, 207–218. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef]
- Sun, M.G.; Williams, J.; Munoz-Pinedo, C.; Perkins, G.A.; Brown, J.M.; Ellisman, M.H.; Green, D.R.; Frey, T.G. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat. Cell Biol. 2007, 9, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in cardiovascular diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef]
- Chaanine, A.H. Autophagy and myocardial remodeling. J. Am. Coll. Cardiol. 2018, 71, 2011–2014. [Google Scholar] [CrossRef]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim. Biophys. Acta (BBA) Gen. Subj. 2008, 1780, 1291–1303. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. The involvement of lysosomes in myocardial aging and disease. Curr. Cardiol. Rev. 2008, 4, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Reasor, M.J. A review of the biology and toxicologic implications of the induction of lysosomal lamellar bodies by drugs. Toxicol. Appl. Pharmacol. 1989, 97, 47–56. [Google Scholar] [CrossRef]
- Gao, J.; Schatton, D.; Martinelli, P.; Hansen, H.; Pla-Martin, D.; Barth, E.; Becker, C.; Altmueller, J.; Frommolt, P.; Sardiello, M.; et al. CLUH regulates mitochondrial biogenesis by binding mRNAs of nuclear-encoded mitochondrial proteins. J. Cell Biol. 2014, 207, 213–223. [Google Scholar] [CrossRef]
- Schatton, D.; Pla-Martin, D.; Marx, M.-C.; Hansen, H.; Mourier, A.; Nemazanyy, I.; Pessia, A.; Zentis, P.; Corona, T.; Kondylis, V.; et al. CLUH regulates mitochondrial metabolism by controlling translation and decay of target mRNAs. J. Cell Biol. 2017, 216, 675–693. [Google Scholar] [CrossRef]
- Wakim, J.; Goudenege, D.; Perrot, R.; Gueguen, N.; Desquiret-Dumas, V.; De La Barca, J.M.C.; Rosa, I.D.; Manero, F.; Le Mao, M.; Chupin, S.; et al. CLUH couples mitochondrial distribution to the energetic and metabolic status. J. Cell Sci. 2017, 130, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Campos, J.C.; Qvit, N.; Qi, X.; Bozi, L.H.M.; Bechara, L.R.G.; Lima, V.M.; Queliconi, B.B.; Disatnik, M.-H.; Dourado, P.M.M.; et al. A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W.; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The Opa1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnauné-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef]
- Nan, J.; Zhu, W.; Rahman, M.; Liu, M.; Li, D.; Su, S.; Zhang, N.; Hu, X.; Yu, H.; Gupta, M.P.; et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 1260–1273. [Google Scholar] [CrossRef]
- Yin, T.; Sandhu, G.; Wolfgang, C.D.; Burrier, A.; Webb, R.L.; Rigel, D.F.; Hai, T.; Whelan, J. Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J. Biol. Chem. 1997, 272, 19943–19950. [Google Scholar] [CrossRef]
- Das, J.; Ghosh, J.; Manna, P.; Sil, P.C. Taurine suppresses doxorubicin-triggered oxidative stress and cardiac apoptosis in rat via up-regulation of PI3-K/Akt and inhibition of p53, p38-JNK. Biochem. Pharmacol. 2011, 81, 891–909. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Fernández-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef]
- Aoki, H.; Kang, P.M.; Hampe, J.; Yoshimura, K.; Noma, T.; Matsuzaki, M.; Izumo, S. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J. Biol. Chem. 2002, 277, 10244–10250. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; De Brito, O.M.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef]
- Berry, J.M.; Le, V.; Rotter, D.; Battiprolu, P.K.; Grinsfelder, B.; Tannous, P.; Burchfield, J.S.; Czubryt, M.; Backs, J.; Olson, E.N.; et al. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circ. Res. 2011, 109, 407–417. [Google Scholar] [CrossRef]
- Kehat, I.; Molkentin, J.D. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010, 122, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rasmussen, T.P.; Koval, O.M.; Joiner, M.L.; Hall, D.D.; Chen, B.; Luczak, E.D.; Wang, Q.; Rokita, A.G.; Wehrens, X.H.; et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 2015, 6, 6081. [Google Scholar] [CrossRef]
- Finkel, T.; Menazza, S.; Holmström, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef]
- Suarez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Diaz-Juarez, J.; Diemer, T.; Dai, A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef]
- Joiner, M.-L.A.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nat. Cell Biol. 2012, 491, 269–273. [Google Scholar] [CrossRef]
- Nguyen, E.K.; Koval, O.M.; Noble, P.; Broadhurst, K.; Allamargot, C.; Wu, M.; Strack, S.; Thiel, W.H.; Grumbach, I.M. CaMKII (Ca2+/calmodulin-dependent kinase II) in mitochondria of smooth muscle cells controls mitochondrial mobility, migration, and neointima formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1333–1345. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2013, 55, 31–41. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Nair, K.S.; Bergen, R.H.; Klaus, K.; Guenzel, A.J.; Hajjar, R.J.; Redfield, M.M. Mitochondrial integrity and function in the progression of early pressure overload–induced left ventricular remodeling. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD + redox balance as a therapy for heart failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef]
- Chen, T.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Li, J.; Zhang, Y.; Bu, P. Correction: Mouse SIRT3 attenuates hypertrophy-related lipid accumulation in the heart through the deacetylation of LCAD. PLoS ONE 2016, 11, e0155173. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart-An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. Circ. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]
- Pound, K.M.; Sorokina, N.; Ballal, K.; Berkich, D.A.; Fasano, M.; LaNoue, K.F.; Taegtmeyer, H.; O’Donnell, J.M.; Lewandowski, E.D. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content. Circ. Res. 2009, 104, 805–812. [Google Scholar] [CrossRef]
- Cheng, C.-F.; Ku, H.-C.; Lin, H. PGC-1α as a pivotal factor in lipid and metabolic regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2009, 86, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Li, D.L.; Hill, J.A. Heart failure and loss of metabolic control. J. Cardiovasc. Pharmacol. 2014, 63, 302–313. [Google Scholar] [CrossRef]
- Wende, A.R.; Brahma, M.K.; McGinnis, G.R.; Young, M.E. Metabolic origins of heart failure. JACC Basic Transl. Sci. 2017, 2, 297–310. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing heart relies on ketone bodies as a fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef]
- Bedi, K.C.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart. Circ. Heart Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.-Y.; Ren, S.; Liu, Y.; et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Z.; Kolwicz, S.C.; Abell, L.; Roe, N.D.; Kim, M.; Zhou, B.; Cao, Y.; Ritterhoff, J.; Gu, H.; et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 2017, 25, 374–385. [Google Scholar] [CrossRef]
- Toczek, M.; Zielonka, D.; Zukowska, P.; Marcinkowski, J.T.; Slominska, E.; Isalan, M.; Smolenski, R.T.; Mielcarek, M. An impaired metabolism of nucleotides underpins a novel mechanism of cardiac remodeling leading to Huntington’s disease related cardiomyopathy. Biochim. Biophys. Acta 2016, 1862, 2147–2157. [Google Scholar] [CrossRef]
- Ramila, K.C.; Jong, C.J.; Pastukh, V.M.; Ito, T.; Azuma, J.; Schaffer, S.W. Role of protein phosphorylation in excitation-contraction coupling in taurine deficient hearts. Am. J. Physiol. Circ. Physiol. 2015, 308, H232–H239. [Google Scholar] [CrossRef]
- Beyranvand, M.R.; Khalafi, M.K.; Roshan, V.D.; Choobineh, S.; Parsa, S.A.; Piranfar, M.A. Effect of taurine supplementation on exercise capacity of patients with heart failure. J. Cardiol. 2011, 57, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Carubelli, V.; Castrini, A.I.; Lazzarini, V.; Gheorghiade, M.; Metra, M.; Lombardi, C.M. Amino acids and derivatives, a new treatment of chronic heart failure? Heart Fail. Rev. 2014, 20, 39–51. [Google Scholar] [CrossRef]
- Tanada, Y.; Kato, T.; Kato, T.; Kawamoto, A.; Okuda, J.; Kimura, T. Branched-chain amino acids ameliorate heart failure with cardiac cachexia in rats. Life Sci. 2015, 137, 20–27. [Google Scholar] [CrossRef]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef]
- Li, X.; Yang, X.; Li, Y.; Yuan, M.; Tian, C.; Yang, Y.; Zhang, X.; Liu, C.; Gao, Y.; Liu, N.; et al. Mitochondria and the pathophysiological mechanism of atrial fibrillation. Curr. Pharm. Des. 2018, 24, 3055–3061. [Google Scholar] [CrossRef]
- Yang, K.-C.; Bonini, M.G.; Dudley, S.C. Mitochondria and arrhythmias. Free. Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Van Der Weg, K.; Prinzen, F.W.; Gorgels, A.P. Editor’s Choice-Reperfusion cardiac arrhythmias and their relation to reperfusion-induced cell death. Eur. Heart J. Acute Cardiovasc. Care 2018, 8, 142–152. [Google Scholar] [CrossRef]
- Wiersma, M.; Van Marion, D.M.; Wüst, R.C.; Houtkooper, R.H.; Zhang, D.; De Groot, N.M.; Henning, R.H.; Brundel, B.J. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar]
- Bowen, T.S.; Rolim, N.P.L.; Fischer, T.; Baekkerud, F.H.; Medeiros, A.; Werner, S.; Brønstad, E.; Rognmo, O.; Mangner, N.; Linke, A.; et al. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur. J. Heart Fail. 2015, 17, 263–272. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef]
- Molina, A.J.A.; Bharadwaj, M.S.; Van Horn, C.; Nicklas, B.J.; Lyles, M.F.; Eggebeen, J.; Haykowsky, M.J.; Brubaker, P.H.; Kitzman, D.W. Skeletal muscle mitochondrial content, oxidative capacity, and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC Heart Fail. 2016, 4, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Linke, A.; Winzer, E. Skeletal muscle alterations in HFrEF vs. HFpEF. Curr. Heart Fail. Rep. 2017, 14, 489–497. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaanine, A.H.; LeJemtel, T.H.; Delafontaine, P. Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure. J. Clin. Med. 2020, 9, 3582. https://doi.org/10.3390/jcm9113582
Chaanine AH, LeJemtel TH, Delafontaine P. Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure. Journal of Clinical Medicine. 2020; 9(11):3582. https://doi.org/10.3390/jcm9113582
Chicago/Turabian StyleChaanine, Antoine H., Thierry H. LeJemtel, and Patrice Delafontaine. 2020. "Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure" Journal of Clinical Medicine 9, no. 11: 3582. https://doi.org/10.3390/jcm9113582
APA StyleChaanine, A. H., LeJemtel, T. H., & Delafontaine, P. (2020). Mitochondrial Pathobiology and Metabolic Remodeling in Progression to Overt Systolic Heart Failure. Journal of Clinical Medicine, 9(11), 3582. https://doi.org/10.3390/jcm9113582