Targeting of the Cancer-Associated Fibroblast—T-Cell Axis in Solid Malignancies

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Cancer-Associated Fibroblast Phenotypes
2.1. Fibroblast Subtyping in Solid Malignancies Identifies a Pan-Tumor Inflammatory CAF Subset
2.2. Inflammatory Fibroblast Subsets in Autoimmunity
3. Mechanisms of CAF-T-Cell Interactions
3.1. Regulation of T-Cell Migration within the Desmoplastic TME
3.1.1. The CAF Secretome
3.1.2. Extracellular Matrix Production (ECM) and CAF Barrier Function
3.2. CAF-Mediated T-Cell Suppression
3.2.1. Modulation of Antigen Presentation
3.2.2. Checkpoint Molecule Expression
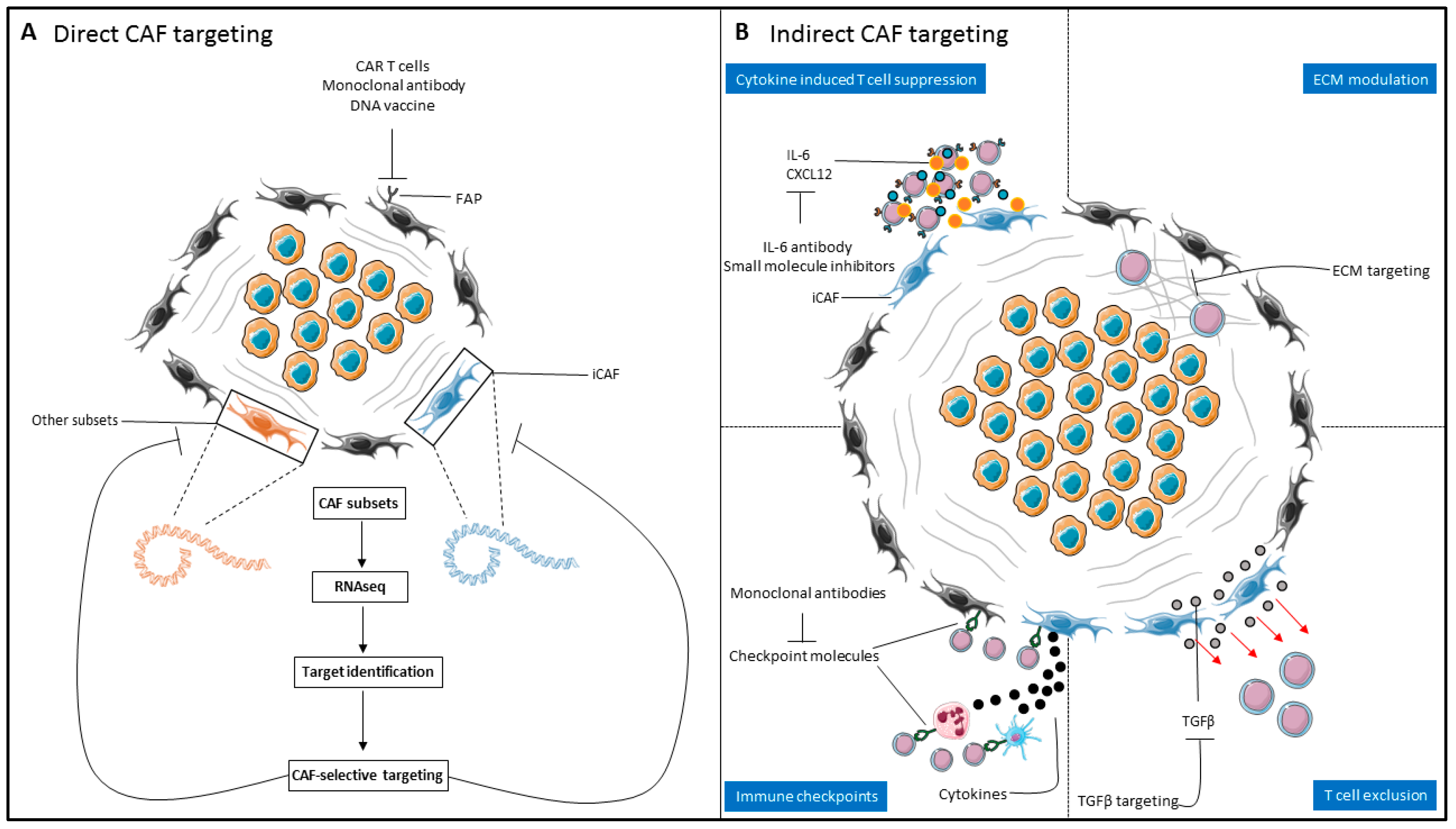
4. Therapeutic Targeting of the CAF–T-Cell Axis in Solid Malignancies
4.1. Direct CAF Targeting
4.2. Indirect CAF targeting
4.2.1. Targeting the CAF Secretome/CAF Signaling
4.2.2. Modulation of the Extracellular Matrix
5. Conclusions
Article highlights
- RNA sequencing has revealed the presence of multiple subsets of cancer-associated fibroblasts (CAFs) in multiple types of solid malignancies
- The inflammatory CAF (iCAF) subset, characterized by the expression of inflammatory cytokines (TGF-β, IL6, CXCL12), is present in a variety of solid malignancies and may shape the tumor immune microenvironment
- The inflammatory state of iCAFs should be viewed as a dynamic response towards cues within the tumor microenvironment, rather than a static, fixed state
- T-cell excluded tumors are strongly associated with Transforming Growth Factor-β (TGF-β) signaling in CAFs
- CAF-mediated regulation of APC function and T cell checkpoint inhibition contributes to the exhausted T cell phenotype
- Targeting of the CAF secretome, TGF-β signaling in CAFs and its ECM-producing functions may improve T cell infiltration and, in combination with checkpoint inhibitors, lead to a release of immune suppression in the TME
- The knowledge gap regarding specific CAF subset function limits the exploitation of direct CAF targeting without significant off-target risks
Author Contributions
Funding
Conflicts of Interest
References
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Lin, D.; Takhar, M.; Ramnarine, V.R.; Dong, X.; Bell, R.H.; Volik, S.V.; Wang, K.; Xue, H.; Wang, Y.; et al. Stromal Gene Expression is Predictive for Metastatic Primary Prostate Cancer. Eur. Urol. 2018, 73, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Winslow, S.; Leandersson, K.; Edsjö, A.; Larsson, C. Prognostic stromal gene signatures in breast cancer. Breast Cancer Res. BCR 2015, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Paulsson, J.; Micke, P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin. Cancer Biol. 2014, 25, 61–68. [Google Scholar] [CrossRef]
- Huijbers, A.; Tollenaar, R.; v Pelt, G.W.; Zeestraten, E.C.M.; Dutton, S.; McConkey, C.C.; Domingo, E.; Smit, V.; Midgley, R.; Warren, B.F. The proportion of tumor-stroma as a strong prognosticator for stage II and III colon cancer patients: Validation in the VICTOR trial. Ann. Oncol. 2012, 24, 179–185. [Google Scholar] [CrossRef]
- Wu, J.; Liang, C.; Chen, M.; Su, W. Association between tumor-stroma ratio and prognosis in solid tumor patients: A systematic review and meta-analysis. Oncotarget 2016, 7, 68954. [Google Scholar] [CrossRef]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.-Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Togo, S.; Polanska, U.M.; Horimoto, Y.; Orimo, A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers 2013, 5, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, Y.; Kashima, T.G.; Nishiyama, T.; Shimazu, K.; Morishita, Y.; Shimazaki, M.; Kii, I.; Horie, H.; Nagai, H.; Kudo, A.; et al. Periostin is expressed in pericryptal fibroblasts and cancer-associated fibroblasts in the colon. J. Histochem. Cytochem. 2008, 56, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Ishii, G.; Hashimoto, H.; Kuwata, T.; Nagai, K.; Date, H.; Ochiai, A. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int. J. Cancer 2015, 137, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Paauwe, M.; Schoonderwoerd, M.J.A.; Helderman, R.F.C.P.; Harryvan, T.J.; Groenewoud, A.; van Pelt, G.W.; Bor, R.; Hemmer, D.M.; Versteeg, H.H.; Snaar-Jagalska, B.E.; et al. Endoglin Expression on Cancer-Associated Fibroblasts Regulates Invasion and Stimulates Colorectal Cancer Metastasis. Clin. Cancer Res. 2018, 24, 6331–6344. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.A.C.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.N.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Räsänen, K.; Vaheri, A. Activation of fibroblasts in cancer stroma. Exp. Cell Res. 2010, 316, 2713–2722. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Avgustinova, A.; Iravani, M.; Robertson, D.; Fearns, A.; Gao, Q.; Klingbeil, P.; Hanby, A.M.; Speirs, V.; Sahai, E.; Calvo, F.; et al. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat. Commun. 2016, 7, 10305. [Google Scholar] [CrossRef]
- Östman, A. PDGF receptors in tumor stroma: Biological effects and associations with prognosis and response to treatment. Adv. Drug Deliv. Rev. 2017, 121, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Man, S.; Duffhues, G.S.; ten Dijke, P.; Baker, D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 2019, 22, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Salati, M.; Sorgentoni, G.; Barbisan, F.; Orciani, M. Characterization of tumor-derived mesenchymal stem cells potentially differentiating into cancer-associated fibroblasts in lung cancer. Clin. Transl. Oncol. 2018, 20, 1582–1591. [Google Scholar] [CrossRef]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y. Pericyte–fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Sainson, R.C.A. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Herrera, M.; Islam, A.B.M.M.K.; Herrera, A.; Martín, P.; García, V.; Silva, J.; Garcia, J.M.; Salas, C.; Casal, I.; de Herreros, A.G.; et al. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin. Cancer Res. 2013, 19, 5914–5926. [Google Scholar] [CrossRef]
- Hussain, A.; Voisin, V.; Poon, S.; Meens, J.; Dmytryshyn, J.; Ho, V.W.; Tang, K.H.; Paterson, J.; Clarke, B.; Bernardini, M.Q.; et al. Distinct cancer-associated fibroblast states drive clinical outcomes in high-grade serous ovarian cancer and are regulated by TCF21. bioRxiv 2019, bioRxiv:519728. [Google Scholar]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Hao, J.; Zeltz, C.; Pintilie, M.; Li, Q.; Sakashita, S.; Wang, T.; Cabanero, M.; Martins-Filho, S.N.; Wang, D.Y.; Pasko, E.; et al. Characterization of Distinct Populations of Carcinoma-Associated Fibroblasts from Non-Small Cell Lung Carcinoma Reveals a Role for ST8SIA2 in Cancer Cell Invasion. Neoplasia 2019, 21, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Hendrayani, S.-F.; Al-Khalaf, H.H.; Aboussekhra, A. The cytokine IL-6 reactivates breast stromal fibroblasts through transcription factor STAT3-dependent up-regulation of the RNA-binding protein AUF1. J. Biol. Chem. 2014, 289, 30962–30976. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Noss, E.H.; Mizoguchi, F.; Huppertz, C.; Wei, K.S.; Watts, G.F.M.; Brenner, M.B. Autocrine Loop Involving IL-6 Family Member LIF, LIF Receptor, and STAT4 Drives Sustained Fibroblast Production of Inflammatory Mediators. Immunity 2017, 46, 220–232. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef]
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 2018, 175, 372–386.e17. [Google Scholar] [CrossRef]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T Cell Migration in Rheumatoid Arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef]
- Kurowska, M.; Rudnicka, W.; Kontny, E.; Janicka, I.; Chorazy, M.; Kowalczewski, J.; Ziółkowska, M.; Ferrari-Lacraz, S.; Strom, T.B.; Maśliński, W. Fibroblast-like synoviocytes from rheumatoid arthritis patients express functional IL-15 receptor complex: Endogenous IL-15 in autocrine fashion enhances cell proliferation and expression of Bcl-x(L) and Bcl-2. J. Immunol. 2002, 169, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Saada, J.I.; Beswick, E.J.; Boya, G.; Qiu, S.M.; Mifflin, R.C.; Raju, G.S.; Reyes, V.E.; Powell, D.W. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology 2008, 135, 1228–1237.e2. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.M.; Okamoto, K.; Komatsu, N.; Sawa, S.; Danks, L.; Penninger, J.M.; Nakashima, T.; Takayanagi, H. Inhibition of the TNF family cytokine RANKL prevents autoimmune inflammation in the central nervous system. Immunity 2015, 43, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Azimi, F.; Scolyer, R.A.; Rumcheva, P.; Moncrieff, M.; Murali, R.; McCarthy, S.W.; Saw, R.P.; Thompson, J.F. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J. Clin. Oncol. 2012, 30, 2678–2683. [Google Scholar] [CrossRef]
- Mahmoud, S.M.A.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.S.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Walter, K.; Omura, N.; Hong, S.-M.; Griffith, M.; Vincent, A.; Borges, M.; Goggins, M. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin. Cancer Res. 2010, 16, 1781–1789. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M.R. Mechanisms of tumor escape from immune system: Role of mesenchymal stromal cells. Immunol. Lett. 2014, 159, 55–72. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; McCoy, G.A.; Folkvord, J.M.; McPherson, J.M. TGF-beta 1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: A fibronectin matrix-dependent event. J. Cell. Physiol. 1997, 170, 69–80. [Google Scholar] [CrossRef]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.G.; Janssen-van Rhijn, C.M.; Morreau, H.; Putter, H.; Tollenaar, R.A.E.M.; van de Velde, C.J.H.; Fleuren, G.J.; Kuppen, P.J.K. Immune system and prognosis in colorectal cancer: A detailed immunohistochemical analysis. Lab. Investig. 2004, 84, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Ohshio, Y.; Teramoto, K.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Daigo, Y.; Ogasawara, K. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. 2015, 106, 134–142. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, C.-T.; Ma, T.-T.; Li, Z.-Y.; Zhou, L.-N.; Mu, B.; Leng, F.; Shi, H.-S.; Li, Y.-O.; Wei, Y.-Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010, 101, 2325–2332. [Google Scholar] [CrossRef]
- MartIn-Fontecha, A.; Sebastiani, S.; Höpken, U.E.; Uguccioni, M.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Regulation of dendritic cell migration to the draining lymph node: Impact on T lymphocyte traffic and priming. J. Exp. Med. 2003, 198, 615–621. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Raker, V.K.; Domogalla, M.P.; Steinbrink, K. Tolerogenic Dendritic Cells for Regulatory T Cell Induction in Man. Front. Immunol. 2015, 6, 569. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.-I.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K.; et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 173, 3844–3854. [Google Scholar] [CrossRef]
- Cheng, J.-T.; Deng, Y.-N.; Yi, H.-M.; Wang, G.-Y.; Fu, B.-S.; Chen, W.-J.; Liu, W.; Tai, Y.; Peng, Y.-W.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schreiner, J.; Müller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of Lung Cancer Is Associated with Increased Dysfunction of T Cells Defined by Coexpression of Multiple Inhibitory Receptors. Cancer Immunol. Res. 2015, 3, 1344–1355. [Google Scholar] [CrossRef]
- Li, J.; Shayan, G.; Avery, L.; Jie, H.-B.; Gildener-Leapman, N.; Schmitt, N.; Lu, B.F.; Kane, L.P.; Ferris, R.L. Tumor-infiltrating Tim-3+ T cells proliferate avidly except when PD-1 is co-expressed: Evidence for intracellular cross talk. Oncoimmunology 2016, 5, e1200778. [Google Scholar] [CrossRef]
- Lu, X.; Yang, L.; Yao, D.; Wu, X.; Li, J.; Liu, X.; Deng, L.; Huang, C.; Wang, Y.; Li, D.; et al. Tumor antigen-specific CD8+ T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell. Immunol. 2017, 313, 43–51. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.J.; Bik, M.A.; Ospelt, C.; Naylor, A.J.; Wehmeyer, C.; Jones, S.W.; Buckley, C.D.; Gay, S.; Filer, A.; Lord, J.M. Endogenous Galectin-9 Suppresses Apoptosis in Human Rheumatoid Arthritis Synovial Fibroblasts. Sci. Rep. 2018, 8, 12887. [Google Scholar] [CrossRef] [PubMed]
- Curran, T.-A.; Jalili, R.B.; Farrokhi, A.; Ghahary, A. IDO expressing fibroblasts promote the expansion of antigen specific regulatory T cells. Immunobiology 2014, 219, 17–24. [Google Scholar] [CrossRef]
- Nazareth, M.R.; Broderick, L.; Simpson-Abelson, M.R.; Kelleher, R.J.; Yokota, S.J.; Bankert, R.B. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J. Immunol. 2007, 178, 5552–5562. [Google Scholar] [CrossRef]
- Ghebeh, H.; Tulbah, A.; Mohammed, S.; Elkum, N.; Bin Amer, S.M.; Al-Tweigeri, T.; Dermime, S. Expression of B7-H1 in breast cancer patients is strongly associated with high proliferative Ki-67-expressing tumor cells. Int. J. Cancer 2007, 121, 751–758. [Google Scholar] [CrossRef]
- Mazanet, M.M.; Hughes, C.C.W. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J. Immunol. 2002, 169, 3581–3588. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef]
- Beyranvand Nejad, E.; Welters, M.J.P.; Arens, R.; van der Burg, S.H. The importance of correctly timing cancer immunotherapy. Expert Opin. Biol. Ther. 2017, 17, 87–103. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.-C.R.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.B. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Chow, K.K.H.; Mata, M.; Shaffer, D.R.; Song, X.-T.; Wu, M.-F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef]
- Lo, A.; Wang, L.-C.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef]
- Pircher, M.; Schuberth, P.; Gulati, P.; Sulser, S.; Weder, W.; Curioni, A.; Renner, C.; Petrausch, U. FAP-specific re-directed T cells first in-man study in malignant pleural mesothelioma: Experience of the first patient treated. J. Immunother. Cancer 2015, 3, P120. [Google Scholar] [CrossRef]
- Hofheinz, R.-D.; al-Batran, S.-E.; Hartmann, F.; Hartung, G.; Jäger, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Frazier, K.; Thomas, R.; Scicchitano, M.; Mirabile, R.; Boyce, R.; Zimmerman, D.; Grygielko, E.; Nold, J.; DeGouville, A.-C.; Huet, S.; et al. Inhibition of ALK5 signaling induces physeal dysplasia in rats. Toxicol. Pathol. 2007, 35, 284–295. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.A.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- de Gramont, A.; Faivre, S.; Raymond, E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology 2017, 6, e1257453. [Google Scholar] [CrossRef]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernández, M.S.; Romero, F.L.; Sepulveda-Sánchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Gueorguieva, I.; Cleverly, A.L.; Stauber, A.; Sada Pillay, N.; Rodon, J.A.; Miles, C.P.; Yingling, J.M.; Lahn, M.M. Defining a therapeutic window for the novel TGF-β inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br. J. Clin. Pharmacol. 2014, 77, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Löffek, S. Transforming of the Tumor Microenvironment: Implications for TGF-Inhibition in the Context of Immune-Checkpoint Therapy. J. Oncol. 2018, 2018, 9732939. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Fujieda, K.; Miyashita, A.; Fukushima, S.; Ikeda, T.; Kubo, Y.; Senju, S.; Ihn, H.; Nishimura, Y.; Oshiumi, H. Combined blockade of IL6 and PD-1/PD-L1 signaling abrogates mutual regulation of their immunosuppressive effects in the tumor microenvironment. Cancer Res. 2018, 78, 5011–5022. [Google Scholar] [CrossRef]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing tumor-infiltrating T cells through inhibition of CXCL12 with NOX-A12 synergizes with PD-1 blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef]
- Di Sario, A.; Bendia, E.; Svegliati Baroni, G.; Ridolfi, F.; Casini, A.; Ceni, E.; Saccomanno, S.; Marzioni, M.; Trozzi, L.; Sterpetti, P.; et al. Effect of pirfenidone on rat hepatic stellate cell proliferation and collagen production. J. Hepatol. 2002, 37, 584–591. [Google Scholar] [CrossRef]
- Shin, J.-M.; Park, J.-H.; Park, I.-H.; Lee, H.-M. Pirfenidone inhibits transforming growth factor β1-induced extracellular matrix production in nasal polyp-derived fibroblasts. Am. J. Rhinol. Allergy 2015, 29, 408–413. [Google Scholar] [CrossRef]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef]
- Miura, Y.; Saito, T.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Inomata, M.; Nei, T.; Saito, Y.; Gemma, A.; Azuma, A. Reduced incidence of lung cancer in patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir. Investig. 2018, 56, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Tumor/Disease Type | Fibroblast Subsets | Human/Mouse | iCAF Subset Characteristics | Reference |
|---|---|---|---|---|
| PDAC | 2 | Mouse | Low αSMA expression, high IL-6, IL-11, LIF expression. | Öhlund D. et al. [36] |
| Ovarian cancer | 2 | Human | FAP+, high IL-6 and CXCL12 expression. | Hussain A. et al. [39] |
| Breast cancer | 4 | Human | CAF-S1 subset, high IL-1β, IL-6, CXCL12 and IL-17 expression. Induces differentiation of Tregs. | Costa A. et al. [40] |
| PDAC | 3 | Human and mouse | Low αSMA expression, high IL-6, IL-8, CXCL1, CXCL2, CXCL12 expression. | Elyada E. et al. [42] |
| Breast and lung cancer | N/a | Human | CD10+GPR77+, high IL-6 and IL-8 expression. | Su S. et al. [44] |
| Head and neck cancer | 4 | Human | 1: PDGF-Rα+, high CXCL12 expression. 2: High αSMA expression, high IL-6 expression. | Puram S. et al. [43] |
| Rheumatoid arthritis | 3 | Human | CD34−THY1+CDH11+: high IL-6 and TNFSF11 expression. Greatly expanded (compared to normal synovium) and associated with perivascular lymphocyte clusters in RA. | Mizoguchi F. et al. [47] |
| Rheumatoid arthritis | 2 | Mouse | THY1+ fibroblast, high expression of IL-6, LIF, IL-33 and IL-34. Linked to enhanced CD4+ T-cell infiltration. | Croft A. et al. [48] |
| Ulcerative colitis | 5 | Human | CAF-S4 subset, high IL-6, TNFS14, IL-33 and lysyl oxidases expression. Greatly expanded (compared to normal colon) in UC. | Kinchen J. et al. [49] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harryvan, T.J.; Verdegaal, E.M.E.; Hardwick, J.C.H.; Hawinkels, L.J.A.C.; van der Burg, S.H. Targeting of the Cancer-Associated Fibroblast—T-Cell Axis in Solid Malignancies. J. Clin. Med. 2019, 8, 1989. https://doi.org/10.3390/jcm8111989
Harryvan TJ, Verdegaal EME, Hardwick JCH, Hawinkels LJAC, van der Burg SH. Targeting of the Cancer-Associated Fibroblast—T-Cell Axis in Solid Malignancies. Journal of Clinical Medicine. 2019; 8(11):1989. https://doi.org/10.3390/jcm8111989
Chicago/Turabian StyleHarryvan, Tom J., Els M. E. Verdegaal, James C. H. Hardwick, Lukas J. A. C. Hawinkels, and Sjoerd H. van der Burg. 2019. "Targeting of the Cancer-Associated Fibroblast—T-Cell Axis in Solid Malignancies" Journal of Clinical Medicine 8, no. 11: 1989. https://doi.org/10.3390/jcm8111989
APA StyleHarryvan, T. J., Verdegaal, E. M. E., Hardwick, J. C. H., Hawinkels, L. J. A. C., & van der Burg, S. H. (2019). Targeting of the Cancer-Associated Fibroblast—T-Cell Axis in Solid Malignancies. Journal of Clinical Medicine, 8(11), 1989. https://doi.org/10.3390/jcm8111989