Oncolytic Virotherapy with Myxoma Virus


Abstract
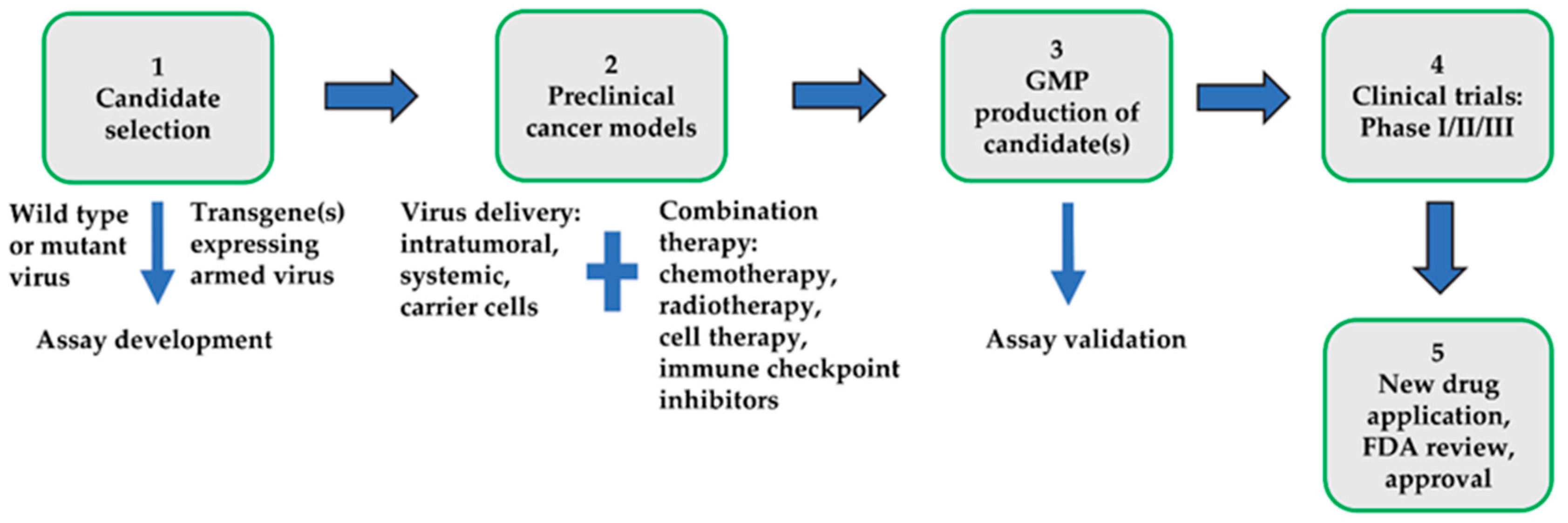
:1. Introduction
2. MYXV Tropism for Cancer Cells
3. Role of Intracellular Signaling in Cancer Cell Tropism
4. MYXV Oncolytic Killing of Cancer Cells
5. Oncolytic Virotherapy for Solid Tumors
5.1. Small Cell Lung Cancer
5.2. Ovarian Cancer
5.3. Glioblastoma
5.4. Gallbladder Cancer
5.5. Melanoma
6. Oncolytic Virotherapy for Hematologic Malignancies
7. Enhancing Oncolytic Activity of MYXV by Genetic Manipulation
8. Conclusions and Future Directions
Author Contributions
Funding
Disclosure
Conflicts of Interest
References
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quintanilla, J.; Seah, I.; Chua, M.; Shah, K. Oncolytic viruses: Overcoming translational challenges. J. Clin. Investig. 2019, 130, 1407–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.; McFadden, G. Viruses for tumor therapy. Cell Host Microbe 2014, 15, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pol, J.G.; Levesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.R.; Fahrner, J.E.; Fend, L.; Bell, J.C.; Mossman, K.L.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7, e1503032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, N.T.; Bell, J.C. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [Green Version]
- Eissa, I.R.; Bustos-Villalobos, I.; Ichinose, T.; Matsumura, S.; Naoe, Y.; Miyajima, N.; Morimoto, D.; Mukoyama, N.; Zhiwen, W.; Tanaka, M.; et al. The Current Status and Future Prospects of Oncolytic Viruses in Clinical Trials against Melanoma, Glioma, Pancreatic, and Breast Cancers. Cancers 2018, 10, 356. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Xu, J.; Liu, X.Y.; Chen, Z.N.; Bian, H. Fighting Cancer with Viruses: Oncolytic Virus Therapy in China. Human Gene Ther. 2018, 29, 151–159. [Google Scholar] [CrossRef]
- Raman, S.S.; Hecht, J.R.; Chan, E. Talimogene laherparepvec: Review of its mechanism of action and clinical efficacy and safety. Immunotherapy 2019, 11, 705–723. [Google Scholar] [CrossRef] [Green Version]
- Stanford, M.M.; Werden, S.J.; McFadden, G. Myxoma virus in the European rabbit: Interactions between the virus and its susceptible host. Vet. Res. 2007, 38, 299–318. [Google Scholar] [CrossRef] [Green Version]
- Kerr, P.J.; Liu, J.; Cattadori, I.; Ghedin, E.; Read, A.F.; Holmes, E.C. Myxoma virus and the Leporipoxviruses: An evolutionary paradigm. Viruses 2015, 7, 1020–1061. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.; Hota-Mitchell, S.; Chen, L.; Barrett, J.; Cao, J.X.; Macaulay, C.; Willer, D.; Evans, D.; McFadden, G. The complete DNA sequence of myxoma virus. Virology 1999, 264, 298–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.W.; Cao, J.X.; Hota-Mitchell, S.; McFadden, G. Immunomodulatory proteins of myxoma virus. Semin. Immunol. 2001, 13, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Seet, B.T.; Johnston, J.B.; Brunetti, C.R.; Barrett, J.W.; Everett, H.; Cameron, C.; Sypula, J.; Nazarian, S.H.; Lucas, A.; McFadden, G. Poxviruses and immune evasion. Annu. Rev. Immunol. 2003, 21, 377–423. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef]
- Bertagnoli, S.; Marchandeau, S. Myxomatosis. Rev. Sci. Tech. (Int. Off. Epizoot.) 2015, 34, 539–547, 549–556. [Google Scholar] [CrossRef]
- Kerr, P.J. Myxomatosis in Australia and Europe: A model for emerging infectious diseases. Antivir. Res. 2012, 93, 387–415. [Google Scholar] [CrossRef]
- Fenner, F.; Day, M.F.; Woodroofe, G.M. Epidemiological consequences of the mechanical transmission of myxomatosis by mosquitoes. J. Hyg. 1956, 54, 284–303. [Google Scholar] [CrossRef] [Green Version]
- Fenner, F.; Woodroofe, G.M. The pathogenesis of infectious myxomatosis; the mechanism of infection and the immunological response in the European rabbit (Oryctolagus cuniculus). Br. J. Exp. Pathol. 1953, 34, 400–411. [Google Scholar]
- Fenner, F. Evolutionary aspects of Myxomatosis in Australia. Mem. Inst. Oswaldo Cruz 1956, 54, 271–278. [Google Scholar] [CrossRef]
- Chan, W.M.; Rahman, M.M.; McFadden, G. Oncolytic myxoma virus: The path to clinic. Vaccine 2013, 31, 4252–4258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellish, P.; Shabashvili, D.; Rahman, M.M.; Nawab, A.; Guijarro, M.V.; Zhang, M.; Cao, C.; Moussatche, N.; Boyle, T.; Antonia, S.; et al. Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. J. Clin. Investig. 2019, 129, 2279–2292. [Google Scholar] [CrossRef] [PubMed]
- Nounamo, B.; Liem, J.; Cannon, M.; Liu, J. Myxoma Virus Optimizes Cisplatin for the Treatment of Ovarian Cancer In Vitro and in a Syngeneic Murine Dissemination Model. Mol. Ther. Oncolytics 2017, 6, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Pisklakova, A.; McKenzie, B.; Zemp, F.; Lun, X.; Kenchappa, R.S.; Etame, A.B.; Rahman, M.M.; Reilly, K.; Pilon-Thomas, S.; McFadden, G.; et al. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro-Oncol. 2016, 18, 1088–1098. [Google Scholar] [CrossRef]
- Weng, M.; Gong, W.; Ma, M.; Chu, B.; Qin, Y.; Zhang, M.; Lun, X.; McFadden, G.; Forsyth, P.; Yang, Y.; et al. Targeting gallbladder cancer: Oncolytic virotherapy with myxoma virus is enhanced by rapamycin in vitro and further improved by hyaluronan in vivo. Mol. Cancer 2014, 13, 82. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.L.; Doty, R.; Tosic, V.; Liu, J.; Kranz, D.M.; McFadden, G.; Macneill, A.L.; Roy, E.J. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol. Immunother. 2011, 60, 1461–1472. [Google Scholar] [CrossRef] [Green Version]
- Doty, R.A.; Liu, J.; McFadden, G.; Roy, E.J.; MacNeill, A.L. Histological evaluation of intratumoral myxoma virus treatment in an immunocompetent mouse model of melanoma. Oncolytic Virotherapy 2013, 2, 1–17. [Google Scholar]
- Tosic, V.; Thomas, D.L.; Kranz, D.M.; Liu, J.; McFadden, G.; Shisler, J.L.; MacNeill, A.L.; Roy, E.J. Myxoma virus expressing a fusion protein of interleukin-15 (IL15) and IL15 receptor alpha has enhanced antitumor activity. PLoS ONE 2014, 9, e109801. [Google Scholar] [CrossRef] [Green Version]
- Lilly, C.L.; Villa, N.Y.; Lemos de Matos, A.; Ali, H.M.; Dhillon, J.S.; Hofland, T.; Rahman, M.M.; Chan, W.; Bogen, B.; Cogle, C.; et al. Ex Vivo Oncolytic Virotherapy with Myxoma Virus Arms Multiple Allogeneic Bone Marrow Transplant Leukocytes to Enhance Graft versus Tumor. Mol. Ther. Oncolytics 2017, 4, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Bartee, E.; Bartee, M.Y.; Bogen, B.; Yu, X.Z. Systemic therapy with oncolytic myxoma virus cures established residual multiple myeloma in mice. Mol. Ther. Oncolytics 2016, 3, 16032. [Google Scholar] [CrossRef] [Green Version]
- Phelps, M.P.; Yang, H.; Patel, S.; Rahman, M.M.; McFadden, G.; Chen, E. Oncolytic Virus-Mediated RAS Targeting in Rhabdomyosarcoma. Mol. Ther. Oncolytics 2018, 11, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, F.I.; Bleck, C.K.; Mercer, J. Poxvirus host cell entry. Curr. Opin. Virol. 2012, 2, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Madlambayan, G.J.; Rahman, M.M.; Smallwood, S.E.; Meacham, A.M.; Hosaka, K.; Scott, E.W.; Cogle, C.R.; McFadden, G. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia 2009, 23, 2313–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, D.; Amet, T.; Hu, N.; Lan, J.; Hu, S.; Yu, Q. Primary human leukocyte subsets differentially express vaccinia virus receptors enriched in lipid rafts. J. Virol. 2013, 87, 9301–9312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, B. Poxvirus cell entry: How many proteins does it take? Viruses 2012, 4, 688–707. [Google Scholar] [CrossRef]
- Moss, B. Membrane fusion during poxvirus entry. Semin. Cell Dev. Biol. 2016, 60, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef]
- Mercer, J.; Knebel, S.; Schmidt, F.I.; Crouse, J.; Burkard, C.; Helenius, A. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. USA 2010, 107, 9346–9351. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, J.C.; Chung, C.S.; Chang, W. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 1999, 73, 8750–8761. [Google Scholar] [CrossRef] [Green Version]
- Chiu, W.L.; Lin, C.L.; Yang, M.H.; Tzou, D.L.; Chang, W. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 2007, 81, 2149–2157. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.S.; Hsiao, J.C.; Chang, Y.S.; Chang, W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 1998, 72, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Chung, C.S.; Heine, H.G.; Chang, W. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 2000, 74, 3353–3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izmailyan, R.; Hsao, J.C.; Chung, C.S.; Chen, C.H.; Hsu, P.W.; Liao, C.L.; Chang, W. Integrin beta1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 2012, 86, 6677–6687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, N.; Chung, C.S.; Chen, C.H.; Liao, C.L.; Chang, W. The lipid raft-associated protein CD98 is required for vaccinia virus endocytosis. J. Virol. 2012, 86, 4868–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.M.; Bartee, E.C.; Moreb, J.S.; Dower, K.; Connor, J.H.; McFadden, G. Myxoma and vaccinia viruses bind differentially to human leukocytes. J. Virol. 2013, 87, 4445–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, N.Y.; Bartee, E.; Mohamed, M.R.; Rahman, M.M.; Barrett, J.W.; McFadden, G. Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology 2010, 401, 266–279. [Google Scholar] [CrossRef] [Green Version]
- Villa, N.Y.; Wasserfall, C.H.; Meacham, A.M.; Wise, E.; Chan, W.; Wingard, J.R.; McFadden, G.; Cogle, C.R. Myxoma virus suppresses proliferation of activated T lymphocytes yet permits oncolytic virus transfer to cancer cells. Blood 2015, 125, 3778–3788. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Barrett, J.W.; Stanford, M.; Werden, S.J.; Johnston, J.B.; Gao, X.; Sun, M.; Cheng, J.Q.; McFadden, G. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. USA 2006, 103, 4640–4645. [Google Scholar] [CrossRef] [Green Version]
- Werden, S.J.; McFadden, G. Pharmacological manipulation of the akt signaling pathway regulates myxoma virus replication and tropism in human cancer cells. J. Virol. 2010, 84, 3287–3302. [Google Scholar] [CrossRef] [Green Version]
- Werden, S.J.; McFadden, G. The role of cell signaling in poxvirus tropism: The case of the M-T5 host range protein of myxoma virus. Biochim. Biophys. Acta 2008, 1784, 228–237. [Google Scholar] [CrossRef]
- Liu, J.; Wennier, S.; Zhang, L.; McFadden, G. M062 is a host range factor essential for myxoma virus pathogenesis and functions as an antagonist of host SAMD9 in human cells. J. Virol. 2011, 85, 3270–3282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Zhang, F.; Yan, B.; Si, C.; Honda, H.; Nagamachi, A.; Sun, L.Z.; Xiang, Y. A paralogous pair of mammalian host restriction factors form a critical host barrier against poxvirus infection. PLoS Pathog. 2018, 14, e1006884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Meng, X.; Townsend, M.B.; Satheshkumar, P.S.; Xiang, Y. Identification of CP77 as the Third Orthopoxvirus SAMD9 and SAMD9L Inhibitor with Unique Specificity for a Rodent SAMD9L. J. Virol. 2019, 93, e00225-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivan, G.; Ormanoglu, P.; Buehler, E.C.; Martin, S.E.; Moss, B. Identification of Restriction Factors by Human Genome-Wide RNA Interference Screening of Viral Host Range Mutants Exemplified by Discovery of SAMD9 and WDR6 as Inhibitors of the Vaccinia Virus K1L-C7L- Mutant. MBio 2015, 6, e01122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Krumm, B.; Li, Y.; Deng, J.; Xiang, Y. Structural basis for antagonizing a host restriction factor by C7 family of poxvirus host-range proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 14858–14863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Liu, J.; Chan, W.M.; Rothenburg, S.; McFadden, G. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 2013, 9, e1003465. [Google Scholar] [CrossRef]
- Rahman, M.M.; Bagdassarian, E.; Ali, M.A.M.; McFadden, G. Identification of host DEAD-box RNA helicases that regulate cellular tropism of oncolytic Myxoma virus in human cancer cells. Sci. Rep. 2017, 7, 15710. [Google Scholar] [CrossRef]
- Kim, M.; Williamson, C.T.; Prudhomme, J.; Bebb, D.G.; Riabowol, K.; Lee, P.W.; Lees-Miller, S.P.; Mori, Y.; Rahman, M.M.; McFadden, G.; et al. The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene 2010, 29, 3990–3996. [Google Scholar] [CrossRef] [Green Version]
- Bartee, E.; McFadden, G. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-beta. Cytokine 2009, 47, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Bartee, E.; Mohamed, M.R.; Lopez, M.C.; Baker, H.V.; McFadden, G. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J. Virol. 2009, 83, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Veyer, D.L.; Carrara, G.; Maluquer de Motes, C.; Smith, G.L. Vaccinia virus evasion of regulated cell death. Immunol. Lett. 2017, 186, 68–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davola, M.E.; Mossman, K.L. Oncolytic viruses: How "lytic" must they be for therapeutic efficacy? Oncoimmunology 2019, 8, e1581528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Myxoma Virus Induces Ligand Independent Extrinsic Apoptosis in Human Myeloma Cells. Clin. Lymphoma Myeloma Leuk. 2016, 16, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Bartee, E.; Chan, W.M.; Moreb, J.S.; Cogle, C.R.; McFadden, G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol. Blood Marrow Transplant. 2012, 18, 1540–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, H.; Barry, M.; Lee, S.F.; Sun, X.; Graham, K.; Stone, J.; Bleackley, R.C.; McFadden, G. M11L: A novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med. 2000, 191, 1487–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvansakul, M.; van Delft, M.F.; Lee, E.F.; Gulbis, J.M.; Fairlie, W.D.; Huang, D.C.; Colman, P.M. A structural viral mimic of prosurvival Bcl-2: A pivotal role for sequestering proapoptotic Bax and Bak. Mol. Cell 2007, 25, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Barrett, J.W.; Nazarian, S.H.; Everett, H.; Gao, X.; Bleackley, C.; Colwill, K.; Moran, M.F.; McFadden, G. Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J. Virol. 2004, 78, 7097–7111. [Google Scholar] [CrossRef] [Green Version]
- Douglas, A.E.; Corbett, K.D.; Berger, J.M.; McFadden, G.; Handel, T.M. Structure of M11L: A myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci. A Publ. Protein Soc. 2007, 16, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Johnston, J.B.; Barrett, J.W.; Nazarian, S.H.; Goodwin, M.; Ricciuto, D.; Wang, G.; McFadden, G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 2005, 23, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Macen, J.L.; Upton, C.; Nation, N.; McFadden, G. SERP1, a serine proteinase inhibitor encoded by myxoma virus, is a secreted glycoprotein that interferes with inflammation. Virology 1993, 195, 348–363. [Google Scholar] [CrossRef]
- Turner, P.C.; Sancho, M.C.; Thoennes, S.R.; Caputo, A.; Bleackley, R.C.; Moyer, R.W. Myxoma virus Serp2 is a weak inhibitor of granzyme B and interleukin-1beta-converting enzyme in vitro and unlike CrmA cannot block apoptosis in cowpox virus-infected cells. J. Virol. 1999, 73, 6394–6404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbasic, A.S.; Hynes, S.; Somrak, A.; Contakos, S.; Rahman, M.M.; Liu, J.; MacNeill, A.L. Oncolysis of canine tumor cells by myxoma virus lacking the serp2 gene. Am. J. Vet. Res. 2012, 73, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Denny, S.K.; Greenside, P.G.; Chaikovsky, A.C.; Brady, J.J.; Ouadah, Y.; Granja, J.M.; Jahchan, N.S.; Lim, J.S.; Kwok, S.; et al. Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov. 2018, 8, 1316–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, A.; Durocher-Allen, L.D.; Ellis, P.M.; Ung, Y.C.; Goffin, J.R.; Ramchandar, K.; Darling, G. Initial management of small-cell lung cancer (limited- and extensive-stage) and the role of thoracic radiotherapy and first-line chemotherapy: A systematic review. Curr. Oncol. 2019, 26, e372–e384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.A.; Liu, S.V. Immune Checkpoint Inhibitors in Small Cell Lung Cancer: A Partially Realized Potential. Adv. Ther. 2019, 36, 1826–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, A.S.; Patel, M.R. Viroimmunotherapy of Thoracic Cancers. Biomedicines 2017, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, B.E.; Park, K.S.; Yiu, G.; Conklin, J.F.; Lin, C.; Burkhart, D.L.; Karnezis, A.N.; Sweet-Cordero, E.A.; Sage, J. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010, 70, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- de Queiroz, N.; Xia, T.; Konno, H.; Barber, G.N. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol. Cancer Res. 2019, 17, 974–986. [Google Scholar] [CrossRef]
- Hoare, J.; Campbell, N.; Carapuca, E. Oncolytic virus immunotherapies in ovarian cancer: Moving beyond adenoviruses. Porto Biomed. J. 2018, 3, e7. [Google Scholar] [CrossRef]
- Correa, R.J.; Komar, M.; Tong, J.G.; Sivapragasam, M.; Rahman, M.M.; McFadden, G.; Dimattia, G.E.; Shepherd, T.G. Myxoma virus-mediated oncolysis of ascites-derived human ovarian cancer cells and spheroids is impacted by differential AKT activity. Gynecol. Oncol. 2012, 125, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.G.; Valdes, Y.R.; Barrett, J.W.; Bell, J.C.; Stojdl, D.; McFadden, G.; McCart, J.A.; DiMattia, G.E.; Shepherd, T.G. Evidence for differential viral oncolytic efficacy in an in vitro model of epithelial ovarian cancer metastasis. Mol. Ther. Oncolytics 2015, 2, 15013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, R.; Yu, Y.; Liu, J.; Luo, T.; Fan, F. Glioblastoma Treatment Modalities besides Surgery. J. Cancer 2019, 10, 4793–4806. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Essand, M. Virus-Based Immunotherapy of Glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, X.; Yang, W.; Alain, T.; Shi, Z.Q.; Muzik, H.; Barrett, J.W.; McFadden, G.; Bell, J.; Hamilton, M.G.; Senger, D.L.; et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005, 65, 9982–9990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, X.; Alain, T.; Zemp, F.J.; Zhou, H.; Rahman, M.M.; Hamilton, M.G.; McFadden, G.; Bell, J.; Senger, D.L.; Forsyth, P.A. Myxoma virus virotherapy for glioma in immunocompetent animal models: Optimizing administration routes and synergy with rapamycin. Cancer Res. 2010, 70, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Zemp, F.J.; Lun, X.; McKenzie, B.A.; Zhou, H.; Maxwell, L.; Sun, B.; Kelly, J.J.; Stechishin, O.; Luchman, A.; Weiss, S.; et al. Treating brain tumor-initiating cells using a combination of myxoma virus and rapamycin. Neuro-Oncology 2013, 15, 904–920. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, B.A.; Zemp, F.J.; Pisklakova, A.; Narendran, A.; McFadden, G.; Lun, X.; Kenchappa, R.S.; Kurz, E.U.; Forsyth, P.A. In vitro screen of a small molecule inhibitor drug library identifies multiple compounds that synergize with oncolytic myxoma virus against human brain tumor-initiating cells. Neuro-Oncology 2015, 17, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Zemp, F.J.; McKenzie, B.A.; Lun, X.; Maxwell, L.; Reilly, K.M.; McFadden, G.; Yong, V.W.; Forsyth, P.A. Resistance to oncolytic myxoma virus therapy in nf1(-/-)/trp53(-/-) syngeneic mouse glioma models is independent of anti-viral type-I interferon. PLoS ONE 2013, 8, e65801. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Thandra, K.C.; Barsouk, A. Epidemiology of gallbladder cancer. Clin. Exp. Hepatol. 2019, 5, 93–102. [Google Scholar] [CrossRef]
- Weng, M.; Zhang, M.; Qin, Y.; Gong, W.; Tang, Z.; Quan, Z.; Wu, K. Targeting gallbladder carcinoma: Bone marrow-derived stem cells as therapeutic delivery vehicles of myxoma virus. Chin. Med. J. 2014, 127, 2350–2356. [Google Scholar]
- Antohe, M.; Nedelcu, R.I.; Nichita, L.; Popp, C.G.; Cioplea, M.; Brinzea, A.; Hodorogea, A.; Calinescu, A.; Balaban, M.; Ion, D.A.; et al. Tumor infiltrating lymphocytes: The regulator of melanoma evolution. Oncol. Lett. 2019, 17, 4155–4161. [Google Scholar] [CrossRef] [PubMed]
- Bayan, C.Y.; Lopez, A.T.; Gartrell, R.D.; Komatsubara, K.M.; Bogardus, M.; Rao, N.; Chen, C.; Hart, T.D.; Enzler, T.; Rizk, E.M.; et al. The Role of Oncolytic Viruses in the Treatment of Melanoma. Curr. Oncol. Rep. 2018, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.M.; Shaban, M.; Barrett, J.W.; Werden, S.J.; Gilbert, P.A.; Bondy-Denomy, J.; Mackenzie, L.; Graham, K.C.; Chambers, A.F.; McFadden, G. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Bais, S.; Bartee, E.; Rahman, M.M.; McFadden, G.; Cogle, C.R. Oncolytic virotherapy for hematological malignancies. Adv. Virol. 2012, 2012, 186512. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Madlambayan, G.J.; Cogle, C.R.; McFadden, G. Oncolytic viral purging of leukemic hematopoietic stem and progenitor cells with Myxoma virus. Cytokine Growth Factor Rev. 2010, 21, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Madlambayan, G.J.; Bartee, E.; Kim, M.; Rahman, M.M.; Meacham, A.; Scott, E.W.; McFadden, G.; Cogle, C.R. Acute myeloid leukemia targeting by myxoma virus in vivo depends on cell binding but not permissiveness to infection in vitro. Leuk. Res. 2012, 36, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.J.; Federspiel, M.J.; Peng, K.W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Valderrama, F.; Cordeiro, J.V.; Schleich, S.; Frischknecht, F.; Way, M. Vaccinia virus-induced cell motility requires F11L-mediated inhibition of RhoA signaling. Science 2006, 311, 377–381. [Google Scholar] [CrossRef]
- Arakawa, Y.; Cordeiro, J.V.; Way, M. F11L-mediated inhibition of RhoA-mDia signaling stimulates microtubule dynamics during vaccinia virus infection. Cell Host Microbe 2007, 1, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Irwin, C.R.; Evans, D.H. Modulation of the myxoma virus plaque phenotype by vaccinia virus protein F11. J. Virol. 2012, 86, 7167–7179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, C.R.; Favis, N.A.; Agopsowicz, K.C.; Hitt, M.M.; Evans, D.H. Myxoma virus oncolytic efficiency can be enhanced through chemical or genetic disruption of the actin cytoskeleton. PLoS ONE 2013, 8, e84134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Tumor-Localized Secretion of Soluble PD1 Enhances Oncolytic Virotherapy. Cancer Res. 2017, 77, 2952–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- De Graaf, J.F.; de Vor, L.; Fouchier, R.A.M.; van den Hoogen, B.G. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.W.; Alston, L.R.; Wang, F.; Stanford, M.M.; Gilbert, P.A.; Gao, X.; Jimenez, J.; Villeneuve, D.; Forsyth, P.; McFadden, G. Identification of host range mutants of myxoma virus with altered oncolytic potential in human glioma cells. J. Neurovirol. 2007, 13, 549–560. [Google Scholar] [CrossRef]
- Liu, J.; Wennier, S.; McFadden, G. The immunoregulatory properties of oncolytic myxoma virus and their implications in therapeutics. Microbes Infect. 2010, 12, 1144–1152. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Type of Cancers | Animal Model | Tumor Establishment | MYXV Constructs and Delivery | Combination Therapy | Outcome (Ref) |
|---|---|---|---|---|---|
| Small cell lung cancer (SCLC) | C57BL/6 (p53lox/loxp p130lox2722/lox2722 RbloxP/loxP) | Intratracheal injection of adenovirus expressing Cre-recombinase | vMyx-M135KO, intranasal instillation | Virus alone and with cisplatin | Prolonged survival of mice when combined with cisplatin [22] |
| NSG | Subcutaneous xenograft of human primary SCLC cells | Wild type (WT), intratumoral | none | Virus replication and extensive tumor necrosis [22] | |
| C57BL/6 | Subcutaneous xenograft of mouse SCLC cells | WT, intratumoral | none | Virus replication, extensive tumor necrosis and CD45+ immune cell infiltration [22] | |
| Ovarian cancer (OC) | C57BL/6 | Murine OC cells in intraperitoneal cavity | WT and vMyx-M062RKO, intraperitoneal cavity | Virus alone and with cisplatin | Prolonged survival of mice when combined with cisplatin [23] |
| Glioblastoma (GBM) | C57BL/6J | Intracranial injection of murine BTICs | WT and vMyx-M11KO, intratumoral | Virus alone and with temozolomide | Prolonged survival of mice when combined with temozolomide [24] |
| Gallbladder cancer (GBC) | CD-1 nude | Subcutaneous xenograft of human GBC cells | WT, intratumoral | Virus alone and with Rapamycin or hyaluronan | Combination treatment with hyaluronan reduced tumor burden and prolonged survival [25] |
| Melanoma | C57BL/6 RAG-/- | Intracranial injection of mouse B16.SIY melanoma cells | WT, intratumoral | Virus alone, with rapamycin and activated T cells | Prolonged survival of mice when combined all the treatments [26] |
| C57BL/6 | Subcutaneous injection of B16F10 cells | WT and vMyx-IL-15, intratumoral injection | none | Prolonged survival of mice treated with vMyx-IL15 [27] | |
| C57BL/6 and C57BL/6 RAG-/- | Subcutaneous injection of B16F10 cells | WT, vMyx-IL-15 and vMyx-IL-15Rα-IL-15 intratumoral injection | none | Prolonged survival of mice with increased infiltration of NK and CD8+ T cells to the tumor bed [28] | |
| Multiple myeloma (MM) | BALB/c | Intravenous injection of mouse MOPC315.BM cells | WT, delivery of virus with total bone marrow | none | Prolonged survival of mice [29] |
| BALB/c | Intravenous injection of mouse MOPC315 cells | WT, systemic delivery | none | Prolonged survival of mice [30] | |
| Embryonal rhabdomyosarcoma (ERMS) | NSG | Subcutaneous xenograft of human ERMS cells | WT and NRAS targeting CRISPR-Cas9 engineered MYXV, intratumoral injection | none | Reduced tumor volume and prolonged mice survival [31] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; McFadden, G. Oncolytic Virotherapy with Myxoma Virus. J. Clin. Med. 2020, 9, 171. https://doi.org/10.3390/jcm9010171
Rahman MM, McFadden G. Oncolytic Virotherapy with Myxoma Virus. Journal of Clinical Medicine. 2020; 9(1):171. https://doi.org/10.3390/jcm9010171
Chicago/Turabian StyleRahman, Masmudur M., and Grant McFadden. 2020. "Oncolytic Virotherapy with Myxoma Virus" Journal of Clinical Medicine 9, no. 1: 171. https://doi.org/10.3390/jcm9010171
APA StyleRahman, M. M., & McFadden, G. (2020). Oncolytic Virotherapy with Myxoma Virus. Journal of Clinical Medicine, 9(1), 171. https://doi.org/10.3390/jcm9010171