Chimeric Antigen Receptor T-Cell Therapy for Colorectal Cancer

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Search Criteria
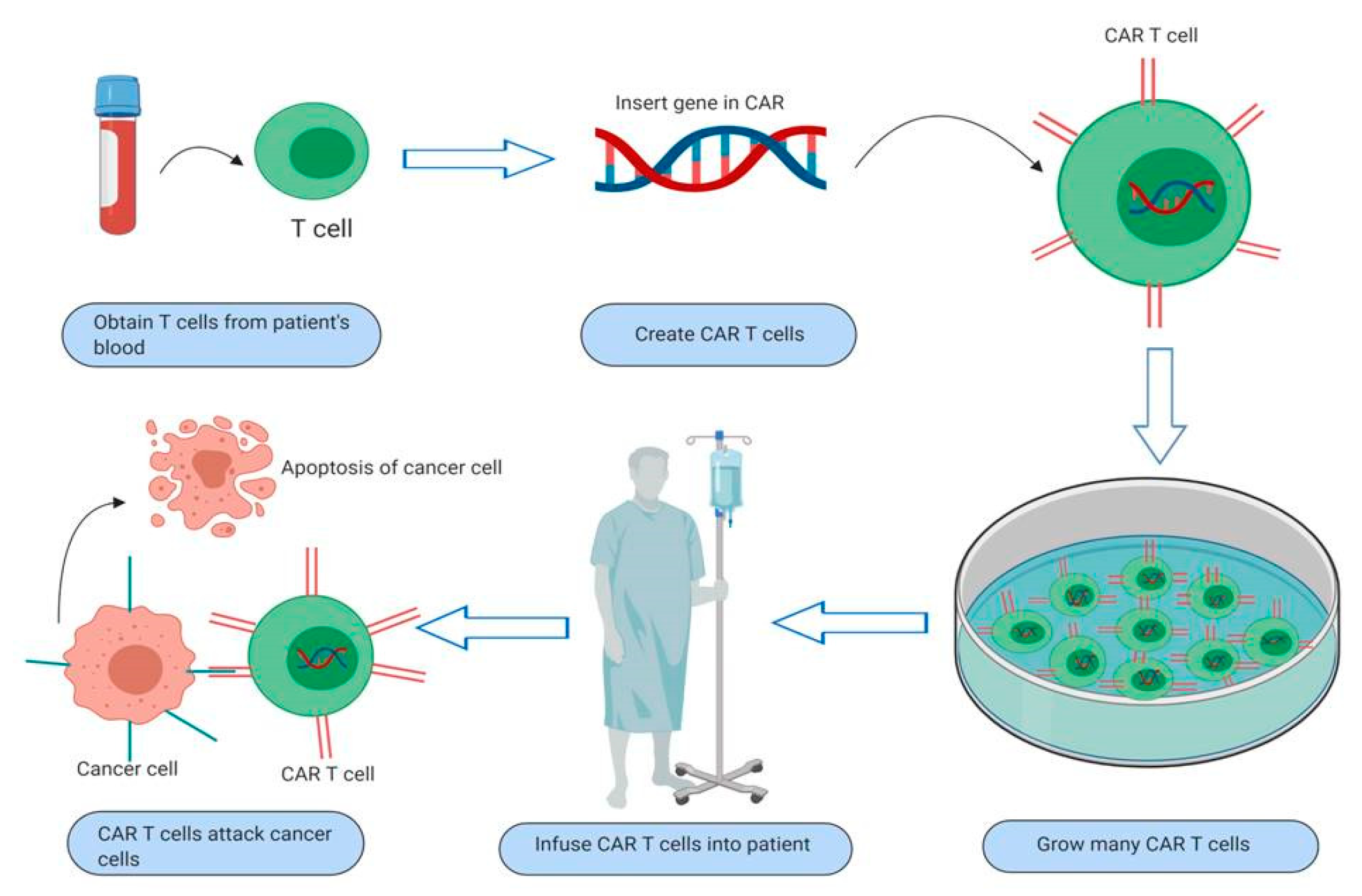
3. Overview and Mechanism of Action of CAR T-Cells
4. Current Toxicities and Administration Difficulties of CAR T-cells
5. CAR T-Cell Therapy in Solid Tumors
6. CAR T-Cell Therapy in Gastrointestinal Malignancies
6.1. Pancreatic Cancer
6.2. Hepatocellular Carcinoma
6.3. Gastric Cancer
6.4. Esophageal Cancer
6.5. Biliary Tract Cancer
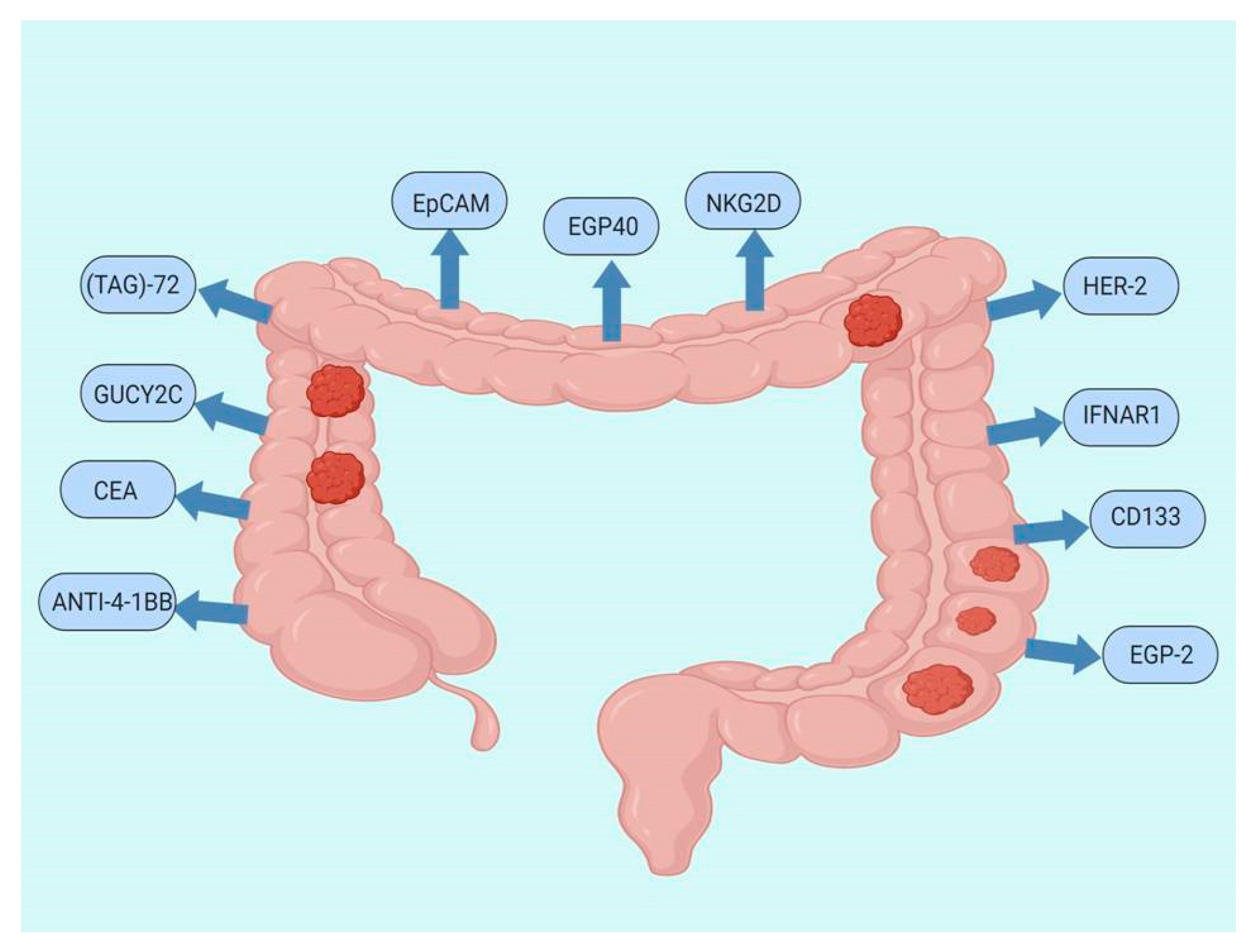
7. CAR T-Cell Therapy Studies for CRC
CAR T-Cells Ongoing Trials in CRC
8. Conclusions and Further Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.; Clarke, S.; Diaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Rittweger, K.; Gilberg, F.; Saltz, L. XELOX vs FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br. J. Cancer 2011, 105, 58–64. [Google Scholar] [CrossRef]
- Yee, N.S. Update in Systemic and Targeted Therapies in Gastrointestinal Oncology. Biomedicines 2018, 6, 34. [Google Scholar] [CrossRef]
- Marques, R.P.; Duarte, G.S.; Sterrantino, C.; Pais, H.L.; Quintela, A.; Martins, A.P.; Costa, J. Triplet (FOLFOXIRI) versus doublet (FOLFOX or FOLFIRI) backbone chemotherapy as first-line treatment of metastatic colorectal cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2017, 118, 54–62. [Google Scholar] [CrossRef]
- Ciombor, K.K.; Bekaii-Saab, T. A Comprehensive Review of Sequencing and Combination Strategies of Targeted Agents in Metastatic Colorectal Cancer. Oncologist 2018, 23, 25–34. [Google Scholar] [CrossRef]
- Koido, S.; Ohkusa, T.; Homma, S.; Namiki, Y.; Takakura, K.; Saito, K.; Ito, Z.; Kobayashi, H.; Kajihara, M.; Uchiyama, K.; et al. Immunotherapy for colorectal cancer. World J. Gastroenterol. 2013, 19, 8531–8542. [Google Scholar] [CrossRef]
- Advani, S.; Kopetz, S. Ongoing and future directions in the management of metastatic colorectal cancer: Update on clinical trials. J. Surg. Oncol. 2019, 119, 642–652. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [PubMed]
- van der Stok, E.P.; Spaander, M.C.W.; Grunhagen, D.J.; Verhoef, C.; Kuipers, E.J. Surveillance after curative treatment for colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 297–315. [Google Scholar] [CrossRef]
- Bonfrate, L.; Altomare, D.F.; Di Lena, M.; Travaglio, E.; Rotelli, M.T.; De Luca, A.; Portincasa, P. MicroRNA in colorectal cancer: New perspectives for diagnosis, prognosis and treatment. J. Gastrointest. Liver Dis. 2013, 22, 311–320. [Google Scholar]
- Mousavi, S.; Moallem, R.; Hassanian, S.M.; Sadeghzade, M.; Mardani, R.; Ferns, G.A.; Khazaei, M.; Avan, A. Tumor-derived exosomes: Potential biomarkers and therapeutic target in the treatment of colorectal cancer. J. Cell Physiol. 2019, 234, 12422–12432. [Google Scholar] [CrossRef]
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal cancer vaccines: Tumor-associated antigens vsneoantigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef] [PubMed]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37 (Suppl. 1), 95–100. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G. The other face of chimeric antigen receptors. Mol. Ther. 2014, 22, 899–900. [Google Scholar] [CrossRef]
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef]
- Quintas-Cardama, A. What CAR Will Win the CD19 Race? Mol. Cancer Ther. 2019, 18, 498–506. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Chimeric antigen receptor T-cell therapies for lymphoma. Nat. Rev. Clin. Oncol. 2018, 15, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Di, S.; Li, Z. Treatment of solid tumors with chimeric antigen receptor-engineered T cells: Current status and future prospects. Sci. China Life Sci. 2016, 59, 360–369. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. AxicabtageneCiloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Prasad, V. Immunotherapy: Tisagenlecleucel-the first approved CAR-T-cell therapy: Implications for payers and policy makers. Nat. Rev. Clin. Oncol. 2018, 15, 11–12. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Dis. 2016, 6, 664–679. [Google Scholar] [CrossRef]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018, 23, 943–947. [Google Scholar] [CrossRef]
- Hay, K.A. Cytokine release syndrome and neurotoxicity after CD19 chimeric antigen receptor-modified (CAR-) T cell therapy. Br. J. Haematol. 2018, 183, 364–374. [Google Scholar] [CrossRef]
- Hunter, B.D.; Jacobson, C.A. CAR T-cell associated neurotoxicity: Mechanisms, clinicopathologic correlates, and future directions. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef]
- Neelapu, S.S. Managing the toxicities of CAR T-cell therapy. Hematol. Oncol. 2019, 37 (Suppl. 1), 48–52. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transpl. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Hanafi, L.A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; Lopez, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, K.; Zhou, X.; Mineishi, S.; Di Stasi, A. Seatbelts in CAR therapy: How Safe Are CARS? Pharmaceuticals 2015, 8, 230–249. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Fuji, S.; Berce, C.; Onaciu, A.; Chira, S.; Petrushev, B.; Micu, W.T.; Moisoiu, V.; Osan, C.; Constantinescu, C.; et al. Chimeric Antigen Receptor T-Cells for the Treatment of B-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 2018, 9, 239. [Google Scholar] [CrossRef]
- Shah, N.N.; Maatman, T.; Hari, P.; Johnson, B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef]
- Gill, S.; Maus, M.V.; Porter, D.L. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016, 30, 157–167. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef]
- Scarfo, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef]
- Yeku, O.; Li, X.; Brentjens, R.J. Adoptive T-Cell Therapy for Solid Tumors. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef]
- Gauthier, J.; Yakoub-Agha, I. Chimeric antigen-receptor T-cell therapy for hematological malignancies and solid tumors: Clinical data to date, current limitations and perspectives. Curr. Res. Transl. Med. 2017, 65, 93–102. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D., Jr.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef]
- Yan, L.; Liu, B. Critical factors in chimeric antigen receptor-modified T-cell (CAR-T) therapy for solid tumors. Onco Targets Ther. 2019, 12, 193–204. [Google Scholar] [CrossRef]
- Xu, J.; Tian, K.; Zhang, H.; Li, L.; Liu, H.; Liu, J.; Zhang, Q.; Zheng, J. Chimeric antigen receptor-T cell therapy for solid tumors require new clinical regimens. Expert Rev. Anticancer Ther. 2017, 17, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Alrifai, D.; Sarker, D.; Maher, J. Prospects for adoptive immunotherapy of pancreatic cancer using chimeric antigen receptor-engineered T-cells. Immunopharmacol. Immunotoxicol. 2016, 38, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhao, G.; Zhao, Y. Combination Immunotherapy Approaches for Pancreatic Cancer Treatment. Can J. Gastroenterol. Hepatol. 2018, 2018, 6240467. [Google Scholar] [CrossRef] [PubMed]
- Jindal, V.; Arora, E.; Masab, M.; Gupta, S. Chimeric antigen receptor T cell therapy in pancreatic cancer: From research to practice. Med. Oncol. 2018, 35, 84. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Tano, Z.E.; Varghese, A.M.; Adusumilli, P.S. CAR T-cell therapy for pancreatic cancer. J. Surg. Oncol. 2017, 116, 63–74. [Google Scholar] [CrossRef]
- O’Hara, M.; Stashwick, C.; Haas, A.R.; Tanyi, J.L. Mesothelin as a target for chimeric antigen receptor-modified T cells as anticancer therapy. Immunotherapy 2016, 8, 449–460. [Google Scholar] [CrossRef]
- He, J.; Zhang, Z.; Lv, S.; Liu, X.; Cui, L.; Jiang, D.; Zhang, Q.; Li, L.; Qin, W.; Jin, H.; et al. Engineered CAR T cells targeting mesothelin by piggyBac transposon system for the treatment of pancreatic cancer. Cell Immunol. 2018, 329, 31–40. [Google Scholar] [CrossRef]
- Heckler, M.; Dougan, S.K. Unmasking Pancreatic Cancer: Epitope Spreading After Single Antigen Chimeric Antigen Receptor T-Cell Therapy in a Human Phase I Trial. Gastroenterology 2018, 155, 11–14. [Google Scholar] [CrossRef]
- Nichetti, F.; Marra, A.; Corti, F.; Guidi, A.; Raimondi, A.; Prinzi, N.; de Braud, F.; Pusceddu, S. The Role of Mesothelin as a Diagnostic and Therapeutic Target in Pancreatic Ductal Adenocarcinoma: A Comprehensive Review. Target Oncol. 2018, 13, 333–351. [Google Scholar] [CrossRef]
- Batchu, R.B.; Gruzdyn, O.V.; Mahmud, E.M.; Chukr, F.; Dachepalli, R.; Manmari, S.K.; Mostafa, G.; Weaver, D.W.; Gruber, S.A. Inhibition of Interleukin-10 in the tumor microenvironment can restore mesothelin chimeric antigen receptor T cell activity in pancreatic cancer in vitro. Surgery 2018, 163, 627–632. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhou, S.; Zhao, J.; Deng, C.; Teng, R.; Zhao, Y.; Chen, J.; Dong, J.; Yin, M.; Bai, Y.; et al. Engineered T lymphocytes eliminate lung metastases in models of pancreatic cancer. Oncotarget 2018, 9, 13694–13705. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef]
- Abate-Daga, D.; Lagisetty, K.H.; Tran, E.; Zheng, Z.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Miermont, A.M.; Teper, Y.; Rudloff, U.; et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 2014, 25, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.M. Chimeric antigen receptor (CAR) T and other T cell strategies for pancreas adenocarcinoma. Chin. Clin. Oncol. 2017, 6, 66. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Berahovich, R.; Zhou, H.; Xu, S.; Harto, H.; Li, L.; Chao, C.C.; Mao, M.M.; Wu, L. CD47-CAR-T Cells Effectively Kill Target Cancer Cells and Block Pancreatic Tumor Growth. Cancers 2017, 9, 139. [Google Scholar] [CrossRef]
- Zhang, E.; Yang, P.; Gu, J.; Wu, H.; Chi, X.; Liu, C.; Wang, Y.; Xue, J.; Qi, W.; Sun, Q.; et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J. Hematol. Oncol. 2018, 11, 102. [Google Scholar] [CrossRef]
- Raj, D.; Yang, M.H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019, 68, 1052–1064. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Ali, A.I.; Oliver, A.J.; Samiei, T.; Chan, J.D.; Kershaw, M.H.; Slaney, C.Y. Genetic Redirection of T Cells for the Treatment of Pancreatic Cancer. Front. Oncol. 2019, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, S.S.; Cheung, N.V. Immunotherapy of hepatocellular carcinoma using chimeric antigen receptors and bispecific antibodies. Cancer Lett. 2017, 399, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wu, L.; Zhou, F.; Hong, Z.; Yuan, Y.; Liu, Z. T Cell-Associated Immunotherapy for Hepatocellular Carcinoma. Cell Physiol. Biochem. 2017, 41, 609–622. [Google Scholar] [CrossRef]
- Chen, Y.; Chang-Yong, E.; Gong, Z.W.; Liu, S.; Wang, Z.X.; Yang, Y.S.; Zhang, X.W. Chimeric antigen receptor-engineered T-cell therapy for liver cancer. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 301–309. [Google Scholar] [CrossRef]
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198–207. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2016, 7, 690. [Google Scholar] [CrossRef]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef]
- Chen, C.; Li, K.; Jiang, H.; Song, F.; Gao, H.; Pan, X.; Shi, B.; Bi, Y.; Wang, H.; Li, Z. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol. Immunother. 2017, 66, 475–489. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Z.; Liu, Z.; Wei, D.; Wu, X.; Bian, H.; Chen, Z. Adoptive cell transfer therapy for hepatocellular carcinoma. Front. Med. 2019, 13, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Kaneko, S. Immune cell therapy for hepatocellular carcinoma. J. Hematol. Oncol. 2019, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Wang, S.; Hu, Y.; Li, Z.; Yang, W.; Lv, Y.; Wang, L.; Zhang, L.; Ji, J. Monoclonal antibody 3H11 chimeric antigen receptors enhance T cell effector function and exhibit efficacy against gastric cancer. Oncol. Lett. 2018, 15, 6887–6894. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Pyo, S.; Kang, C.H.; Lee, C.O.; Lee, H.K.; Choi, S.U.; Park, C.H. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS ONE 2018, 13, e0198347. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; He, M.; Tao, F.; Xu, G.; Ye, M.; Zheng, Y.; Li, Y. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemother. Pharmacol. 2018, 82, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shi, Z.; Wang, P.; Wang, C.; Yang, L.; Du, G.; Zhang, H.; Shi, B.; Jia, J.; Li, Q.; et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J. Natl. Cancer Inst. 2019, 111, 409–418. [Google Scholar] [CrossRef]
- Kiesgen, S.; Chicaybam, L.; Chintala, N.K.; Adusumilli, P.S. Chimeric Antigen Receptor (CAR) T-Cell Therapy for Thoracic Malignancies. J. Thorac. Oncol. 2018, 13, 16–26. [Google Scholar] [CrossRef]
- Shi, H.; Yu, F.; Mao, Y.; Ju, Q.; Wu, Y.; Bai, W.; Wang, P.; Xu, R.; Jiang, M.; Shi, J. EphA2 chimeric antigen receptor-modified T cells for the immunotherapy of esophageal squamous cell carcinoma. J. Thorac. Dis. 2018, 10, 2779–2788. [Google Scholar] [CrossRef]
- DeLeon, T.T.; Zhou, Y.; Nagalo, B.M.; Yokoda, R.T.; Ahn, D.H.; Ramanathan, R.K.; Salomao, M.A.; Aqel, B.A.; Mahipal, A.; Bekaii-Saab, T.S.; et al. Novel immunotherapy strategies for hepatobiliary cancers. Immunotherapy 2018, 10, 1077–1091. [Google Scholar] [CrossRef]
- Xu, J.Y.; Ye, Z.L.; Jiang, D.Q.; He, J.C.; Ding, Y.M.; Li, L.F.; Lv, S.Q.; Wang, Y.; Jin, H.J.; Qian, Q.J. Mesothelin-targeting chimeric antigen receptor-modified T cells by piggyBac transposon system suppress the growth of bile duct carcinoma. Tumour Biol. 2017, 39, 1010428317695949. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor-Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 expression status in diverse cancers: Review of results from 37,992 patients. Cancer Metastasis Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Magee, M.S.; Kraft, C.L.; Abraham, T.S.; Baybutt, T.R.; Marszalowicz, G.P.; Li, P.; Waldman, S.A.; Snook, A.E. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. Oncoimmunology 2016, 5, e1227897. [Google Scholar] [CrossRef] [PubMed]
- Magee, M.S.; Abraham, T.S.; Baybutt, T.R.; Flickinger, J.C., Jr.; Ridge, N.A.; Marszalowicz, G.P.; Prajapati, P.; Hersperger, A.R.; Waldman, S.A.; Snook, A.E. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol. Res. 2018, 6, 509–516. [Google Scholar] [CrossRef]
- Sheen, A.J.; Irlam, J.; Kirillova, N.; Guest, R.D.; Sherlock, D.J.; Hawkins, R.E.; Gilham, D.E. Gene therapy of patient-derived T lymphocytes to target and eradicate colorectal hepatic metastases. Dis. Colon. Rectum. 2003, 46, 793–804. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA (+) Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef]
- Blat, D.; Zigmond, E.; Alteber, Z.; Waks, T.; Eshhar, Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. 2014, 22, 1018–1028. [Google Scholar] [CrossRef]
- Chi, X.; Yang, P.; Zhang, E.; Gu, J.; Xu, H.; Li, M.; Gao, X.; Li, X.; Zhang, Y.; Hu, J. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med. 2019, 8, 4753–4765. [Google Scholar] [CrossRef]
- Darcy, P.K.; Haynes, N.M.; Snook, M.B.; Trapani, J.A.; Cerruti, L.; Jane, S.M.; Smyth, M.J. Redirected perforin-dependent lysis of colon carcinoma by ex vivo genetically engineered CTL. J. Immunol. 2000, 164, 3705–3712. [Google Scholar] [CrossRef] [PubMed]
- Simmons, A.; Whitehead, R.P.; Kolokoltsov, A.A.; Davey, R.A. Use of recombinant lentiviruspseudotyped with vesicular stomatitis virus glycoprotein G for efficient generation of human anti-cancer chimeric T cells by transduction of human peripheral blood lymphocytes in vitro. Virol. J. 2006, 3, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mardiana, S.; John, L.B.; Henderson, M.A.; Slaney, C.Y.; von Scheidt, B.; Giuffrida, L.; Davenport, A.J.; Trapani, J.A.; Neeson, P.J.; Loi, S.; et al. A Multifunctional Role for Adjuvant Anti-4-1BB Therapy in Augmenting Antitumor Response by Chimeric Antigen Receptor T Cells. Cancer Res. 2017, 77, 1296–1309. [Google Scholar] [CrossRef] [PubMed]
- Daly, T.; Royal, R.E.; Kershaw, M.H.; Treisman, J.; Wang, G.; Li, W.; Herlyn, D.; Eshhar, Z.; Hwu, P. Recognition of human colon cancer by T cells transduced with a chimeric receptor gene. Cancer Gene Ther. 2000, 7, 284–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, Q.; Xi, J.; Wang, L.; Wang, X.; Ma, X.; Deng, Q.; Lu, Y.; Kumar, M.; Zhou, Z.; Li, L.; et al. Correction to: miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J. Hematol. Oncol. 2018, 11, 90. [Google Scholar] [CrossRef]
- Katz, S.C.; Point, G.R.; Cunetta, M.; Thorn, M.; Guha, P.; Espat, N.J.; Boutros, C.; Hanna, N.; Junghans, R.P. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016, 23, 142–148. [Google Scholar] [CrossRef]
- Ang, W.X.; Li, Z.; Chi, Z.; Du, S.H.; Chen, C.; Tay, J.C.; Toh, H.C.; Connolly, J.E.; Xu, X.H.; Wang, S. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget 2017, 8, 13545–13559. [Google Scholar] [CrossRef]
- Zhang, B.L.; Li, D.; Gong, Y.L.; Huang, Y.; Qin, D.Y.; Jiang, L.; Liang, X.; Yang, X.; Gou, H.F.; Wang, Y.S.; et al. Preclinical Evaluation of Chimeric Antigen Receptor-Modified T Cells Specific to Epithelial Cell Adhesion Molecule for Treating Colorectal Cancer. Hum. Gene Ther. 2019, 30, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gao, F.; Li, N.; Li, Q.; Zhou, Y.; Yang, T.; Cai, Z.; Du, P.; Chen, F.; Cai, J. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am. J. Cancer Res. 2019, 9, 945–958. [Google Scholar]
- Teng, R.; Zhao, J.; Zhao, Y.; Gao, J.; Li, H.; Zhou, S.; Wang, Y.; Sun, Q.; Lin, Z.; Yang, W.; et al. Chimeric Antigen Receptor-modified T Cells Repressed Solid Tumors and Their Relapse in an Established Patient-derived Colon Carcinoma Xenograft Model. J. Immunother. 2019, 42, 33–42. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed]
- Kuczma, M.P.; Ding, Z.C.; Li, T.; Habtetsion, T.; Chen, T.; Hao, Z.; Bryan, L.; Singh, N.; Kochenderfer, J.N.; Zhou, G. The impact of antibiotic usage on the efficacy of chemoimmunotherapy is contingent on the source of tumor-reactive T cells. Oncotarget 2017, 8, 111931–111942. [Google Scholar] [CrossRef] [PubMed]
- Ren-Heidenreich, L.; Mordini, R.; Hayman, G.T.; Siebenlist, R.; LeFever, A. Comparison of the TCR zeta-chain with the FcR gamma-chain in chimeric TCR constructs for T cell activation and apoptosis. Cancer Immunol. Immunother. 2002, 51, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Ikeda, H.; Sato, M.; Ohkuri, T.; Abe, H.; Kuroki, M.; Onodera, M.; Miyamoto, M.; Kondo, S.; Nishimura, T. Antitumor activity of chimeric immunoreceptor gene-modified Tc1 and Th1 cells against autologous carcinoembryonic antigen-expressing colon cancer cells. Cancer Sci. 2006, 97, 920–927. [Google Scholar] [CrossRef]
- alloSHRINK-Standard Chemotherapy Regimen and Immunotherapy with Allogeneic NKG2D-based CYAD-101 Chimeric Antigen Receptor T-cell-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03692429 (accessed on 13 August 2019).
- EGFR-IL12-CART Cells for Patients with Metastatic Colorectal Cancer-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03542799 (accessed on 20 December 2019).
- EGFR CART Cells for Patients with Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03152435 (accessed on 20 December 2019).
- A Study of Chimeric Antigen Receptor T Cells Combined with Interventional Therapy in Advanced Liver Malignancy. Available online: https://clinicaltrials.gov/ct2/show/NCT02959151 (accessed on 20 December 2019).
- Hepatic Transarterial Administrations of NKR-2 in Patients with Unresectable Liver Metastases from Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03370198 (accessed on 20 December 2019).
- Dose Escalation and Dose Expansion Phase I Study to Assess the Safety and Clinical Activity of Multiple Doses of NKR-2 Administered Concurrently with FOLFOX in Colorectal Cancer with Potentially Resectable Liver Metastases. Available online: https://clinicaltrials.gov/ct2/show/NCT03310008 (accessed on 20 December 2019).
- CAR-T Cell Immunotherapy in MUC1 Positive Solid Tumor-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02617134 (accessed on 14 August 2019).
- A Clinical Research of CAR T Cells Targeting HER2 Positive Cancer-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02713984 (accessed on 14 August 2019).
- A Clinical Research of CAR T Cells Targeting CEA Positive Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02349724 (accessed on 20 December 2019).
- CAR-T Intraperitoneal Infusions for CEA-Expressing Adenocarcinoma Peritoneal Metastases or Malignant Ascites (IPC). Available online: https://clinicaltrials.gov/ct2/show/NCT03682744 (accessed on 18 December 2019).
- Autologous CAR-T/TCR-T Cell Immunotherapy for Malignancies-Tabular View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03638206 (accessed on 14 August 2019).
- A Clinical Research of CAR T Cells Targeting EpCAM Positive Cancer-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03013712 (accessed on 14 August 2019).
- Treatment of Relapsed and/or Chemotherapy Refractory Advanced Malignancies by CART133 [Internet]. Available online: https://clinicaltrials.gov/ct2/show/record/NCT02541370?cond=Colorectal+Cancer&intr=chimeric+antigen+receptor+t-cell&rank=13 (accessed on 9 July 2019).
- CAR-T Hepatic Artery Infusions or Pancreatic Venous Infusions for CEA-Expressing Liver Metastases or Pancreas Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02850536 (accessed on 19 December 2019).
- Binary Oncolytic Adenovirus in Combination with HER2-Specific CAR VST, Advanced HER2 Positive Solid Tumors (VISTA)-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03740256 (accessed on 14 August 2019).
- CAR-T Hepatic Artery Infusions and Sir-Spheres for Liver Metastases. Available online: https://clinicaltrials.gov/ct2/show/NCT02416466 (accessed on 20 December 2019).



{kind=link}
{kind=link}
| Target | Pathology | Trial ID | Study Phase | Administration | Patient Number | Year | Reference |
|---|---|---|---|---|---|---|---|
| EGFR IL-12 | Metastatic colorectal cancer | NCT03542799 | I/II | Systemic | 20 | 2018 | [127] |
| EGFR | EGFR-positive colorectal Cancer | NCT03152435 | I/II | Systemic | 20 | 2017 | [128] |
| NKG2D | Metastatic colorectal cancer | NCT03692429 | I | Systemic | 36 | 2018 | [126] |
| CEA | Metastatic colorectal cancer | NCT02959151 | I/II | Vascular interventional therapy or intratumoral injection | 20 | 2016 | [129] |
| NKR-2 | Unresectable liver metastasis of colorectal cancer | NCT03370198 | I | Hepatic transarterial | 18 | 2017 | [130] |
| NKR-2 | Potentially resectable liver metastasis of colorectal cancer | NCT03310008 | I | Systemic | 36 | 2017 | [131] |
| MUC 1 | Colorectal cancer | NCT02617134 | I/II | Systemic | 20 | 2015 | [132] |
| HER2 | Colorectal cancer | NCT02713984 | I/II | Systemic | 60 | 2016 | [133] |
| CEA | Colorectal cancer | NCT02349724 | I | Systemic | 75 | 2015 | [134] |
| CEA | Peritoneal metastases or malignant ascites of Colorectal cancer | NCT03682744 | I | Intraperitoneal infusion | 18 | 2018 | [135] |
| C-MET | Colorectal cancer | NCT03638206 | I/II | Systemic | 73 | 2018 | [136] |
| EpCAM | Colorectal cancer | NCT03013712 | I/II | Vascular interventional therapy | 60 | 2017 | [137] |
| Endoscopy mediated infusion | |||||||
| CD133 | Colorectal cancer | NCT02541370 | I/II | Systemic | 20 | 2015 | [138] |
| CEA | CEA + liver metastases from gastrointestinal tumors including colorectal cancer | NCT02850536 | I | Hepatic transarterial | 5 | 2015 | [139] |
| Intrapancreatic retrograde venous infusion | |||||||
| HER2 | Colorectal cancer | NCT03740256 | I | Systemic &Intratumoral | 39 | 2018 | [140] |
| CEA | CEA + adenocarcinoma with liver metastases from gastrointestinal tumors including colorectal cancer | NCT02416466 | I | Hepatic transarterial administration | 8 | 2015 | [141] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sur, D.; Havasi, A.; Cainap, C.; Samasca, G.; Burz, C.; Balacescu, O.; Lupan, I.; Deleanu, D.; Irimie, A. Chimeric Antigen Receptor T-Cell Therapy for Colorectal Cancer. J. Clin. Med. 2020, 9, 182. https://doi.org/10.3390/jcm9010182
Sur D, Havasi A, Cainap C, Samasca G, Burz C, Balacescu O, Lupan I, Deleanu D, Irimie A. Chimeric Antigen Receptor T-Cell Therapy for Colorectal Cancer. Journal of Clinical Medicine. 2020; 9(1):182. https://doi.org/10.3390/jcm9010182
Chicago/Turabian StyleSur, Daniel, Andrei Havasi, Calin Cainap, Gabriel Samasca, Claudia Burz, Ovidiu Balacescu, Iulia Lupan, Diana Deleanu, and Alexandru Irimie. 2020. "Chimeric Antigen Receptor T-Cell Therapy for Colorectal Cancer" Journal of Clinical Medicine 9, no. 1: 182. https://doi.org/10.3390/jcm9010182
APA StyleSur, D., Havasi, A., Cainap, C., Samasca, G., Burz, C., Balacescu, O., Lupan, I., Deleanu, D., & Irimie, A. (2020). Chimeric Antigen Receptor T-Cell Therapy for Colorectal Cancer. Journal of Clinical Medicine, 9(1), 182. https://doi.org/10.3390/jcm9010182