Platelet Depletion is Effective in Ameliorating Anxiety-Like Behavior and Reducing the Pro-Inflammatory Environment in the Hippocampus in Murine Experimental Autoimmune Encephalomyelitis

 , ,
, ,
Abstract
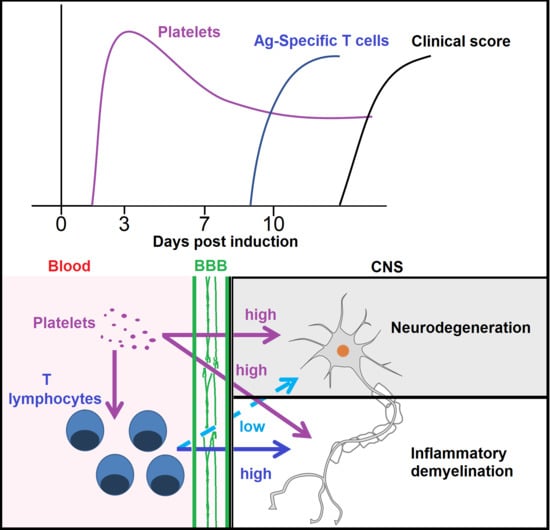
1. Introduction
2. Experimental Section
2.1. EAE Induction
2.2. Estimation of Platelet Numbers and Platelet Depletion
2.3. EPM Test
2.4. Intracellular Cytokine Staining (ICS)
2.5. RNA Isolation, cDNA Synthesis, and qPCR Analysis
2.6. Histology and Immunochemistry
2.7. Quantification of Plasma Soluble P-Selectin (sP-Selectin) by Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Statistical Analyses
3. Results
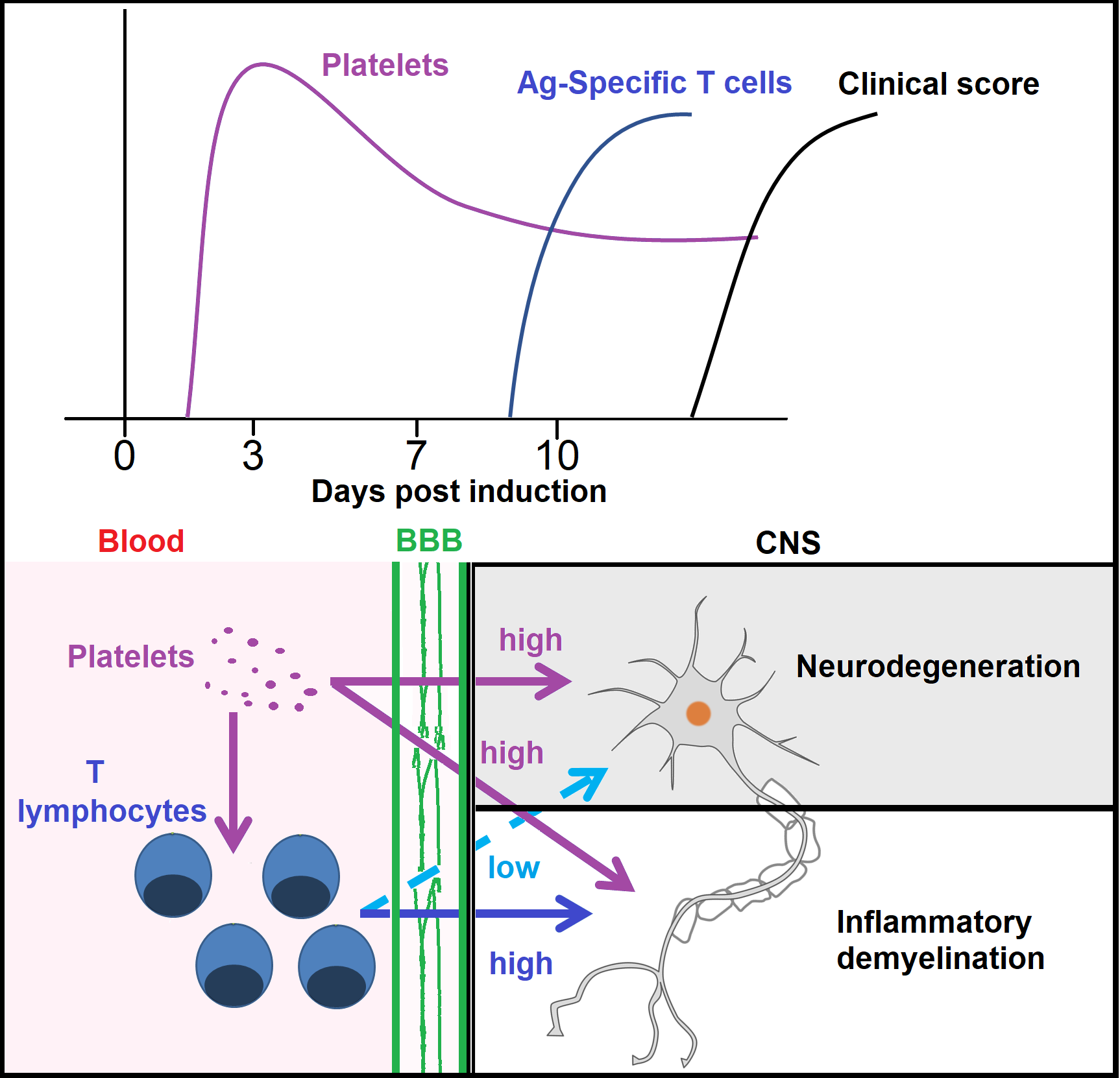
3.1. There Is a Direct Relationship between Platelet Accumulation and That of Antigen-Specific T Cells in Neuroinflammation
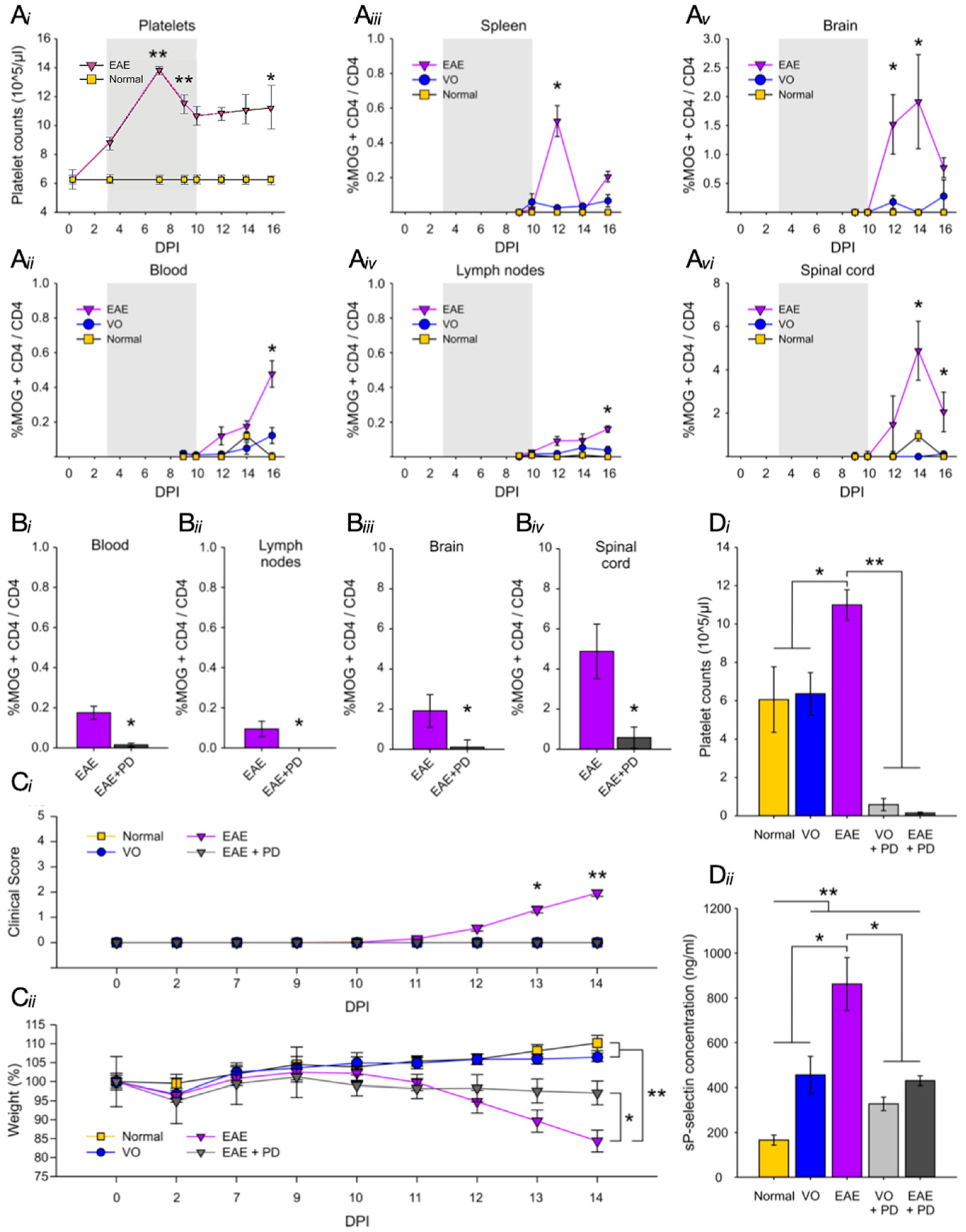
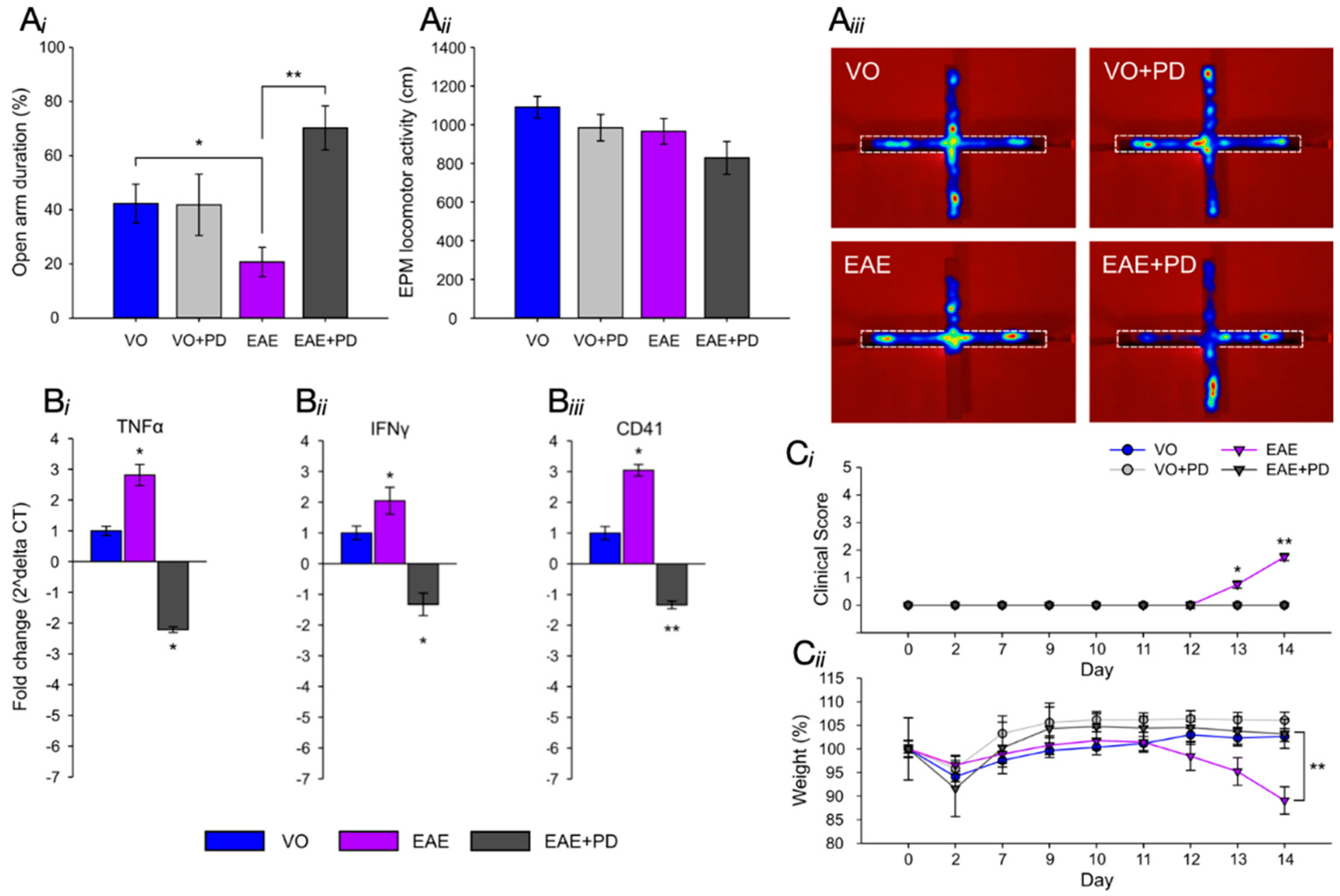
3.2. Platelet Depletion Significantly Reduces Anxiety-Like Behavior in EAE-Induced Mice
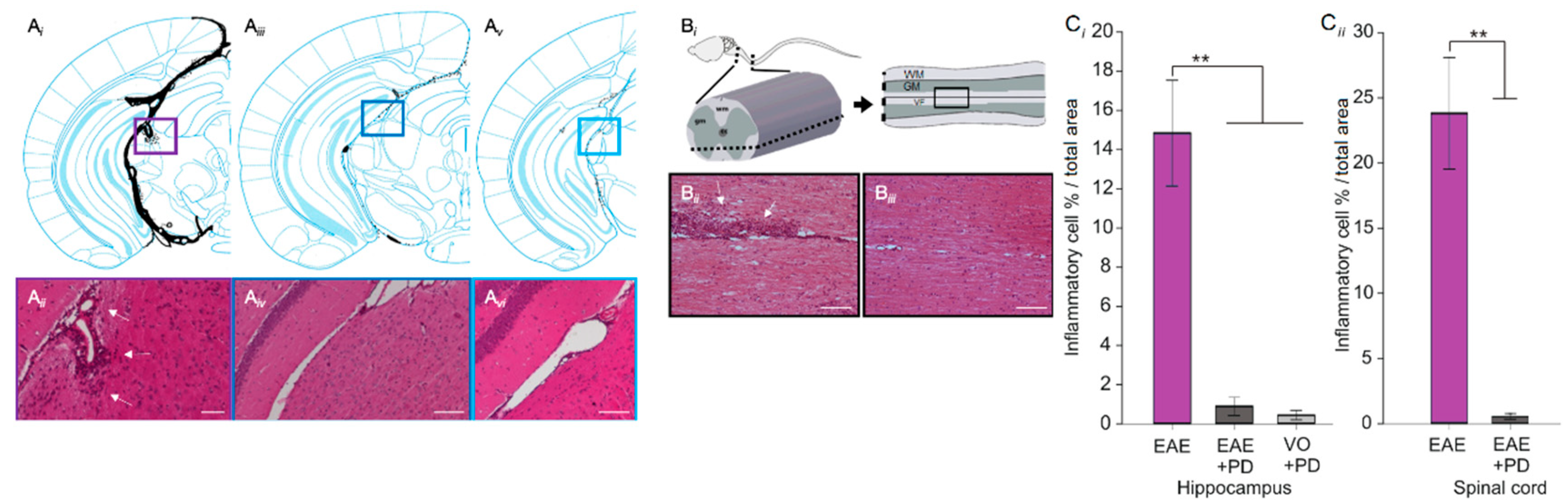
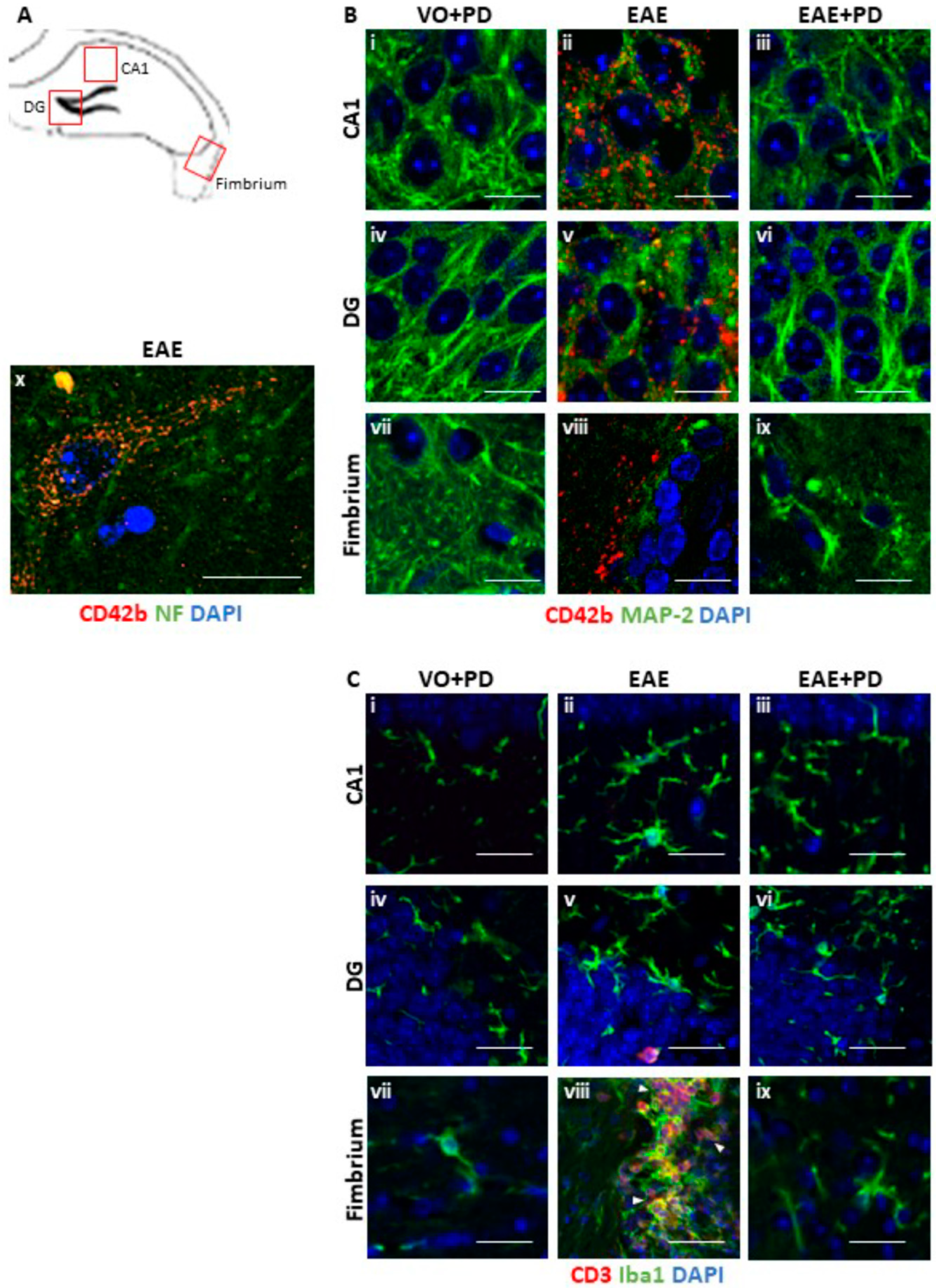
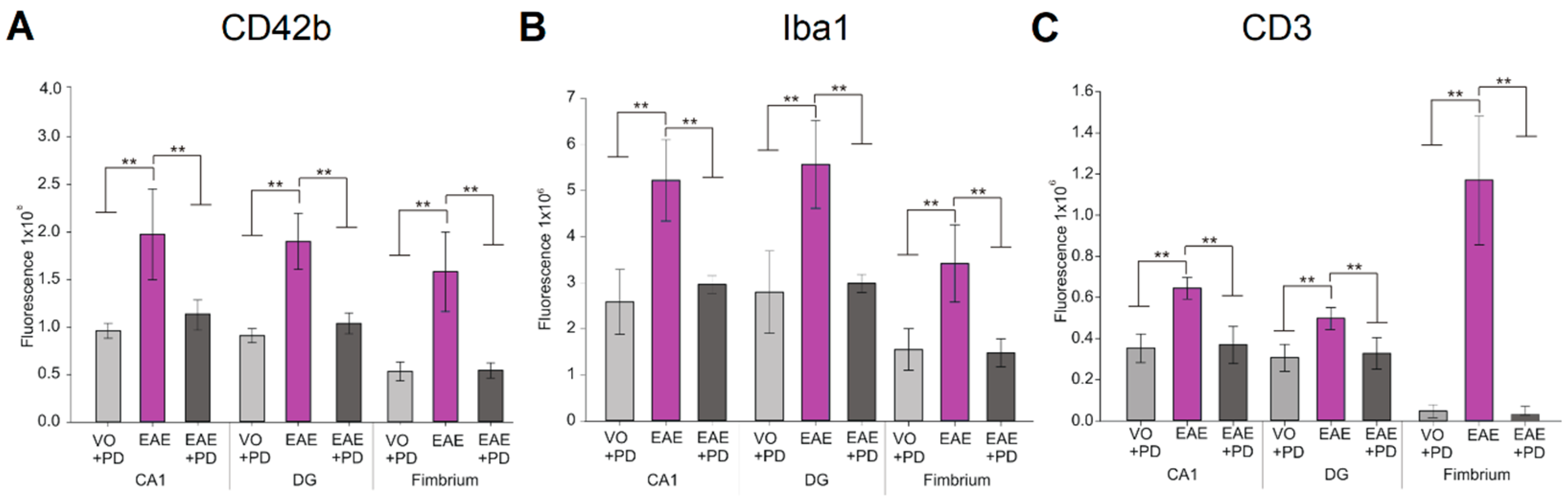
3.3. Anxiety-Like Behavior and the Pro-Inflammatory Environment in the Hippocampus are Characterized by Platelet–Neuron Association
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Confavreux, C.; Vukusic, S. The clinical course of multiple sclerosis. In Multiple Sclerosis and Related Disorders, 3rd ed.; Goodin, D.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 343–369. ISBN 9780444520012. [Google Scholar]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple sclerosis. New Engl. J. Med. 2018, 378, 160–180. [Google Scholar] [CrossRef]
- Lassmann, H. Mechanisms of white matter damage in multiple sclerosis. Glia 2014, 62, 1816–1830. [Google Scholar] [CrossRef]
- Friese, M.A.; Shattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.A.; Urrutia, R.; Kubes, P. Platelets: Bridging hemostasis, inflammation and immunity. Int. J. Lab. Hematol. 2013, 35, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Rainger, G.E.; Chimen, M.; Harrison, M.J.; Yates, C.M.; Harrison, P.; Watson, S.P.; Lordkipanidze, M.; Nash, G.B. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 2015, 26, 507–520. [Google Scholar] [CrossRef] [PubMed]
- McFayden, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving hemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.R.; Storey, R.F. The role of platelets in neuroinflammation. Thromb. Hemost. 2015, 1114, 449–458. [Google Scholar] [CrossRef]
- Langer, H.; Chevakis, T. Platelets and neurovascular inflammation. Thromb. Hemost. 2013, 110, 888–893. [Google Scholar] [CrossRef]
- Xu, X.R.; Zhang, D.; Oswald, D.E.; Carrim, N.; Wang, X.; Hou, Y.; Zhang, Q.; Lavalle, C.; McKeown, T.; Marshall, A.H.; et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumour metastasis and beyond. Crit. Rev. Clin. Lab. Sci. 2016, 53, 409–430. [Google Scholar] [CrossRef]
- Pankratz, S.; Bittner, S.; Kehrel, B.E.; Langer, H.E.; Kleinshnitz, C.; Meuth, S.; Gobel, K. The inflammatory role of platelets: Translational insights from experimental studies of autoimmune disorders. Int. J. Mol. Sci. 2016, 17, 1723. [Google Scholar] [CrossRef] [PubMed]
- Sheremata, W.A.; Jy, W.; Horstman, L.L.; Ahn, Y.S.; Alexander, J.S.; Minagar, A. Evidence of platelet activation in multiple sclerosis. J. Neuroinflammation 2008, 5, 27–321. [Google Scholar] [CrossRef] [PubMed]
- Wachowicz, B.; Morel, A.; Miller, E.; Saluk, J. The physiology of blood platelets and changes in their biological activities in multiple sclerosis. Acta Neurobiol. Exp. 2016, 76, 269–281. [Google Scholar] [CrossRef]
- Steinman, L. Platelets provide a bounty of potential targets for therapy in multiple sclerosis. Circ. Res. 2012, 110, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Amor, S. Experimental autoimmune encephalomyelitis is a good model of multiple sclerosis if used wisely. Mult. Scler. Relat. Disord. 2014, 3, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Mix, E.; Meyer-Rienecker, H.; Hartung, H.P.; Zettl, Z.K. Animal models of multiple sclerosis—Potentials and limitations. Prog. Neurobiol. 2010, 92, 386–404. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Ramiro, B.; Olivia-Nacarino, P.; Serrano-Pertierra, E.; Blanco-Gelaz, M.A.; Weksler, B.B.; Romero, J.A.; Couraud, P.O.; Tunon, A.; Lopez-Larrea, C.; Millan, J.; et al. Microparticles in multiple sclerosis and clinically isolated syndrome: Effect on endothelial barrier function. BMC Neurosci. 2014, 15, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.; Bijak, M.; Miller, E.; Rywaniak, J.; Miller, S.; Saluk, J. Relationship between the increased haemostatic properties of blood platelets and oxidative stress levels in multiple sclerosis patients with in secondary progressive stage. Oxid. Med. Cell Longev. 2015, 240918. [Google Scholar] [CrossRef]
- Morel, A.; Miller, E.; Bijak, M.; Saluk, K. The increased level of COX-dependent arachidonic acid metabolism in blood platelets from secondary progressive multiple sclerosis patients. Mol. Cell. Biochem. 2016, 420, 85–94. [Google Scholar] [CrossRef]
- Morel, A.; Rywaniak, J.; Bijak, M.; Miller, E.; Niwald, M.; Saluk, J. Flow cytometric analysis reveals the high levels of platelet activation parameters in the circulation of multiple sclerosis patients. Mol. Cell. Biochem. 2017, 430, 69–80. [Google Scholar] [CrossRef]
- Langer, H.F.; Choi, E.Y.; Zhou, H.; Scleicher, R.; Chung, K.-J.; Tang, Z.; Gobel, K.; Bdeir, K.; Chatzigeorgiou, A.; Wong, C.; et al. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ. Res. 2012, 110, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Starossom, S.C.; Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Au, C.; Lau, A.Y.; Weiner, H.L.; Poniomarev, E.D. Platelets play differential roles during the initiation and progression of autoimmune neuroinflammation. Circ. Res. 2015, 117, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Sotnikov, I.; Veremeyko, T.; Starossom, S.C.; Barteneva, N.; Weiner, H.L.; Poniomarev, E.D. Platelets recognize brain-specific glycolipid structures, respond to neurovascular damage and promote neuroinflammation. PLoS ONE 2013, 8, e58979. [Google Scholar] [CrossRef]
- D’Souza, C.S.; Li, Z.; Maxwell, D.L.; Trusler, O.; Murphy, M.; Crewther, S.; Peter, K.; Orian, J.M. Platelets drive inflammation and target grey matter and the retina in autoimmune-mediated encephalomyelitis. J. Neuropathol. Exp. Neurol. 2018, 77, 567–576. [Google Scholar] [CrossRef]
- Geurts, J.J.; Barkhof, F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008, 7, 841–851. [Google Scholar] [CrossRef]
- Peterson, J.W.; Bo, L.; Mork, S.; Chang, A.; Trapp, B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef]
- Vercellino, M.; Plano, F.; Votta, B.; Mutani, R.; Giordana, M.T.; Cavalla, P. Grey matter pathology in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2005, 64, 1101–1107. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Faber-Rod, J.C.; Bauer, J.; Lucchinetti, C.F.; Sorensen, P.S.; Laursen, H.; Stadelmann, C.; Brück, W.; Rauschka, H.; Schmidbauer, M.; et al. Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol. 2007, 17, 38–44. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical demyelination and diffuse WM injury in multiple sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef]
- Calabrese, M.; De Stefano, N.; Atzori, M.; Bernardi, V.; Mattisi, I.; Barachino, L.; Morra, A.; Rinaldi, L.; Romualdi, C.; Perini, P.; et al. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch. Neurol. 2007, 64, 1416–1422. [Google Scholar] [CrossRef]
- Geurts, J.J.; Bo, L.; Roosendaal, S.D.; Hazes, T.; Daniëls, R.; Barkhof, F.; Witter, M.P.; Huitinga, I.; van der Valk, P. Extensive hippocampal demyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, C.P.; Donaldson, I.; Bo, L.; Owens, T.; Lowe, J.; Evangelou, N. Regional variations in the extent and pattern of grey matter demyelination in multiple sclerosis: A comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord. J. Neurol. Neurosurg. Psychiatry 2009, 80, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Dukes, S.; Patel, R.; Nicholas, R.; Vora, A.; Reynolds, R. Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol. 2009, 19, 238–253. [Google Scholar] [CrossRef] [PubMed]
- Poppenk, J.; Evensmoen, H.R.; Moscovitch, M.; Nadel, L. Long-axis specialization of the human hippocampus. Trends Cognit. Sci. 2013, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; O’Donoghue, S.; Counihan, T.; McDonald, C.; Calabresi, P.A.; Ahmed, M.A.S.; Kaplin, A.; Hallahan, B. Neuropsychiatric syndromes in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Politte, L.C.; Huffman, J.C.; Stern, T.A. Neuropsychiatric manifestations of multiple sclerosis. J. Clin. Psychiatry 2018, 10, 318–324. [Google Scholar] [CrossRef]
- Morris, G.; Reiche, E.M.V.; Murru, A.; Carvalho, A.F.; Maes, M.; Berk, B.; Puri, B.K. Multiple immune-inflammatory and oxidative and nitrosative stress pathways explain the frequent presence of depression in multiple sclerosis. Mol. Neurobiol. 2018, 55, 6282–6306. [Google Scholar] [CrossRef]
- Rocca, M.A.; Barkhof, F.; De Luca, J.; Frisén, J.; Geurts, J.J.G.; Hulst, H.E.; Sastre-Garriga, J.; Filippi, M. The hippocampus in multiple sclerosis. Lancet Neurol. 2018, 17, 918–926. [Google Scholar] [CrossRef]
- Hyncicova, E.; Kalina, A.; Vyhnalek, M.; Nikolao, T.; Martinkovic, L.; Lisy, J.; Hort, J.; Meluzinova, E.; Laczo, J. Health-related quality of life, neuropsychiatric symptoms and structural brain changes in clinically isolated syndrome. PLoS ONE 2018, 13, e0200254. [Google Scholar] [CrossRef]
- Kyran, E.L.; Robinson, C.; Kocovski, P.; Li, Z.; Dang, P.T.; Hale, M.W.; Orian, J.M. Multiple pathological mechanisms contribute to hippocampal damage in the experimental autoimmune encephalomyelitis model of multiple sclerosis. NeuroReport 2017, 29, 19–24. [Google Scholar] [CrossRef]
- Pollak, Y.; Orion, E.; Goshen, I.; Ovadia, H.; Yirmiya, R. Experimental autoimmune encephalomyelitis-associated behavioral syndrome as a model of ‘depression due to multiple sclerosis’. Brain Behav. Immun. 2002, 16, 533–543. [Google Scholar] [CrossRef]
- Carobroz, A.P.; Bertoglio, L.J. Ethological and temporal analyses of anxiety-like behavior: The elevated plus-maze model 20 years on. Neurosci. Biobehav. Rev. 2005, 29, 1193–1205. [Google Scholar] [CrossRef]
- Kocovski, P.; Dang, P.T.; D’Souza, C.S.; Stamper, C.E.; Hale, M.W.; Orian, J.M. Differential anxiety-like responses in NOD/ShiLtJ and C57BL/6J mice following experimental autoimmune encephalomyelitis induction and oral gavage. Lab. Anim. 2018, 52, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Pham, H.; Doerbecker, J.; Ramp, A.; Ayers, M.M.; Orian, J.M. Experimental autoimmune encephalomyeltis (EAE) in C57bl/6 mice is not associated with astrogliosis. J. Neuroimmunol. 2011, 232, 51–62. [Google Scholar] [CrossRef]
- Costa, A.A.; Morato, S.; Roque, C.; Tinós, R. A computational model for exploratory activity of rats with different anxiety levels in elevated plus-maze. J. Neurosci. Method. 2014, 236, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Pino, P.A.; Cardona, A.E. Isolation of brain and spinal cord mononuclear cells using percoll gradients. J. Vis. Exp. 2011, 48, e2348. [Google Scholar] [CrossRef] [PubMed]
- Lawther, A.J.; Clissold, M.L.; Ma, S.; Kent, S.; Lowry, C.A.; Gundlach, A.L.; Hale, M.W. Anxiogenic drug administration and elevated plus-maze exposure in rats activate populations of relaxin-3 neurons in the nucleus incertus and serotonergic neurons in the dorsal raphe nucleus. Neuroscience 2015, 303, 270–284. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S.S. How to successfully apply animal studies in experimental autoimmune encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 2006, 60, 12–21. [Google Scholar] [CrossRef]
- Haji, N.; Mandelosi, G.; Gentile, A.; Sacchetti, L.; Fresegna, D.; Rossi, S.; Musella, A.; Seoman, H.; Motta, C.; Studer, V.; et al. TNF-α-mediated anxiety in a mouse model of multiple sclerosis. Exp. Neurol. 2013, 237, 296–303. [Google Scholar] [CrossRef]
- Olechowski, C.; Tenorio, G.; Sauve, Y.; Kerr, B.J. Changes in nociceptive sensitivity and object recognition in experimental autoimmune encephalomyelitis (EAE). Exp. Neurol. 2013, 241, 113–121. [Google Scholar] [CrossRef]
- Bolton, C.; Smith, P. Defining and regulating acute inflammatory lesion formation during the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. CNS Neurol. Disord. Drug Targets 2015, 14, 915–935. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer Sequence (5′–3′) | Reverse Primer Sequence (5′–3′) |
|---|---|---|
| IFN-γ | TCATGGCTGTTTCTGGCTGT | CCCAGATACAACCCCGCAAT |
| TNF-α | AAGCCTGTAGCCCACGTCGTA | GGCACCACTAGTTGGTTGTCTTTG |
| CD41 | TTTCTGCAGCCTAAGGGCC | GGCAGCCACAGCAATATCATT |
| HSP90 | GCTTTCCCGTCAAGATGCCT | CACCACTTCCTTGACCCTCC |
| GAPDH | GCTCATGACCACAGTCCATGC | GTTGGGATAGGGCCTCTCTTG |
| β-actin | AGTGTGACGTTGACATCCGT | GCAGCTCAGTAACAGTCCGC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocovski, P.; Jiang, X.; D’Souza, C.S.; Li, Z.; Dang, P.T.; Wang, X.; Chen, W.; Peter, K.; Hale, M.W.; Orian, J.M. Platelet Depletion is Effective in Ameliorating Anxiety-Like Behavior and Reducing the Pro-Inflammatory Environment in the Hippocampus in Murine Experimental Autoimmune Encephalomyelitis. J. Clin. Med. 2019, 8, 162. https://doi.org/10.3390/jcm8020162
Kocovski P, Jiang X, D’Souza CS, Li Z, Dang PT, Wang X, Chen W, Peter K, Hale MW, Orian JM. Platelet Depletion is Effective in Ameliorating Anxiety-Like Behavior and Reducing the Pro-Inflammatory Environment in the Hippocampus in Murine Experimental Autoimmune Encephalomyelitis. Journal of Clinical Medicine. 2019; 8(2):162. https://doi.org/10.3390/jcm8020162
Chicago/Turabian StyleKocovski, Pece, Xiangrui Jiang, Claretta S. D’Souza, Zhenjiang Li, Phuc T. Dang, Xiaowei Wang, Weisan Chen, Karlheinz Peter, Matthew W. Hale, and Jacqueline M. Orian. 2019. "Platelet Depletion is Effective in Ameliorating Anxiety-Like Behavior and Reducing the Pro-Inflammatory Environment in the Hippocampus in Murine Experimental Autoimmune Encephalomyelitis" Journal of Clinical Medicine 8, no. 2: 162. https://doi.org/10.3390/jcm8020162
APA StyleKocovski, P., Jiang, X., D’Souza, C. S., Li, Z., Dang, P. T., Wang, X., Chen, W., Peter, K., Hale, M. W., & Orian, J. M. (2019). Platelet Depletion is Effective in Ameliorating Anxiety-Like Behavior and Reducing the Pro-Inflammatory Environment in the Hippocampus in Murine Experimental Autoimmune Encephalomyelitis. Journal of Clinical Medicine, 8(2), 162. https://doi.org/10.3390/jcm8020162