Peptide Antagonists for P-selectin Discriminate between Sulfatide-Dependent Platelet Aggregation and PSGL-1-Mediated Cell Adhesion

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mice
2.3. Preparation of Sulfatide Containing Liposomes
2.4. Liposome Interaction with Selectins
2.5. Surface Plasmon Resonance (Biacore) Analysis
2.6. Blood Collection and Platelet Isolation
2.7. Static Adhesion
2.8. Perfusion Studies
2.9. Immunofluorescence Studies
2.10. Analysis of Platelet Aggregation
2.11. Statistical Analysis
3. Results
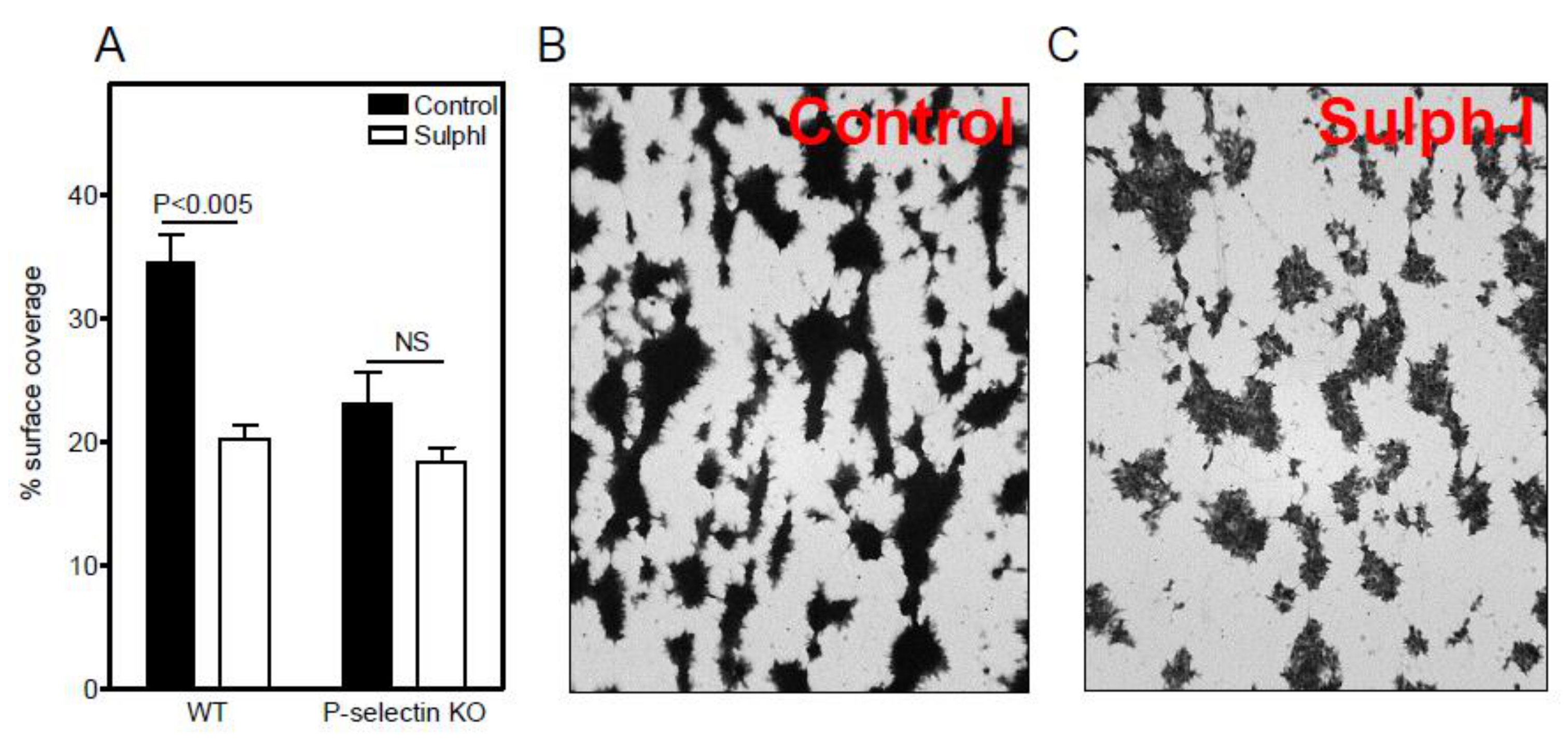
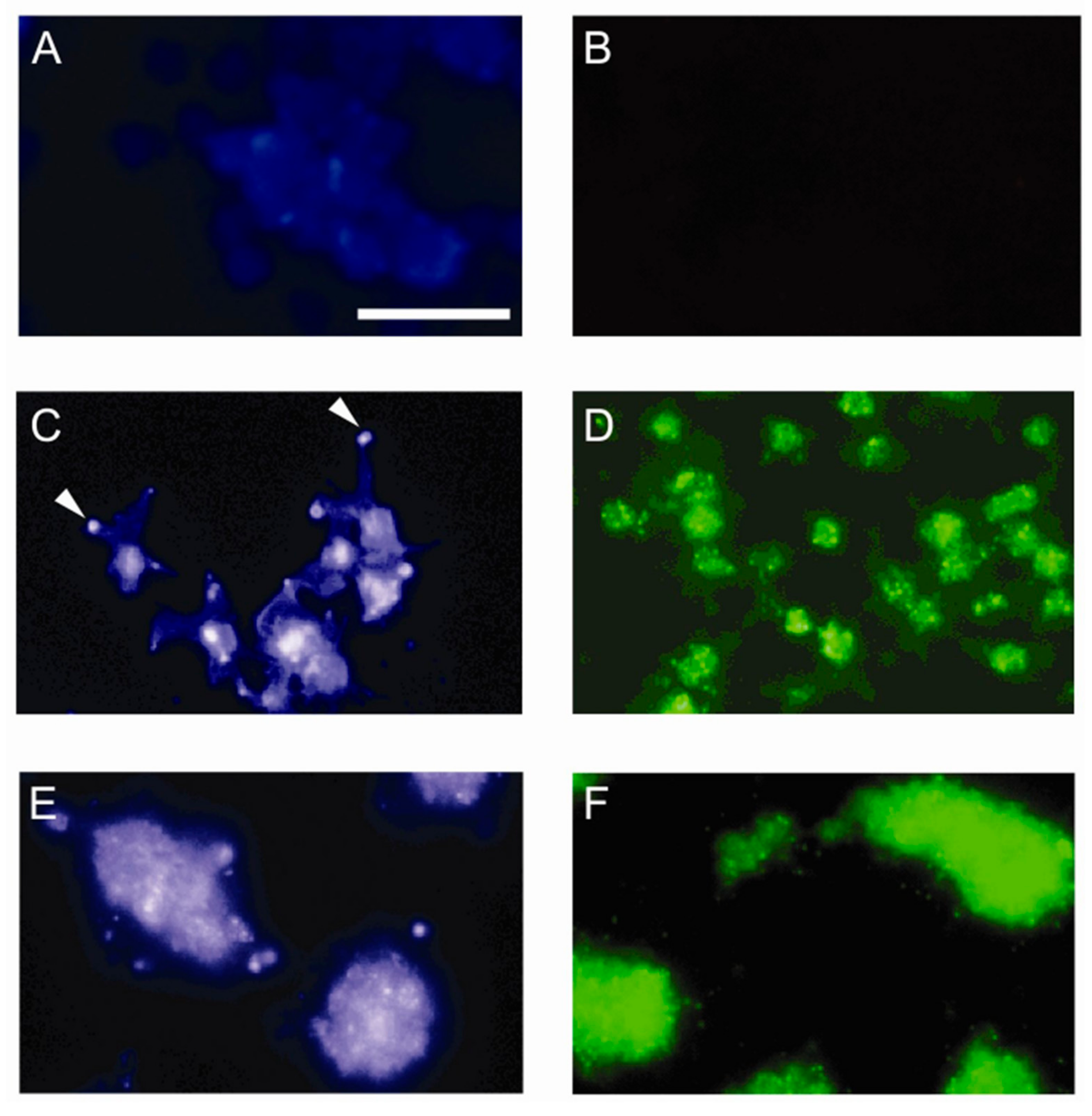
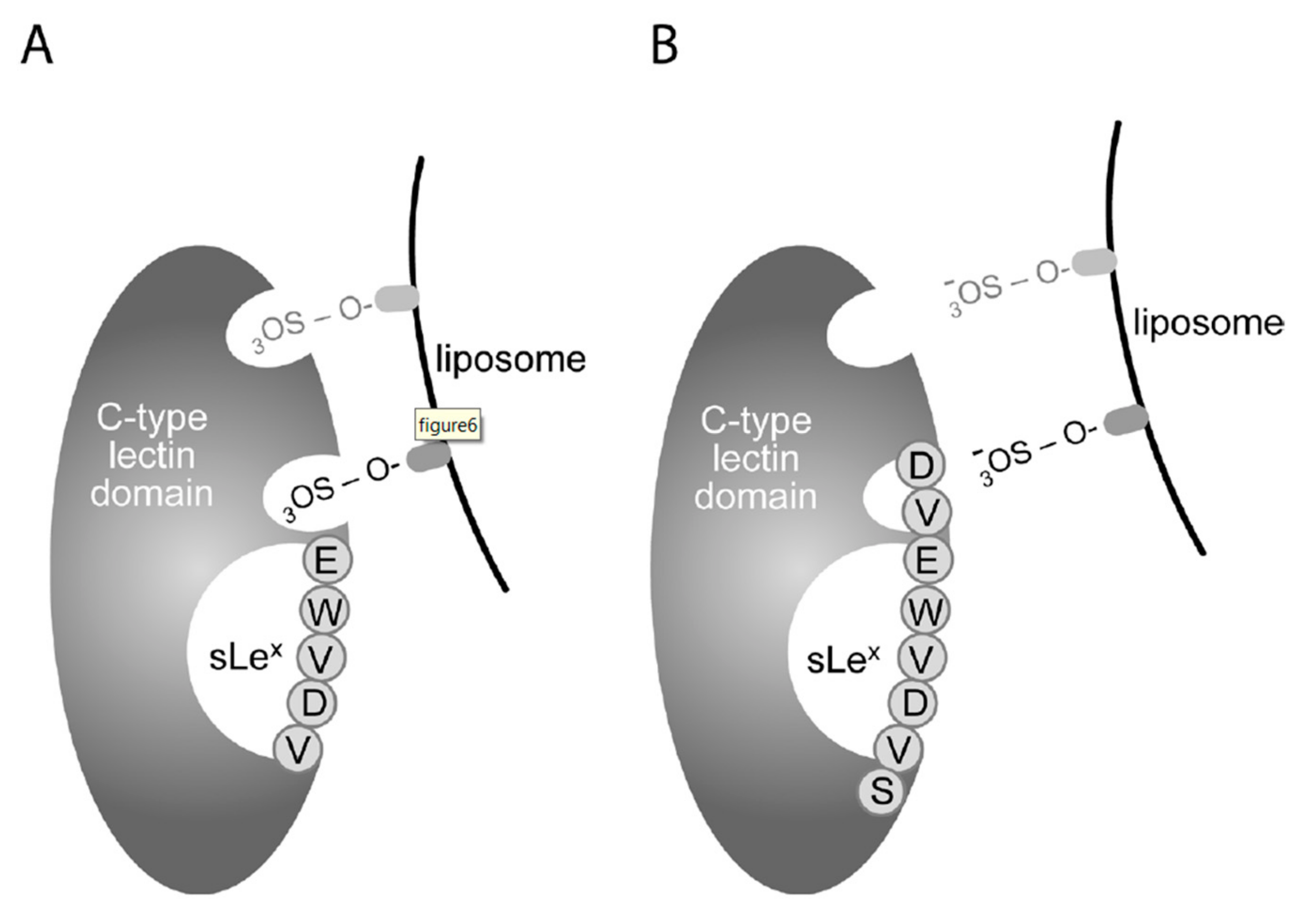
3.1. Sulfatide P-selectin Tethering
3.2. Sulfatide Surface Density is Important for Interaction with P-selectin
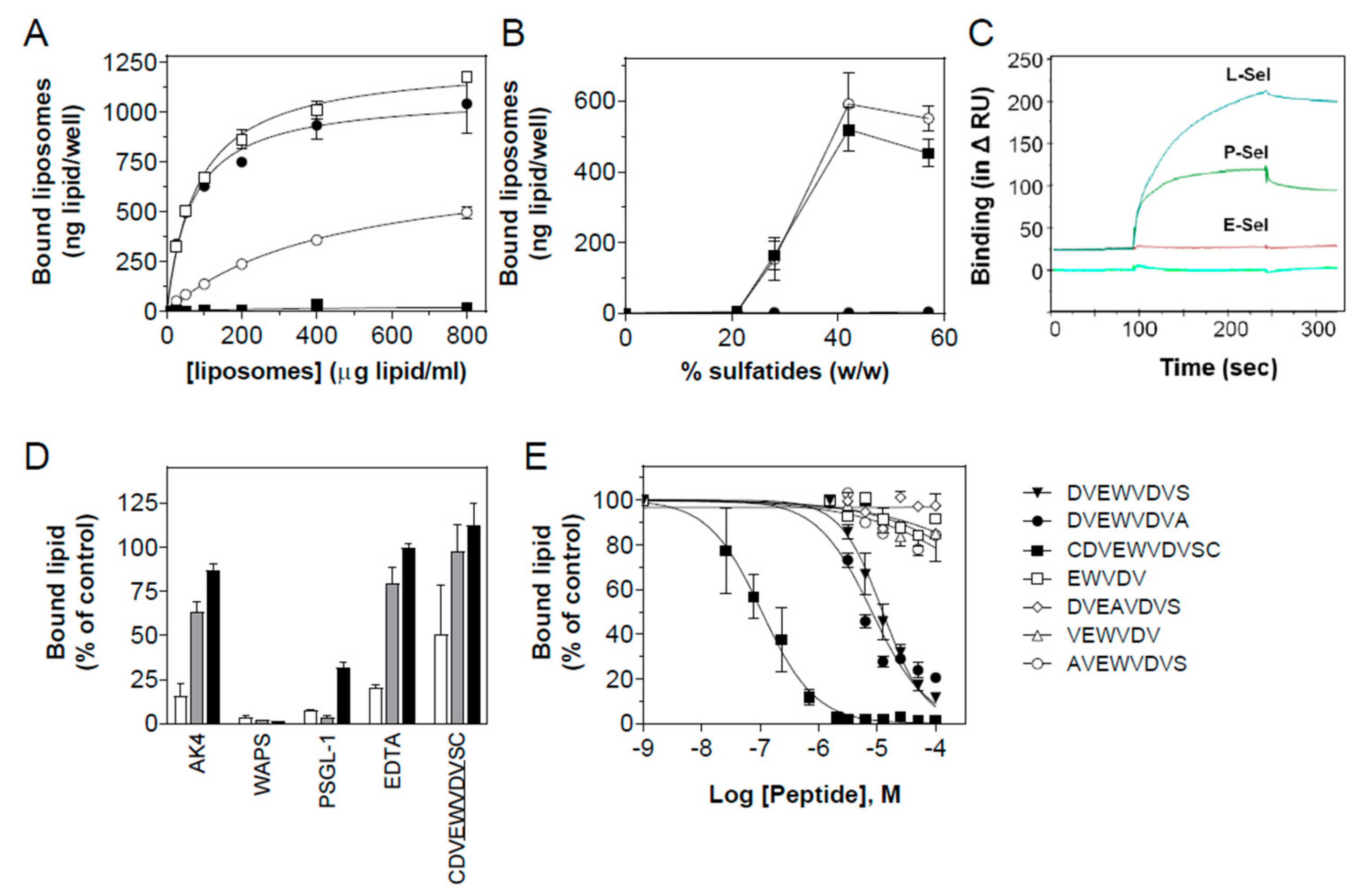
3.3. Characterization of the Interaction of Sulfatide Liposomes with P-selectin
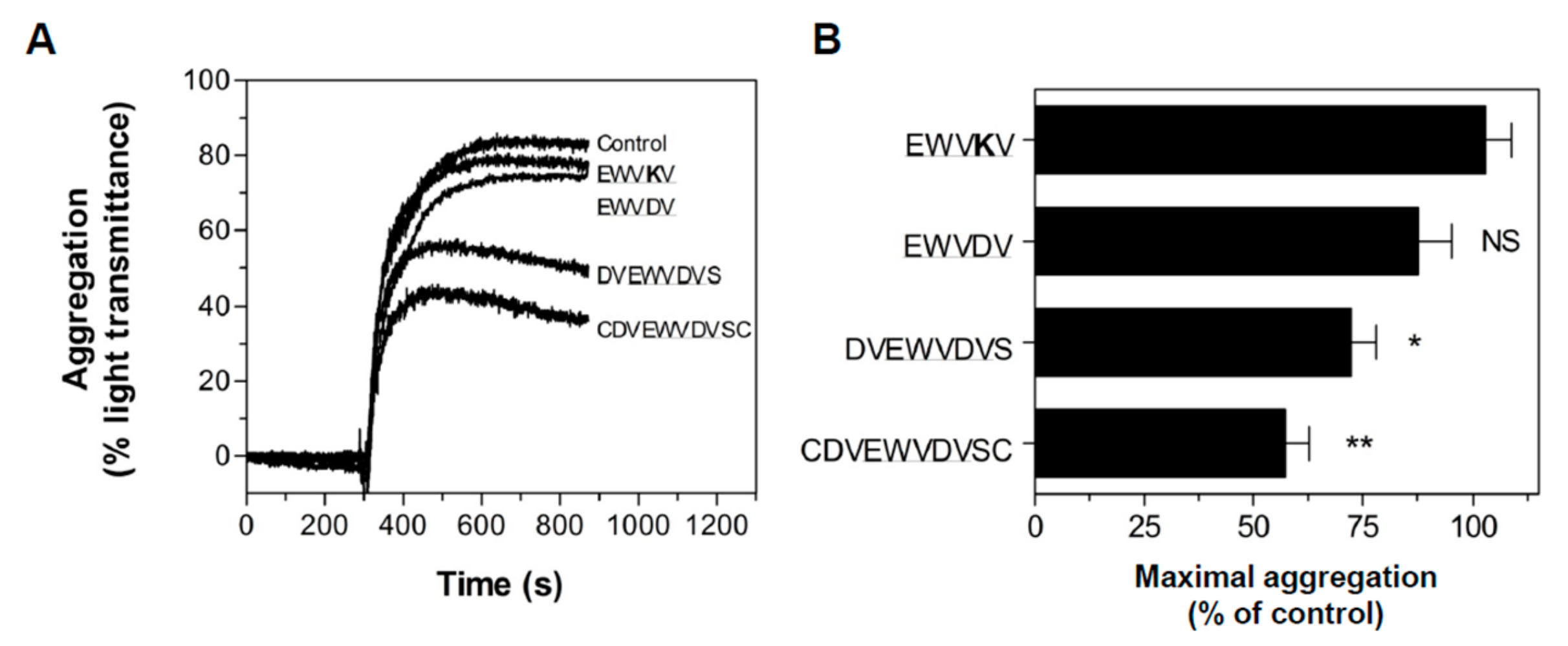
3.4. Peptide Antagonists Inhibit Platelet Aggregation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ley, K. The role of selectins in inflammation and disease. Trends Mol. Med. 2003, 9, 263–268. [Google Scholar] [CrossRef]
- Lutters, B.C.H.; Leeuwenburgh, M.A.; Appeldoorn, C.C.M.; Molenaar, T.J.; van Berkel, T.J.; Biessen, E.A. Blocking endothelial adhesion molecules: A potential therapeutic strategy to combat atherogenesis. Curr. Opin. Lipidol. 2004, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Berman, C.L.; Yeo, E.L.; Wencel Drake, J.D.; Furie, B.C.; Ginsberg, M.H.; Furie, B. A platelet alpha granule membrane protein that is associated with the plasma membrane after activation. Characterization and subcellular localization of platelet activation-dependent granule-external membrane protein. J. Clin. Investig. 1986, 78, 130–137. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Beckstead, J.H.; Moore, K.L.; Marshall-Carlson, L.; Bainton, D.F. GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J. Clin. Investig. 1989, 84, 92–99. [Google Scholar] [CrossRef]
- Burns, A.R.; Bowden, R.A.; Abe, Y.; Walker, D.C.; Simon, S.I.; Entman, M.L.; Smith, C.W. P-selectin mediates neutrophil adhesion to endothelial cell borders. J. Leukoc. Biol. 1999, 65, 299–306. [Google Scholar] [CrossRef]
- Kameda, H.; Morita, I.; Handa, M.; Kaburaki, J.; Yoshida, T.; Mimori, T.; Murota, S.; Ikeda, Y. Re-expression of functional P-selectin molecules on the endothelial cell surface by repeated stimulation with thrombin. Br. J. Haematol. 1997, 97, 348–355. [Google Scholar] [CrossRef]
- Larsen, E.; Celi, A.; Gilbert, G.E.; Furie, B.C.; Erban, J.K.; Bonfanti, R.; Wagner, D.D.; Furie, B. PADGEM protein: A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 1989, 59, 305–312. [Google Scholar] [CrossRef]
- Frenette, P.S.; Denis, C.V.; Weiss, L.; Jurk, K.; Subbarao, S.; Kehrel, B.; Hartwig, J.H.; Vestweber, D.; Wagner, D.D. P-selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J. Exp. Med. 2000, 191, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, V.R.; Sardar, M.Y.; Ying, Y.; Song, X.; Haller, C.; Dai, E.; Wang, X.; Hanjaya-Putra, D.; Sun, L.; Morikis, V.; et al. Glycopeptide analogues of PSGL-1 inhibit P-selectin in vitro and in vivo. Nat. Commun. 2015, 6, 6387. [Google Scholar] [CrossRef]
- Théorêt, J.F.; Yacoub, D.; Hachem, A.; Gillis, M.A.; Merhi, Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb. Res. 2011, 128, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Needham, L.K.; Schnaar, R.L. The HNK-1 reactive sulfoglucunoryl glycolipids are ligands for L-selectin and P-selectin but not E-selectin. Proc. Natl. Acad. Sci. USA 1993, 90, 1359–1363. [Google Scholar] [CrossRef]
- Aruffo, A.; Kolanus, W.; Walz, G.; Fredman, P.; Seed, B. CD62/P-selectin recognition of myeloid and tumor cell sulfatides. Cell 1991, 67, 35–44. [Google Scholar] [CrossRef]
- Kushi, Y.; Arita, M.; Ishizuka, I.; Kasama, T.; Fredman, P.; Handa, S. Sulfatide is expressed in both erythrocytes and platelets of bovine origin. Biochim. Biophys. Acta 1996, 13, 254–262. [Google Scholar] [CrossRef]
- Roberts, D.D.; Ginsburg, V. Sulfated glycolipids and cell adhesion. Arch. Biochem. Biophys. 1988, 267, 405–415. [Google Scholar] [CrossRef]
- Kurosawa, N.; Kadomatsu, K.; Ikematsu, S.; Sakuma, S.; Kimura, T.; Muramatsu, T. Midkine binds specificically to sulfatide. The role of sulfatide in cell attachment to midkine-coated surfaces. Eur. J. Biochem. 2000, 267, 344–351. [Google Scholar] [CrossRef]
- Ida, M.; Satoh, A.; Matsumoto, I.; Kojima-Aikawa, K. Human annexin V binds to sulfatide: Contribution to regulation of blood coagulation. J. Biochem. 2004, 135, 583–588. [Google Scholar] [CrossRef]
- Galustian, C.; Childs, R.A.; Stoll, M.; Ishida, H.; Kiso, M.; Feizi, T. Synergistic interactions of the two classes of ligand, sialyl-Lewis (a/x) fuco-oligosaccherides and short sulpho-motifs, with the P- and L-selectins: Implications for therapeutic inhibitor design. Immunology 2002, 105, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Revelle, B.M.; Scott, D.; Beck, P.J. Single amino acid residues in the E- and P-selectin epidermal growth factor domains can determine carbohydrate binding specificity. J. Biol. Chem. 1996, 271, 16160–16170. [Google Scholar] [CrossRef] [PubMed]
- Erbe, D.V.; Watson, S.R.; Presta, L.G.; Wolitzky, B.A.; Foxall, C.; Brandley, B.K.; Lasky, L.A. P- and E-selectin use common sites for carbohydrate ligand recognition and cell adhesion. J. Cell Biol. 1993, 120, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Merten, M.; Chow, T.; Hellums, J.D.; Thiagarajan, P. A new role for P-selectin in shear-induced platelet aggregation. Circulation 2000, 102, 2045–2050. [Google Scholar] [CrossRef]
- Merten, M.; Thiagarajan, P. P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation 2000, 102, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Merten, M.; Thiagarajan, P. Role for sulfatides in platelet aggregation. Circulation 2001, 104, 2955–2960. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Merten, M.; Beythien, C.; Gutensohn, K.; Kühnl, P.; Meinertz, T.; Thiagarajan, P. Sulfatides activate platelets through P-selectin and enhance platelet and platelet-leukocyte aggregation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 258–263. [Google Scholar] [CrossRef]
- Welsh, J.D.; Charonko, J.J.; Salmanzadeh, A.; Drahos, K.E.; Shafiee, H.; Stremler, M.A.; Davalos, R.V.; Capelluto, D.G.; Vlachos, P.P.; Finkielstein, C.V. Disabled-2 modulates homotypic and heterotypic platelet interactions by binding to sulfatides. Br. J. Haematol. 2011, 154, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, M.; Frenette, P.S.; Saffaripour, S.; Johnson, R.C.; Hynes, R.O.; Wagner, D.D. Defects in hemostasis in P-selectin-deficient mice. Blood 1996, 87, 1238–1242. [Google Scholar]
- Inoue, T.; Taguchi, I.; Abe, S.; Li, G.; Hu, R.; Nakajima, T.; Hara, A.; Aoyama, T.; Kannagi, R.; Kyogashima, M.; et al. Sulfatides are associated with neointimal thickening after vascular injury. Atherosclerosis 2010, 211, 291–296. [Google Scholar] [CrossRef]
- Suchanski, J.; Grzegrzolka, J.; Owczarek, T.; Pasikowski, P.; Piotrowska, A.; Kocbach, B.; Nowak, A.; Dziegiel, P.; Wojnar, A.; Ugorski, M. Sulfatide decreases the resistance to stress-induced apoptosis and increases P-selectin-mediated adhesion: A two-edged sword in breast cancer progression. Breast Cancer Res. 2018, 20, 133. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, T.J.M.; Appeldoorn, C.C.M.; de Haas, S.A.M.; Michon, I.N.; Bonnefoy, A.; Hoylaerts, M.F.; Pannekoek, H.; van Berkel, T.J.; Kuiper, J.; Biessen, E.A. Specific inhibition of P-selectin-mediated adhesion by phage display-derived peptide antagonists. Blood 2002, 100, 3570–3577. [Google Scholar] [CrossRef] [PubMed]
- Appeldoorn, C.C.M.; Molenaar, T.J.M.; Bonnefoy, A.; van Leeuwen, S.H.; Vandervoort, P.A.; Hoylaerts, M.F.; van Berkel, T.J.; Biessen, E.A. Rational optimization of a short human P-selectin-binding peptide leads to nanomolar affinity antagonists. J. Biol. Chem. 2003, 278, 10201–10207. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J.; Hollenbaugh, D.; King, G.; Harte, W., Jr.; Eustice, D.C.; Darveau, R.P.; Aruffo, A. CD62/P-selectin binding sites for myeloid cells and sulfatides are overlapping. Biochemistry 1994, 33, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Camphausen, R.T.; Sullivan, F.X.; Cumming, D.A. Core 2 ß-1,6-N-acetylglucosaminyltransferase enzyme activity is critical for P-selectin glycoprotein ligand-1 binding to P-selectin. Blood 1996, 88, 3872–3879. [Google Scholar] [PubMed]
- Heijnen, H.F.; van Lier, M.; Waaijenborg, S.; Ohno-Iwashita, Y.; Waheed, A.A.; Inomata, M.; Gorter, G.; Möbius, W.; Akkerman, J.W.; Slot, J.W. Concentration of rafts in platelet filopodia correlates with recruitment of c-Src and CD63 to these domains. J. Thromb. Haemostasis 2003, 1, 1161–1173. [Google Scholar] [CrossRef]
- Pernber, Z.; Molander-Melin, M.; Berthold, C.H.; Hansson, E.; Fredman, P. Expression of the myelin and oligodendrocyte progenitor marker sulfatide in neurons and astrocytes of adult rat brain. J. Neurosci. Res. 2002, 69, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.P.; Fournier, D.J.; Andrews, R.K.; Gorman, J.J.; Chesterman, C.N.; Berndt, M.C. Characterization of human platelet GMP-140 as a heparin-binding protein. Biochem. Biophys. Res. Commun. 1989, 164, 1373–1379. [Google Scholar] [CrossRef]
- Skinner, M.P.; Lucas, C.M.; Burns, G.F.; Chesterman, C.N.; Berndt, M.C. GMP-140 binding to neutrophils is inhibited by sulfated glycans. J. Biol. Chem. 1991, 266, 5371–5374. [Google Scholar] [PubMed]
- Noda, H.; Kurono, M.; Ohishi, N.; Yagi, K. Stabilization of egg yolk phosphatidylcholine liposomes by the insertion of sulfatide. Biochim. Biophys. Acta 1993, 21, 127–131. [Google Scholar] [CrossRef]
- Greenspan, P.; Gutman, R.L. Endocytosis of sulfatides by macrophages: Relationship to cellular uptake of phosphatidylserine. J. Leukoc. Biol. 1994, 55, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Ido, T. Liposome targeting to rat brain: Effect of osmotic opening of the blood-brain barrier. Brain Res 1993, 629, 171–175. [Google Scholar] [CrossRef]
- Guchhait, P.; Shrimpton, C.N.; Honke, K.; Rumbaut, R.E.; Lopez, J.A.; Thiagarajan, P. Effect of an anti-sulfatide single-chain antibody probe on platelet function. Thromb. Haemostasis 2008, 99, 552–557. [Google Scholar]
- Leppanen, A.; White, S.P.; Helin, J.; McEver, R.P.; Cummings, R.D. Binding of glycosulfopeptides to P-selectin requires stereospecific contributions of individual tyrosine sulfate and sugar residues. J. Biol. Chem. 2000, 275, 39569–39578. [Google Scholar] [CrossRef]
- Somers, W.S.; Tang, J.; Shaw, G.D.; Camphausen, R.T. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to sLe(X) and PSGL-1. Cell 2000, 103, 467–479. [Google Scholar] [CrossRef]
- Ramachandran, V.; Nollert, M.U.; Qiu, H.; Liu, W.-J.; Cummings, R.D.; Zhu, C.; McEver, R.P. Tyrosine replacement in P-selectin glycoprotein ligand-1 affects distinct kinetic and mechanical properties of bonds with P- and L-selectin. Proc. Natl. Acad. Sci. USA 1999, 96, 13771–13776. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ramachandran, V.; Kang, J.; Kishimoto, T.K.; Cummings, R.D.; McEver, R.P. Identification of N-terminal residues on P-selectin glycoprotein ligand-1 required for binding to P-selectin. J. Biol. Chem. 1998, 273, 7078–7087. [Google Scholar] [CrossRef] [PubMed]
- Pouyani, T.; Seed, B. PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell 1995, 83, 333–343. [Google Scholar] [CrossRef]
- Sako, D.; Comess, K.M.; Barone, K.M.; Barone, K.M.; Camphausen, R.T.; Cumming, D.A.; Shaw, G.D. A sulfated peptide segment at the amino terminus of PSGL-1 is critical for P-selectin binding. Cell 1995, 83, 323–331. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | PSGL-1 * EC50 (μM) | Sulfatide EC50 (μM) | Aggregation † |
|---|---|---|---|
| EWVKV | >1000 | >1000 | 0% (NS ‡) |
| EWVDV | 12 | >1000 | 12% (NS) |
| VEWVDV | 13 | >1000 | Nd § |
| AVEWVDVS | 12 | >1000 | Nd |
| DAEWVDVS | 19 | >1000 | Nd |
| DVAWVDVS | 41 | >1000 | Nd |
| DVE AVDVS | >1000 | >1000 | Nd |
| DVEWADVS | >1000 | >1000 | Nd |
| DVEWVAVS | >1000 | >1000 | Nd |
| DVEWVDAS | >1000 | >1000 | Nd |
| DVEWVDVA | 7 | 8 | Nd |
| DVEWVDVS | 11 | 12 | 28% (p<0.05) |
| CDVEWVDVSC | 9 | 0.2 | 43% (p<0.001) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korporaal, S.J.A.; Molenaar, T.J.M.; Lutters, B.C.H.; Meurs, I.; Drost-Verhoef, S.; Kuiper, J.; van Berkel, T.J.C.; Biessen, E.A.L. Peptide Antagonists for P-selectin Discriminate between Sulfatide-Dependent Platelet Aggregation and PSGL-1-Mediated Cell Adhesion. J. Clin. Med. 2019, 8, 1266. https://doi.org/10.3390/jcm8081266
Korporaal SJA, Molenaar TJM, Lutters BCH, Meurs I, Drost-Verhoef S, Kuiper J, van Berkel TJC, Biessen EAL. Peptide Antagonists for P-selectin Discriminate between Sulfatide-Dependent Platelet Aggregation and PSGL-1-Mediated Cell Adhesion. Journal of Clinical Medicine. 2019; 8(8):1266. https://doi.org/10.3390/jcm8081266
Chicago/Turabian StyleKorporaal, Suzanne J.A., Tom J.M. Molenaar, Bianca C.H. Lutters, Illiana Meurs, Sandra Drost-Verhoef, Johan Kuiper, Theo J.C. van Berkel, and Erik A.L. Biessen. 2019. "Peptide Antagonists for P-selectin Discriminate between Sulfatide-Dependent Platelet Aggregation and PSGL-1-Mediated Cell Adhesion" Journal of Clinical Medicine 8, no. 8: 1266. https://doi.org/10.3390/jcm8081266
APA StyleKorporaal, S. J. A., Molenaar, T. J. M., Lutters, B. C. H., Meurs, I., Drost-Verhoef, S., Kuiper, J., van Berkel, T. J. C., & Biessen, E. A. L. (2019). Peptide Antagonists for P-selectin Discriminate between Sulfatide-Dependent Platelet Aggregation and PSGL-1-Mediated Cell Adhesion. Journal of Clinical Medicine, 8(8), 1266. https://doi.org/10.3390/jcm8081266