Osteochondritis Dissecans (OCD)-Derived Chondrocytes Display Increased Senescence, Oxidative Stress, Chaperone-Mediated Autophagy and, in Co-Culture with Adipose-Derived Stem Cells (ASCs), Enhanced Expression of MMP-13

 ,
,
Abstract
1. Introduction
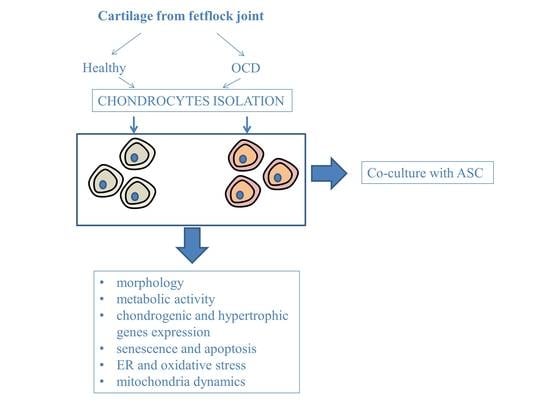
2. Experimental Section
2.1. Animals and Collection of Cartilage Samples
2.2. Adipose-Derived Stem Cells (ASC) and Chondrocytes’ Isolation and Culture
2.3. Evaluation of Viability and Proliferation Rate
2.4. Evaluation of Cellular Morphology
2.5. Measurement of Superoxide Dismutase (SOD) Activity and Nitric Oxide (NO) Concentration
2.6. Chondrocytes-ASC Co-Culture
2.7. Western Blotting
2.8. Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.9. Statistics
3. Results
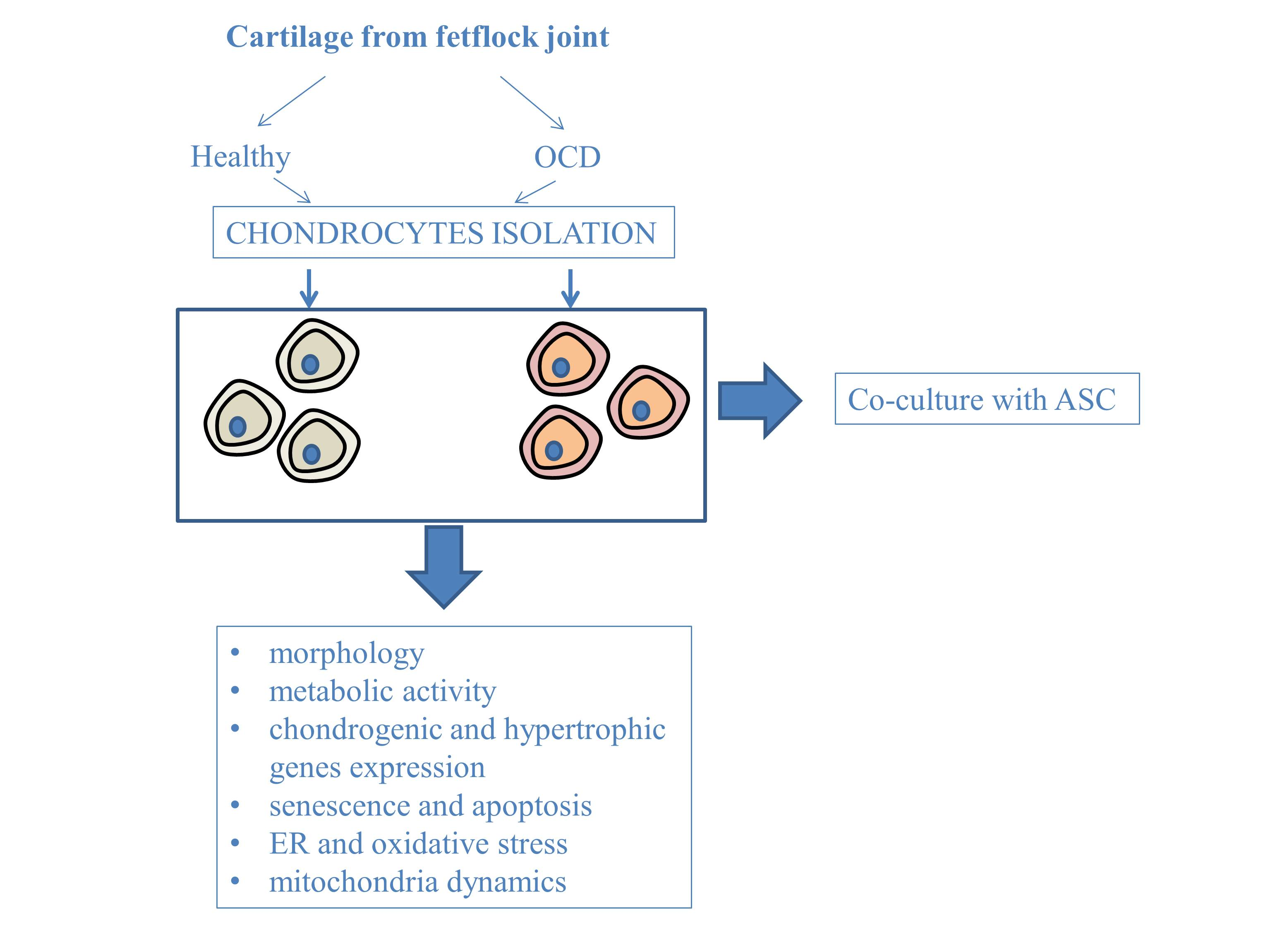
3.1. Metabolic Activity and Morphology
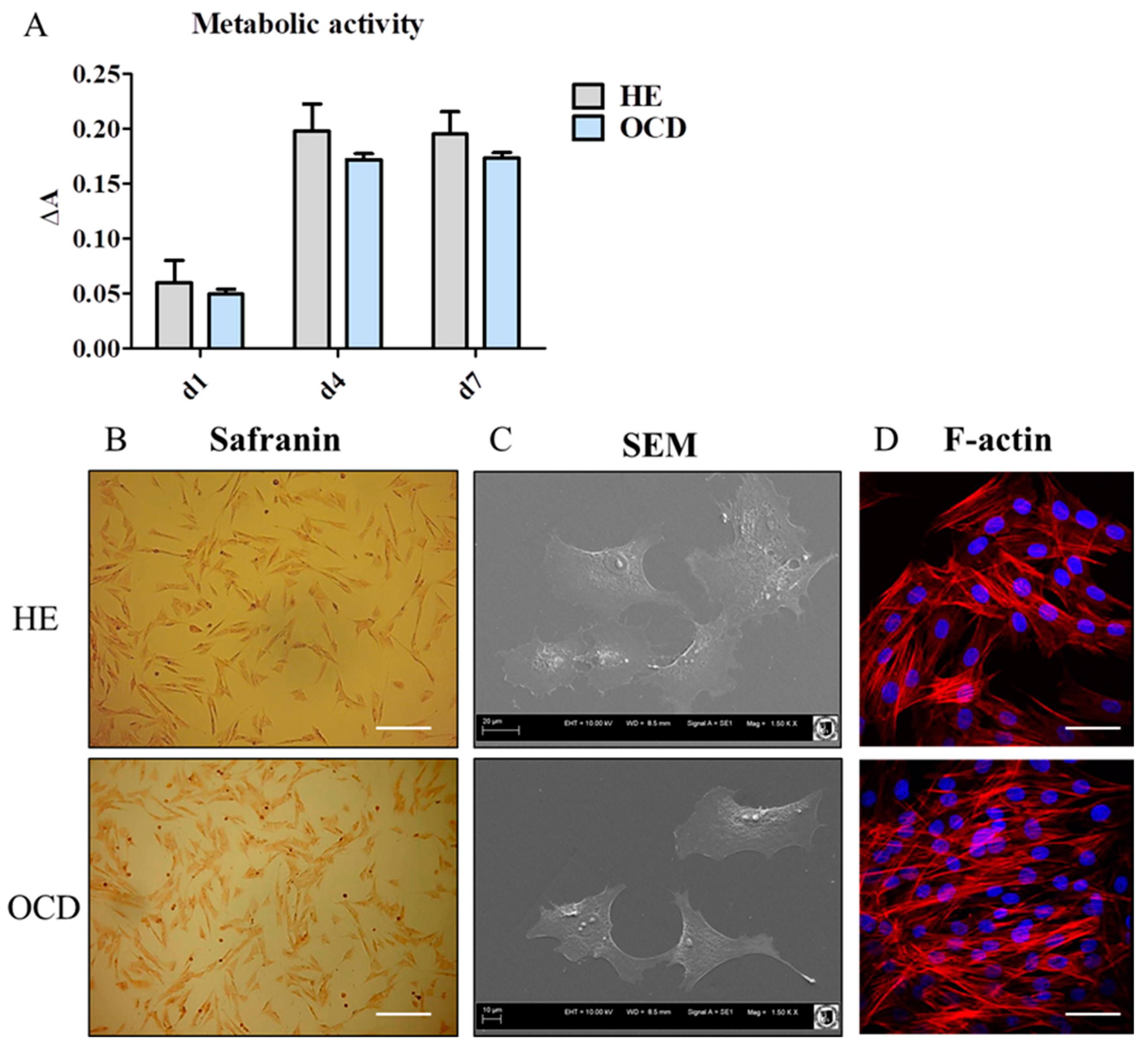
3.2. Expression of Chondrogenic and Hypertrophic Genes
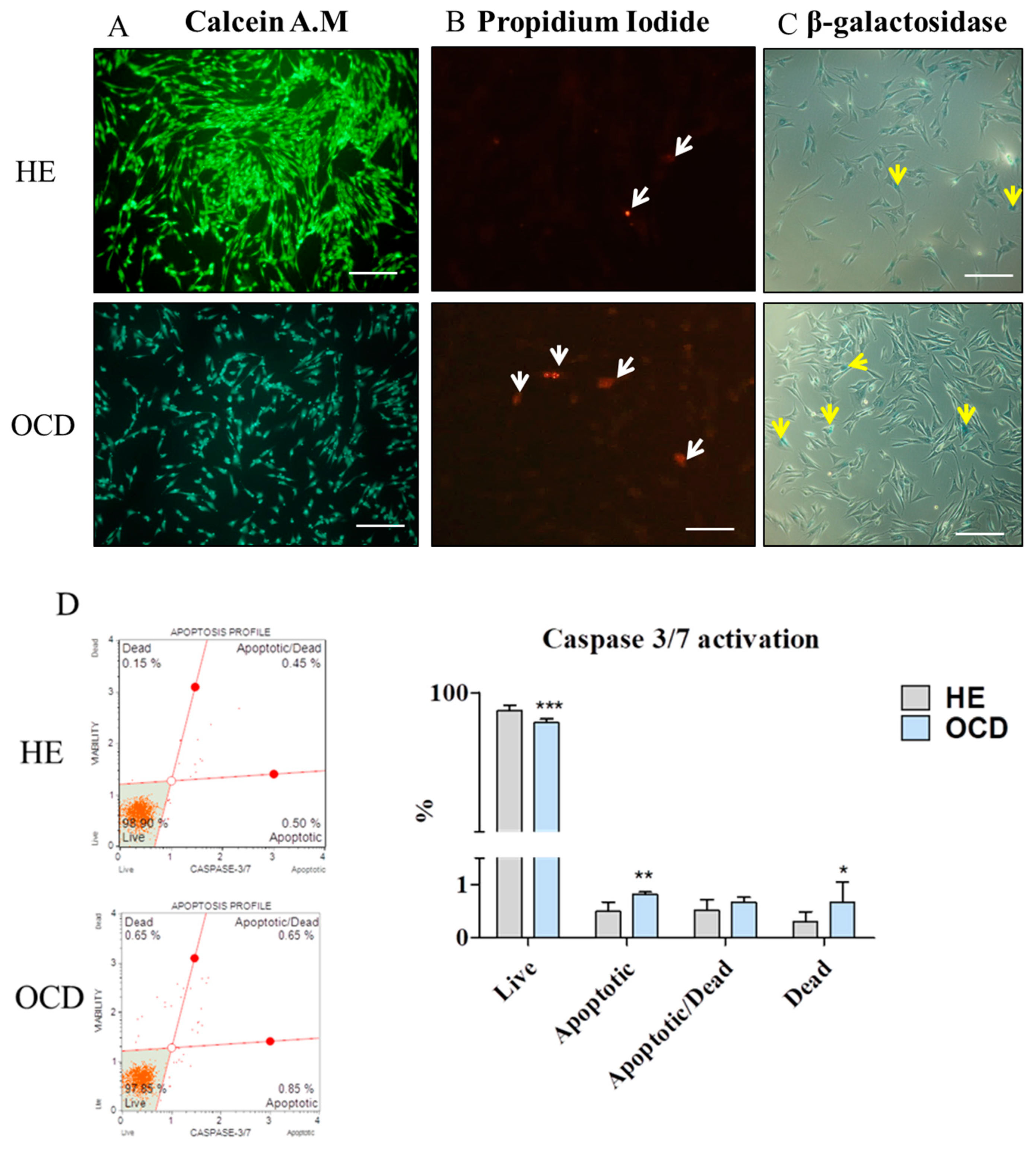
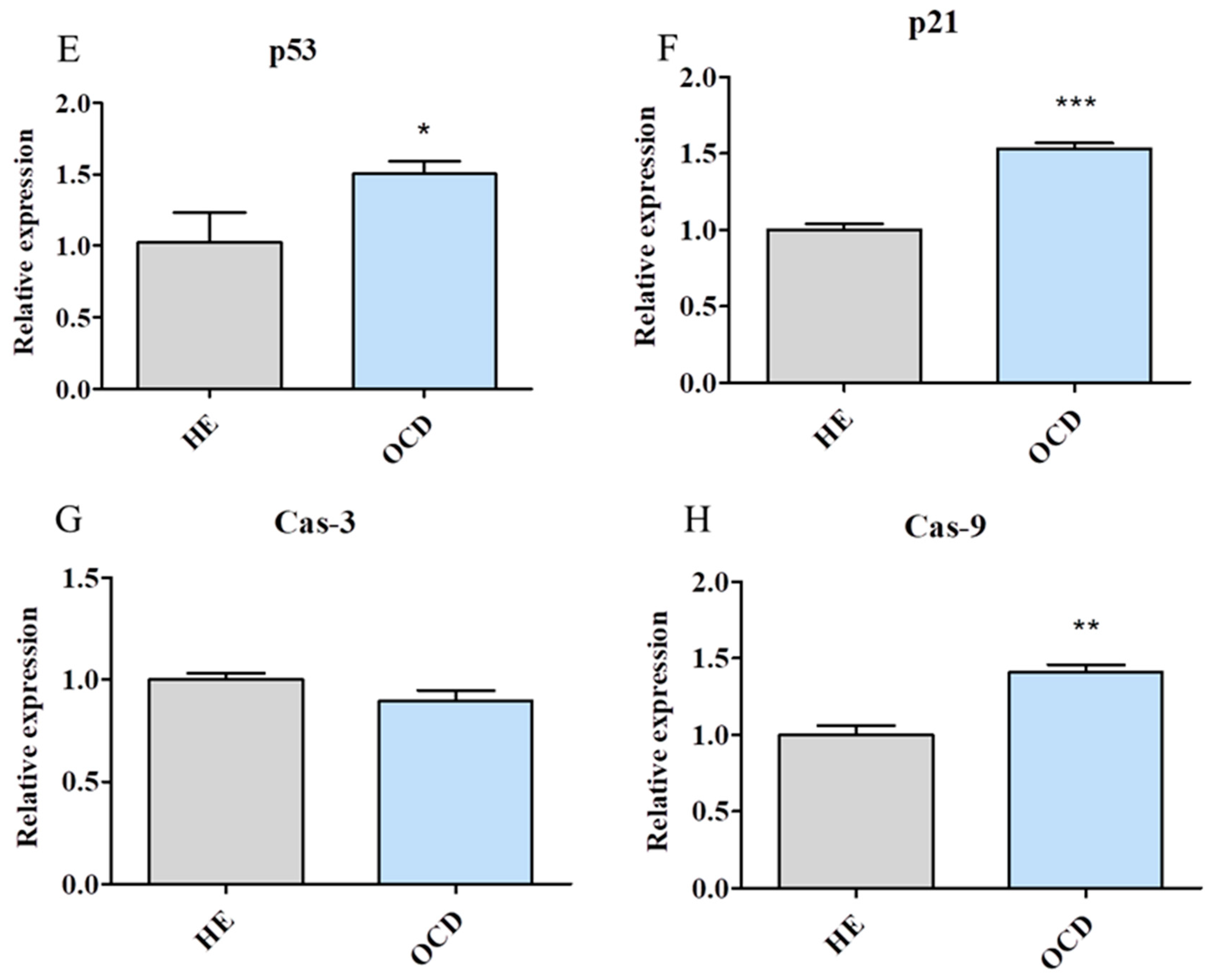
3.3. Apoptosis and Senescence
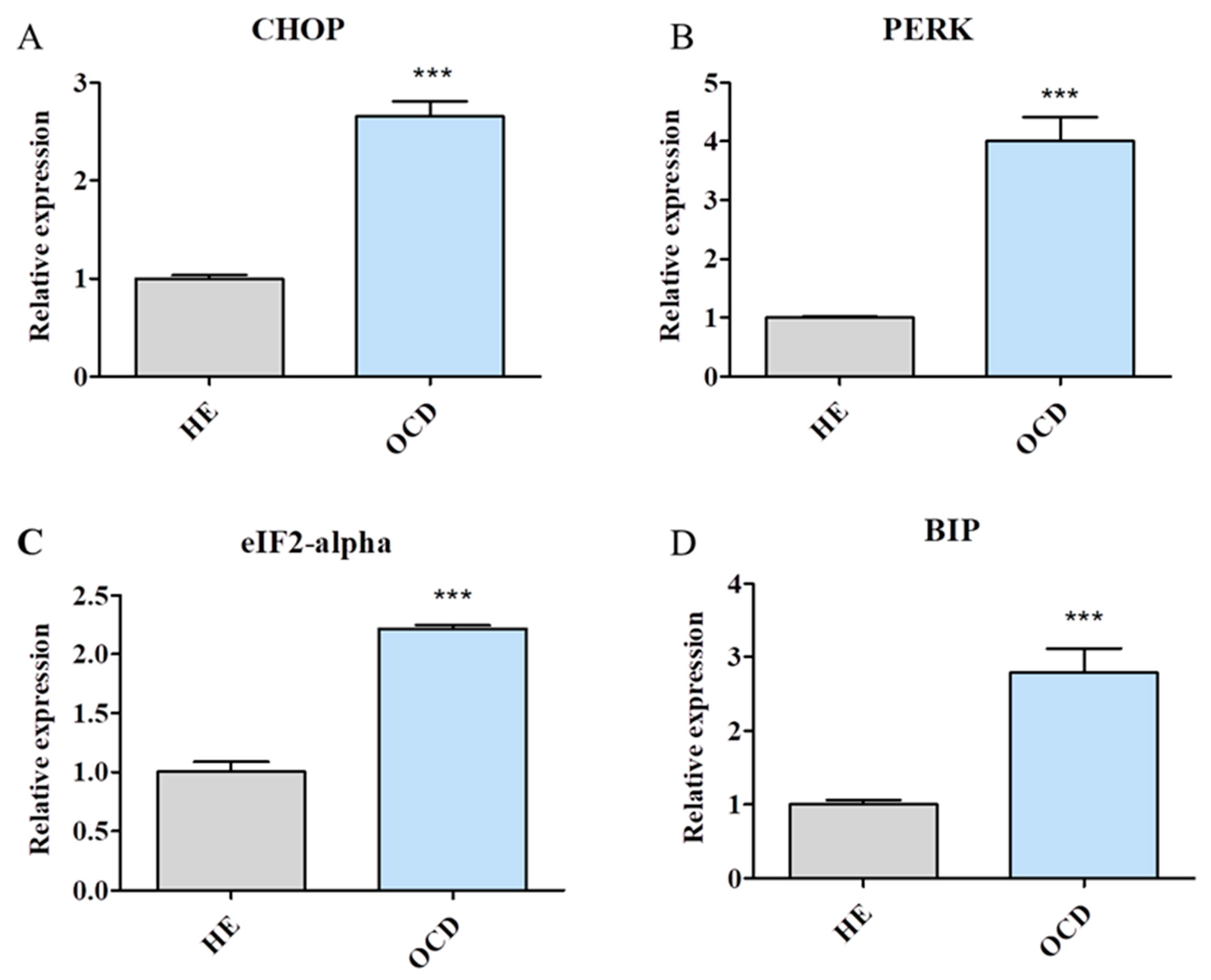
3.4. Expression of Endoplasmic Reticulum (ER) Stress-Related Genes
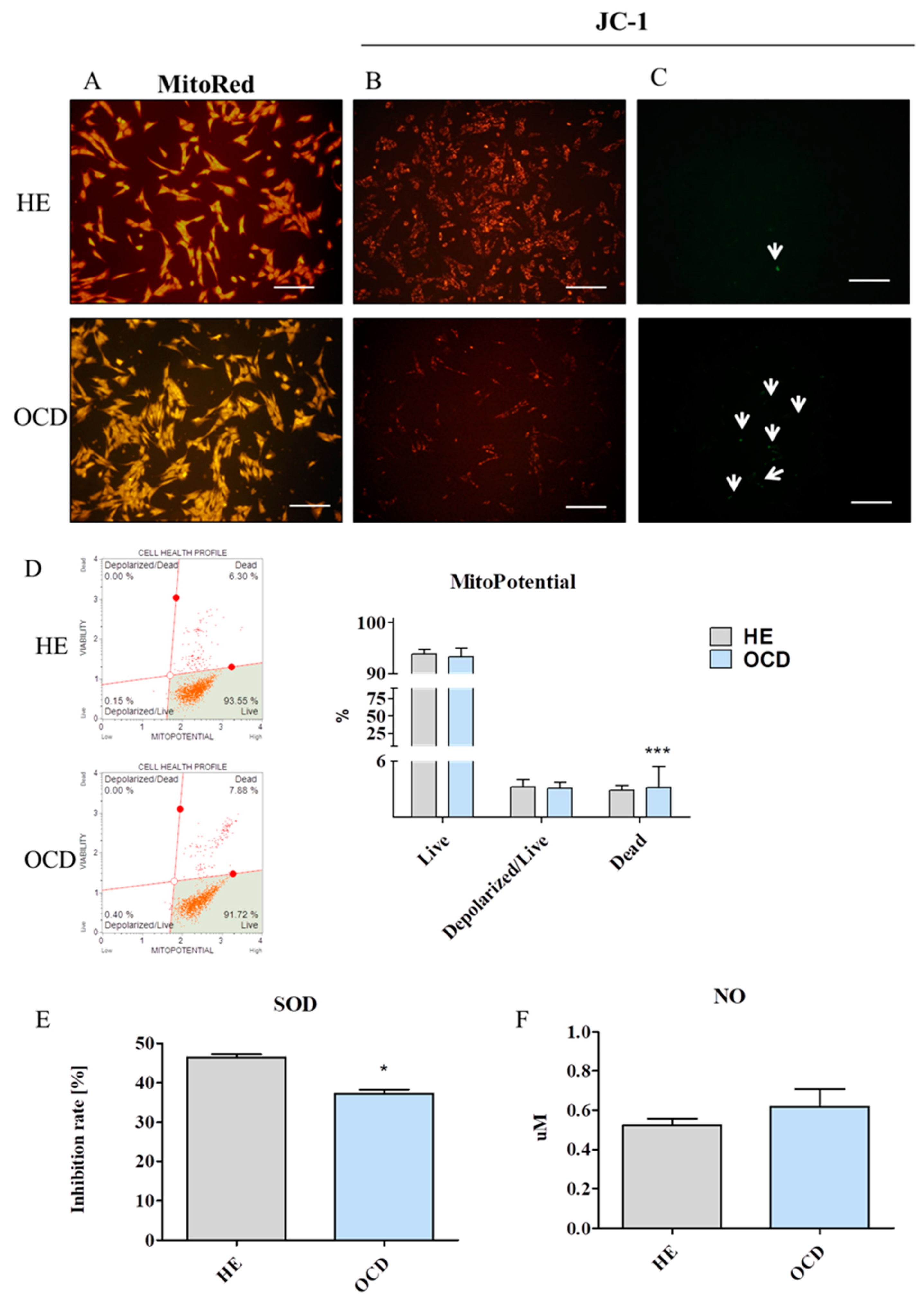
3.5. Assessment of Mitochondria Condition
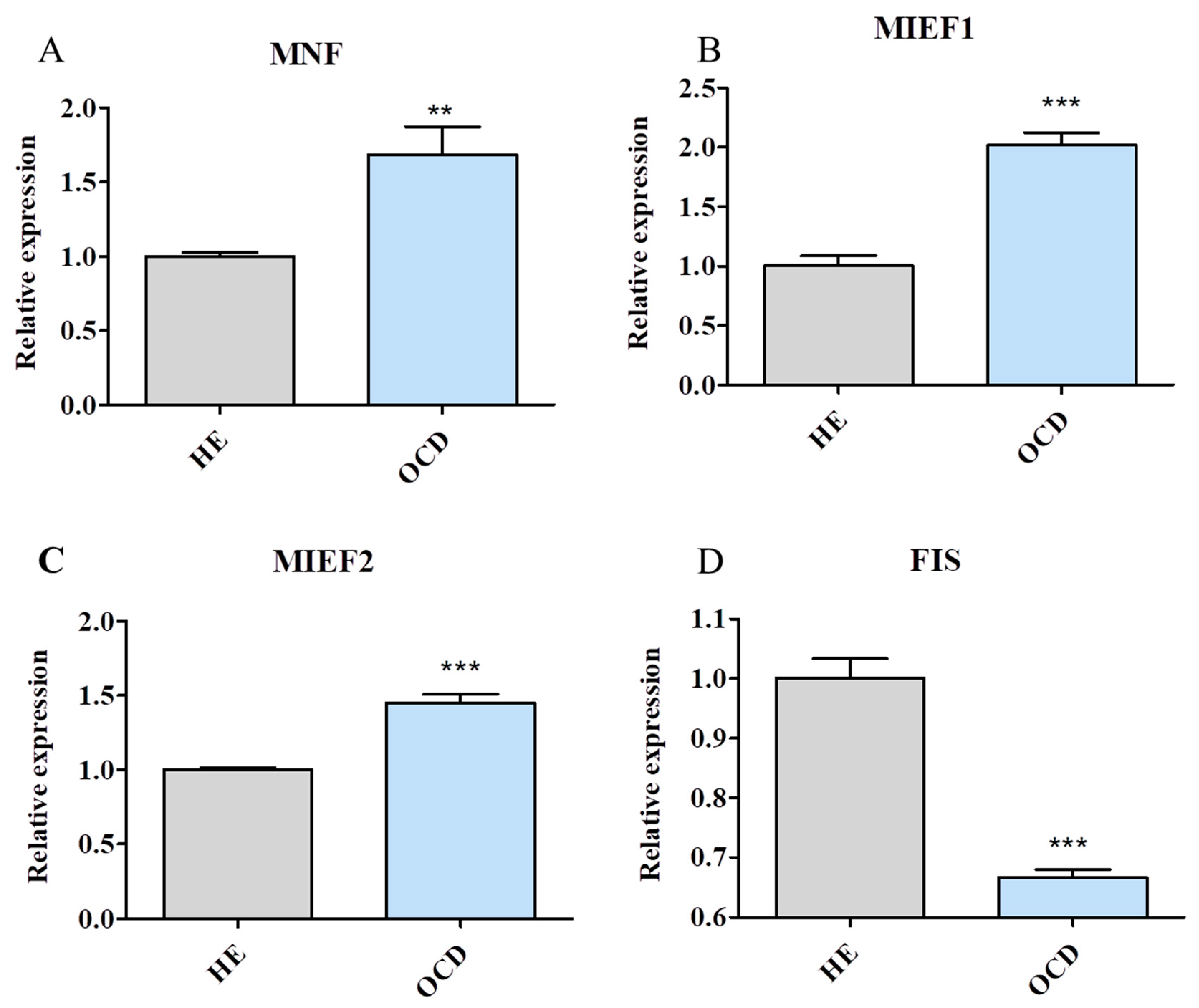
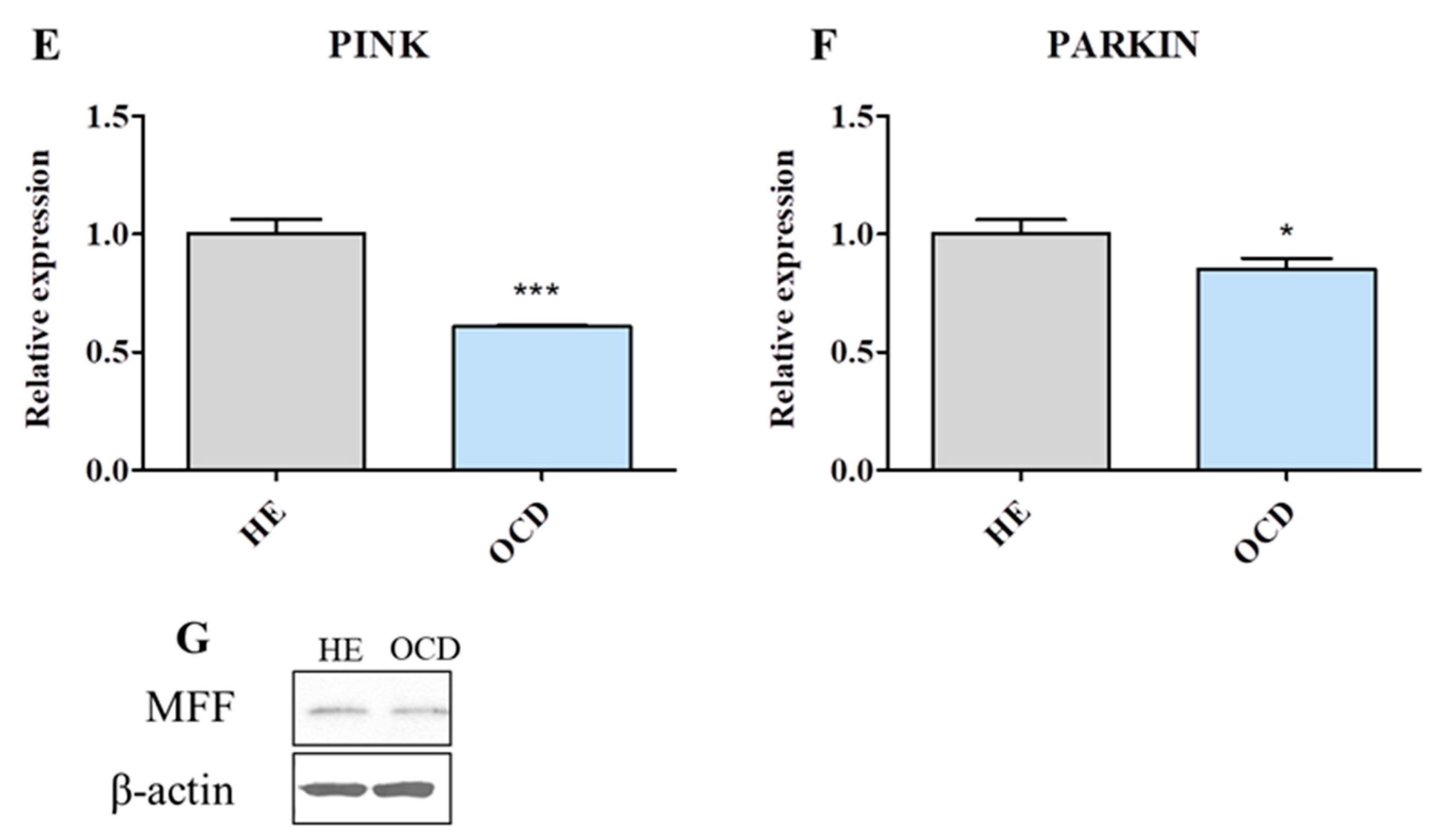
3.6. Mitochondrial Dynamics and Autophagy
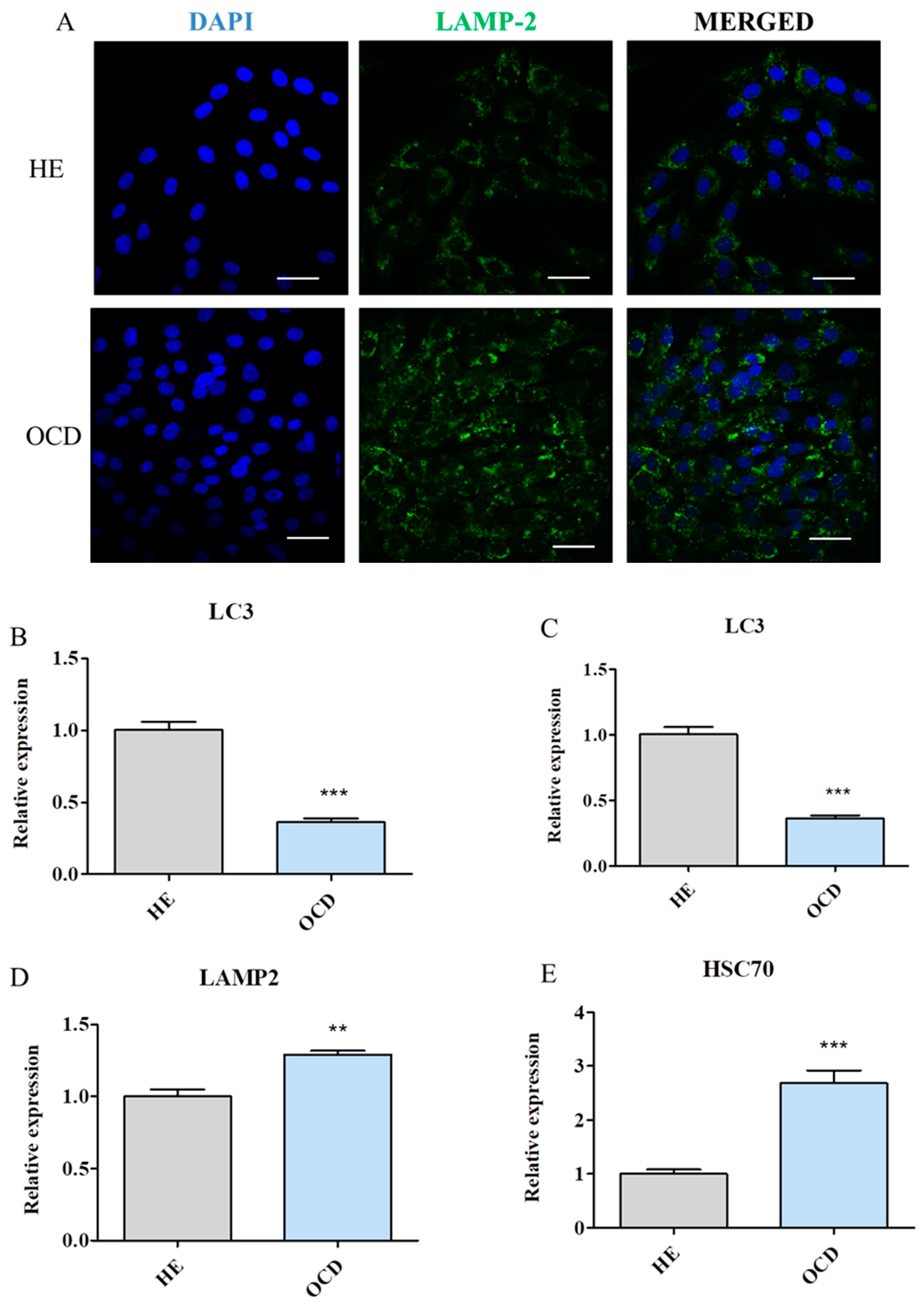
3.7. Evaluation of Autophagy
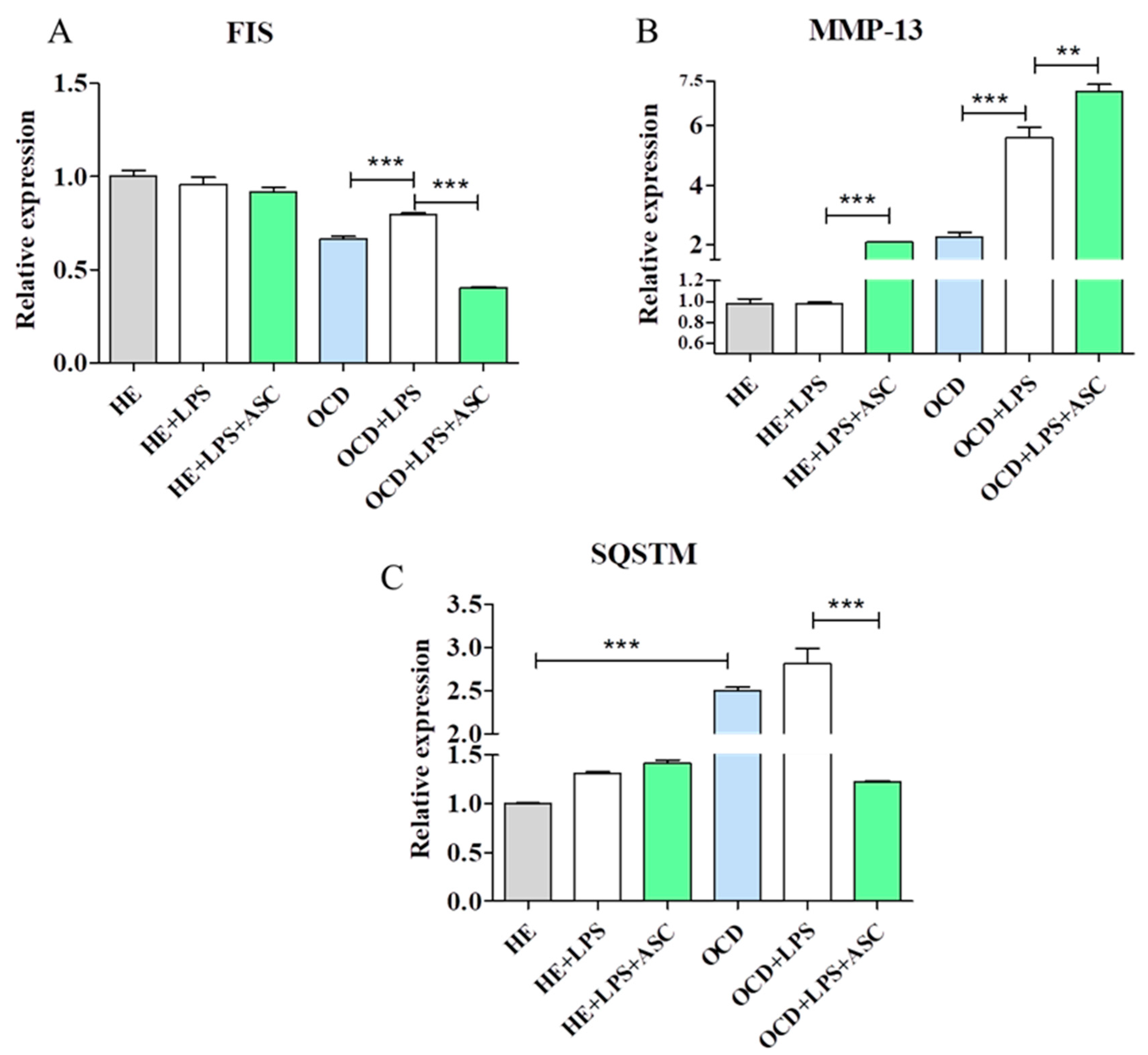
3.8. Chondrocytes-ASC Co-Culture
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Weeren, P.R. Etiology, Diagnosis, and Treatment of OC(D). Clin. Tech. Equine Pract. 2006, 5, 248–258. [Google Scholar] [CrossRef]
- Foland, J.W.; McIlwraith, C.W.; Trotter, G.W. Arthroscopic surgery for osteochondritis dissecans of the femoropatellar joint of the horse. Equine Vet. J. 1992, 24, 419–423. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, C.W.; Frisbie, D.D.; Kawcak, C.E. The horse as a model of naturally occurring osteoarthritis. Bone Jt. Res. 2012, 1, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Frisbie, D.D.; Cross, M.W.; McIlwraith, C.W. A comparative study of articular cartilage thickness in the stifle of animal species used in human pre-clinical studies compared to articular cartilage thickness in the human knee. Vet. Comp. Orthop. Traumatol. VCOT 2006, 19, 142–146. [Google Scholar] [CrossRef]
- Chu, C.R.; Szczodry, M.; Bruno, S. Animal models for cartilage regeneration and repair. Tissue Eng. Part B Rev. 2010, 16, 105–115. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, C.W.; Fortier, L.A.; Frisbie, D.D.; Nixon, A.J. Equine Models of Articular Cartilage Repair. Cartilage 2011, 2, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, K.; Cook, J.L.; Stoker, A.M.; Turnquist, S.E.; Kreeger, J.M.; Tomlinson, J.L. Characterizing osteochondrosis in the dog: Potential roles for matrix metalloproteinases and mechanical load in pathogenesis and disease progression. Osteoarthr. Cartil. 2005, 13, 225–234. [Google Scholar] [CrossRef]
- Semevolos, S.A.; Nixon, A.J.; Brower-Toland, B.D. Changes in molecular expression of aggrecan and collagen types I, II, and X, insulin-like growth factor-I, and transforming growth factor-beta1 in articular cartilage obtained from horses with naturally acquired osteochondrosis. Am. J. Vet. Res. 2001, 62, 1088–1094. [Google Scholar] [CrossRef]
- Garvican, E.R.; Vaughan-Thomas, A.; Redmond, C.; Clegg, P.D. Chondrocytes harvested from osteochondritis dissecans cartilage are able to undergo limited in vitro chondrogenesis despite having perturbations of cell phenotype in vivo. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2008, 26, 1133–1140. [Google Scholar] [CrossRef]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a Protective Mechanism in Normal Cartilage and its Aging-related Loss is Linked with Cell Death and Osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef]
- Cheng, N.-T.; Meng, H.; Ma, L.-F.; Zhang, L.; Yu, H.-M.; Wang, Z.-Z.; Guo, A. Role of autophagy in the progression of osteoarthritis: The autophagy inhibitor, 3-methyladenine, aggravates the severity of experimental osteoarthritis. Int. J. Mol. Med. 2017, 39, 1224. [Google Scholar] [CrossRef] [PubMed]
- Héraud, F.; Héraud, A.; Harmand, M.F. Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 2000, 59, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Hemmel, M.; Neureiter, D.; Gebhard, P.M.; Zeiler, G.; Kirchner, T.; McKenna, L. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: A study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001, 44, 1304–1312. [Google Scholar] [PubMed]
- Aigner, T.; Kim, H.A.; Roach, H.I. Apoptosis in osteoarthritis. Rheum. Dis. Clin. N. Am. 2004, 30, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Lotz, M.; Stoltz, J.M. Osteoarthritis, Inflammation and Degradation: A Continuum; Blanco, F.J., López-Armada, M.J., Rego, I., Eds.; IOS Press, Inc.: Clifton, VA, USA, 2017; pp. 192–205. [Google Scholar]
- Maneiro, E.; Martín, M.A.; de Andres, M.C.; López-Armada, M.J.; Fernández-Sueiro, J.L.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003, 48, 700–708. [Google Scholar] [CrossRef]
- Kim, D.; Song, J.; Kang, Y.; Park, S.; Kim, Y.I.; Kwak, S.; Lim, D.; Park, R.; Chun, C.H.; Choe, S.K.; et al. Fis1 depletion in osteoarthritis impairs chondrocyte survival and peroxisomal and lysosomal function. J. Mol. Med. 2016, 94, 1373–1384. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Khan, N.M.; Ahmad, I.; Haqqi, T.M. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr. Cartil. 2018, 26, 1087–1097. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, H.; Yan, X.; Gu, H.; Gu, Z.; Liu, F. Endoplasmic reticulum stress participates in the progress of senescence and apoptosis of osteoarthritis chondrocytes. Biochem. Biophys. Res. Commun. 2017, 491, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.E.; McBurney, D.L.; Horton, W.E. The presence of extracellular matrix alters the chondrocyte response to endoplasmic reticulum stress. J. Cell. Biochem. 2011, 112, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Wang, H.; Sun, S. Taurine alleviates endoplasmic reticulum stress in the chondrocytes from patients with osteoarthritis. Redox Rep. Commun. Free Radic. Res. 2018, 23, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.; Oxford, A.E.; Tawara, K.; Jorcyk, C.L.; Oxford, J.T. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 665. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Kornicka, K.; Basinska, K.; Czyrek, A. Equine Metabolic Syndrome Affects Viability, Senescence, and Stress Factors of Equine Adipose-Derived Mesenchymal Stromal Stem Cells: New Insight into EqASCs Isolated from EMS Horses in the Context of Their Aging. Oxid. Med. Cell. Longev. 2016, 2016, 4710326. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Marycz, K.; Kornicka, K.; Szlapka-Kosarzewska, J.; Weiss, C. Excessive Endoplasmic Reticulum Stress Correlates with Impaired Mitochondrial Dynamics, Mitophagy and Apoptosis, in Liver and Adipose Tissue, but Not in Muscles in EMS Horses. Int. J. Mol. Sci. 2018, 19, 165. [Google Scholar] [CrossRef] [PubMed]
- Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Semevolos, S.A.; Duesterdieck-Zellmer, K.F.; Larson, M.; Kinsley, M.A. Expression of pro-apoptotic markers is increased along the osteochondral junction in naturally occurring osteochondrosis. Bone Rep. 2018, 9, 19–26. [Google Scholar] [CrossRef]
- Xu, M.; Bradley, E.W.; Weivoda, M.M.; Hwang, S.M.; Pirtskhalava, T.; Decklever, T.; Curran, G.L.; Ogrodnik, M.; Jurk, D.; Johnson, K.O.; et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 780–785. [Google Scholar] [CrossRef]
- Kim, D.Y.; Taylor, H.W.; Moore, R.M.; Paulsen, D.B.; Cho, D.-Y. Articular chondrocyte apoptosis in equine osteoarthritis. Vet. J. 2003, 166, 52–57. [Google Scholar] [CrossRef]
- Johnson, K.A.; van Etten, D.; Nanda, N.; Graham, R.M.; Terkeltaub, R.A. Distinct transglutaminase 2-independent and transglutaminase 2-dependent pathways mediate articular chondrocyte hypertrophy. J. Biol. Chem. 2003, 278, 18824–18832. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.-M.; Shan, Z.-Z.; Nakamura, H.; Masuko-Hongo, K.; Kato, T.; Nishioka, K.; Yudoh, K. Catabolic stress induces features of chondrocyte senescence through overexpression of caveolin 1: Possible involvement of caveolin 1-induced down-regulation of articular chondrocytes in the pathogenesis of osteoarthritis. Arthritis Rheum. 2006, 54, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Nishiyama, T.; Hayashi, S.; Fujishiro, T.; Takebe, K.; Kanzaki, N.; Kuroda, R.; Kurosaka, M. Role of p53 in human chondrocyte apoptosis in response to shear strain. Arthritis Rheum. 2009, 60, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.; Whitehouse, A.; Sharman, P.; Perry, M.; Adams, M. Increased Apoptosis in Human Osteoarthritic Cartilage Corresponds to Reduced Cell Density and Expression of Caspase-3. Arthritis Rheum. 2004, 50, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Mirams, M.; Ayodele, B.A.; Tatarczuch, L.; Henson, F.M.; Pagel, C.N.; Mackie, E.J. Identification of novel osteochondrosis—Associated genes. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2016, 34, 404–411. [Google Scholar] [CrossRef]
- Blanco, F.J.; Guitian, R.; Vázquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Wang, M.; Sampson, E.R.; Jin, H.; Li, J.; Ke, Q.H.; Im, H.-J.; Chen, D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 2013, 15, R5. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.M.; Rowan, A.D.; Cawston, T.E.; Young, D.A. Metalloproteinase and inhibitor expression profiling of resorbing cartilage reveals pro-collagenase activation as a critical step for collagenolysis. Arthritis Res. Ther. 2006, 8, R142. [Google Scholar] [CrossRef]
- Laverty, S.; Okouneff, S.; Ionescu, M.; Reiner, A.; Pidoux, I.; Webber, C.; Rossier, Y.; Billinghurst, R.C.; Poole, A.R. Excessive degradation of type II collagen in articular cartilage in equine osteochondrosis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2002, 20, 1282–1289. [Google Scholar] [CrossRef]
- Tetsunaga, T.; Nishida, K.; Furumatsu, T.; Naruse, K.; Hirohata, S.; Yoshida, A.; Saito, T.; Ozaki, T. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthr. Cartil. 2011, 19, 222–232. [Google Scholar] [CrossRef]
- Wang, M.; Tang, D.; Shu, B.; Wang, B.; Jin, H.; Hao, S.; Dresser, K.A.; Shen, J.; Im, H.-J.; Sampson, E.R.; et al. Conditional activation of β-catenin signaling in mice leads to severe defects in intervertebral disc tissue. Arthritis Rheum. 2012, 64, 2611–2623. [Google Scholar] [CrossRef]
- Mirams, M.; Tatarczuch, L.; Ahmed, Y.A.; Pagel, C.N.; Jeffcott, L.B.; Davies, H.M.S.; Mackie, E.J. Altered gene expression in early osteochondrosis lesions. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2009, 27, 452–457. [Google Scholar] [CrossRef]
- Wang, X.; Manner, P.A.; Horner, A.; Shum, L.; Tuan, R.S.; Nuckolls, G.H. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 963–973. [Google Scholar] [CrossRef]
- Bonyadi Rad, E.; Musumeci, G.; Pichler, K.; Heidary, M.; Szychlinska, M.A.; Castrogiovanni, P.; Marth, E.; Böhm, C.; Srinivasaiah, S.; Krönke, G.; et al. Runx2 mediated Induction of Novel Targets ST2 and Runx3 Leads to Cooperative Regulation of Hypertrophic Differentiation in ATDC5 Chondrocytes. Sci. Rep. 2017, 7, 17947. [Google Scholar] [CrossRef]
- Maldonado, M.; Nam, J. The Role of Changes in Extracellular Matrix of Cartilage in the Presence of Inflammation on the Pathology of Osteoarthritis. BioMed Res. Int. 2013, 2013, 284873. [Google Scholar] [CrossRef]
- Del Carlo, M.; Loeser, R.F. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002, 46, 394–403. [Google Scholar] [CrossRef]
- Stadler, J.; Stefanovic-Racic, M.; Billiar, T.R.; Curran, R.D.; McIntyre, L.A.; Georgescu, H.I.; Simmons, R.L.; Evans, C.H. Articular chondrocytes synthesize nitric oxide in response to cytokines and lipopolysaccharide. J. Immunol. 1991, 147, 3915–3920. [Google Scholar]
- Oh, M.; Fukuda, K.; Asada, S.; Yasuda, Y.; Tanaka, S. Concurrent generation of nitric oxide and superoxide inhibits proteoglycan synthesis in bovine articular chondrocytes: Involvement of peroxynitrite. J. Rheumatol. 1998, 25, 2169–2174. [Google Scholar]
- Toussaint, O.; Medrano, E.E.; von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Fu, Y.; Kinter, M.; Hudson, J.; Humphries, K.M.; Lane, R.S.; White, J.R.; Hakim, M.; Pan, Y.; Verdin, E.; Griffin, T.M. Aging Promotes Sirtuin 3-Dependent Cartilage Superoxide Dismutase 2 Acetylation and Osteoarthritis. Arthritis Rheumatol. 2016, 68, 1887–1898. [Google Scholar] [CrossRef]
- Takada, K.; Hirose, J.; Senba, K.; Yamabe, S.; Oike, Y.; Gotoh, T.; Mizuta, H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int. J. Exp. Pathol. 2011, 92, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Uehara, Y.; Hirose, J.; Yamabe, S.; Okamoto, N.; Okada, T.; Oyadomari, S.; Mizuta, H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthr. Cartil. 2014, 22, 1007–1017. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, D.L.; Ravussin, E. The role of mitochondria in health and disease. Curr. Opin. Pharmacol. 2009, 9, 780–786. [Google Scholar] [CrossRef]
- Yoneda, M.; Miyatake, T.; Attardi, G. Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol. Cell. Biol. 1994, 14, 2699–2712. [Google Scholar] [CrossRef]
- Srinivas, V.; Shapiro, I.M. Chondrocytes embedded in the epiphyseal growth plates of long bones undergo autophagy prior to the induction of osteogenesis. Autophagy 2006, 2, 215–216. [Google Scholar] [CrossRef]
- Chang, J.; Wang, W.; Zhang, H.; Hu, Y.; Wang, M.; Yin, Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 2013, 32, 1311–1318. [Google Scholar] [CrossRef]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis Int. J. Program. Cell Death 2010, 15, 631–638. [Google Scholar] [CrossRef]
- Jiang, L.-B.; Lee, S.; Wang, Y.; Xu, Q.-T.; Meng, D.-H.; Zhang, J. Adipose-derived stem cells induce autophagic activation and inhibit catabolic response to pro-inflammatory cytokines in rat chondrocytes. Osteoarthr. Cartil. 2016, 24, 1071–1081. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward and Reverse Primer Sequence (5′-3′) | Size of Amplicon (bp) | Accession No. |
|---|---|---|---|
| ACAN | F: TGGTGTCCTCTTCTTGTCGCTTTC | 160 | XM_014733894.1 |
| R: ACGATACATTTGCTGTGCTTCGGC | |||
| DCN | F: GATGCAGCTAGCCTGAGAGG | 248 | XM 014841263.1 |
| R: GTGTTGTATCCAGGTGGGCA | |||
| COMP | F: AGTGTCGCAAGGATAACTGCGTGA | 238 | NM_001081856.1 |
| R: TCCTGATCTGTGTCCTTCTGGTCA | |||
| MMP-13 | F: TGCAGCTGTTCACTTTGAGGA | 200 | NM_001081804.1 |
| R: ACTGTATGGGCCCGTTGAAA | |||
| p53 | F: TACTCCCCTGCCCTCAACAA | 252 | U37120.1 |
| R: AGGAATCAGGGCCTTGAGGA | |||
| p21 | F: GAAGAGAAACCCCCAGCTCC | 241 | XM_014853747.1 |
| R: TGACTGCATCAAACCCCACA | |||
| Cas-3 | F: GGCAGACTTCCTGTATGCGT | 167 | XM_023630401.1 |
| R: CCATGGCTACCTTGCGGTTA | |||
| Cas-9 | F: CACCTTCCCAGGCTTTGTCT | 224 | XM_005607504.3 |
| R: GGCTCTGGCCTCAGTAAGTT | |||
| CHOP | F: AGCCAAAATCAGAGCCGGAA | 272 | XM 014844003.1 |
| R: GGGGTCAAGAGTGGTGAAGG | |||
| PERK | F: GTGACTGCAATGGACCAGGA | 283 | XM 014852775.1 |
| R: TCACGTGCTCACGAGGATATT | |||
| eIF2-alpha | F: AGTCTTCAGGCATTGGCTCC | 489 | XM_001488848.6 |
| R: CCGAGTGGGACATGTATCGG | |||
| BECLIN | F: GATGCGTTATGCCCAGATGC | 233 | XM 014833759.1 |
| R: AACGGCAGCTCCTCTGAAAT | |||
| LAMP-2 | F: GCACCCCTGGGAAGTTCTTA | 139 | XM 014733098.1 |
| R: ATCCAGCGAACACTCTTGGG | |||
| SQSTM | F: CATCGGAGGATCCCAGTGTG | 207 | XM_005599173.3 |
| R: CCGGTTTGTTAGGGTCGGAA | |||
| LC-3 | F: TTCTGAGACACAGTCGGAGC | 128 | XM_001493613.6 |
| R: CTTTGTTCGAAGGTGTGGCG | |||
| FIS | F: GGTGCGAAGCAAGTACAACG | 118 | XM 001504462.4 |
| R: GTTGCCCACAGCCAGATAGA | |||
| PINK | F: GCACAATGAGCCAGGAGCTA | 298 | XM 014737247.1 |
| R: GGGGTATTCACGCGAAGGTA-0 | |||
| PARKIN | F: TCCCAGTGGAGGTCGATTCT | 218 | XM 014858374.1 |
| R: CCCTCCAGGTGTGTTCGTTT | |||
| MNF | F: AAGTGGCATTTTTCGGCAGG | 217 | XM 001495170.5 |
| R: TCCATATGAAGGGCATGGGC | |||
| Coll-9 | F: CGGTGAAAAGGGCAACACAG | 260 | XM_014734623.1 |
| R: GTTCACCGACTACACCCCTG | |||
| RUNX-2 | F: ACTTTGCAGAGATGGGCCTC | 76 | XM_005603968.2 |
| R: CTAGGAAGTCGGGATGGGGA | |||
| RUNX-3 | F: ATCAAGGTGACCGTGGATGG | 219 | XM_008532790.1 |
| R: GGAGAAAGGGTTCAGGTCCG | |||
| MIEF-1 | F: ATGCTGGGCATCGCTACAC | 284 | XM_023631522.1 |
| R: CGGAGCCGTGACTTCTTCAA | |||
| MIEF-2 | F: CGTTCTATTATCAGGCAGGTCC | 108 | XM_005597824.3 |
| R: AGAACTCTGCCATGGTCTTCT | |||
| HSC70 | F: GATTAACAAGAGGGCTGTCCGTC | 122 | XM_023628864.1 |
| R: GCCTGGGTGCTAGAAGAGAGA | |||
| BIP | F: CTGTAGCGTATGGTGCTGCT | 122 | XM_005606029.2 |
| R: CATGACACCTCCCACGGTTT | |||
| GAPDH | F: GATGCCCCAATGTTTGTGA | 250 | NM 001163856.1 |
| R: AAGCAGGGATGATGTTCTGG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornicka, K.; Al Naem, M.; Röcken, M.; Zmiertka, M.; Marycz, K. Osteochondritis Dissecans (OCD)-Derived Chondrocytes Display Increased Senescence, Oxidative Stress, Chaperone-Mediated Autophagy and, in Co-Culture with Adipose-Derived Stem Cells (ASCs), Enhanced Expression of MMP-13. J. Clin. Med. 2019, 8, 328. https://doi.org/10.3390/jcm8030328
Kornicka K, Al Naem M, Röcken M, Zmiertka M, Marycz K. Osteochondritis Dissecans (OCD)-Derived Chondrocytes Display Increased Senescence, Oxidative Stress, Chaperone-Mediated Autophagy and, in Co-Culture with Adipose-Derived Stem Cells (ASCs), Enhanced Expression of MMP-13. Journal of Clinical Medicine. 2019; 8(3):328. https://doi.org/10.3390/jcm8030328
Chicago/Turabian StyleKornicka, Katarzyna, Mohamad Al Naem, Michael Röcken, Marta Zmiertka, and Krzysztof Marycz. 2019. "Osteochondritis Dissecans (OCD)-Derived Chondrocytes Display Increased Senescence, Oxidative Stress, Chaperone-Mediated Autophagy and, in Co-Culture with Adipose-Derived Stem Cells (ASCs), Enhanced Expression of MMP-13" Journal of Clinical Medicine 8, no. 3: 328. https://doi.org/10.3390/jcm8030328
APA StyleKornicka, K., Al Naem, M., Röcken, M., Zmiertka, M., & Marycz, K. (2019). Osteochondritis Dissecans (OCD)-Derived Chondrocytes Display Increased Senescence, Oxidative Stress, Chaperone-Mediated Autophagy and, in Co-Culture with Adipose-Derived Stem Cells (ASCs), Enhanced Expression of MMP-13. Journal of Clinical Medicine, 8(3), 328. https://doi.org/10.3390/jcm8030328