Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome

 , ,
, ,  and
and
Abstract
1. Introduction
2. Methods
2.1. Patients
2.2. Clinical Manifestations
2.3. Genetic Analyses
2.4. Clinical Events
2.5. Statistical Analyses
3. Results
3.1. Clinical Manifestations
3.2. Death of Any Cause
3.3. Proximal Aortic Surgery
3.4. Distal Aortic Repair
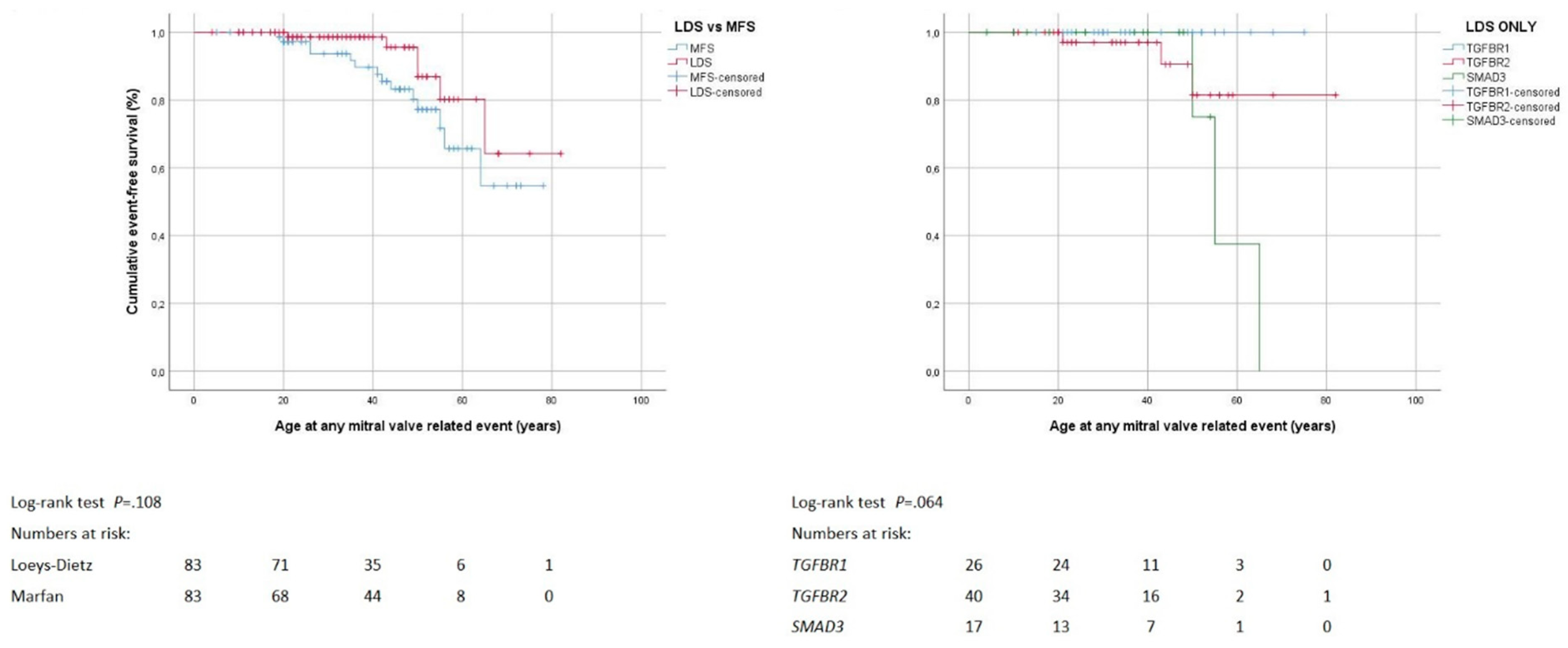
3.5. Mitral Valve Surgery
3.6. Combined Procedures and Reinterventions
4. Discussion
4.1. Clinical Manifestations
4.2. Death of Any Cause
4.3. Proximal Aortic Surgery
4.4. Distal Aortic Repair
4.5. Mitral Valve Surgery
4.6. Combined Procedures and Reinterventions
4.7. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pyeritz, R.E. Heritable thoracic aortic disorders. Curr. Opin. Cardiol. 2014, 29, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Marfan, A.B.-J. Un cas de déformation congènital des quatre membres plus pronouncée aux extrémitiés charactérisée par l’allongement des os avec un certain degré d’amonassesment. Bull. Mem. Soc. Med. Hop. 1896, 13, 220–226. [Google Scholar]
- Beighton, P.; De Paepe, A.; Danks, D.; Finidori, G.; Gedde-Dahl, T.; Goodman, R.; Hall, J.G.; Hillister, D.W.; Horton, W.; McKusick, V.A.; et al. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am. J. Med. Genet. 1988, 29, 581–594. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, A.; Devereux, R.B.; Dietz, H.C.; Hennekam, R.C.; Pyeritz, R.E. Revised diagnostic criteria for the Marfan syndrome. Am. J. Med. Genet. 1996, 62, 417–426. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef]
- von Kodolitsch, Y.; De Backer, J.; Schüler, H.; Bannas, P.; Behzadi, C.; Bernhardt, A.M.; Hillebrand, M.; Fuisting, B.; Sheikhzadeh, S.; Rybczynski, M.; et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl. Clin. Genet. 2015, 8, 137–155. [Google Scholar] [CrossRef]
- Loeys, B.L.; Chen, J.; Neptune, E.R.; Judge, D.P.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Micha, D.; Guo, D.C.; Hilhorst-Hofstee, Y.; van Kooten, F.; Atmaja, D.; Overwater, E.; Cayami, F.K.; Regalado, E.S.; van Uffelen, R.; Venselaar, H.; et al. SMAD2 Mutations are associated with arterial aneurysms and dissections. Hum. Mutat. 2015, 36, 1145–1149. [Google Scholar] [CrossRef]
- van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; Roos-Hesselink, J.W.; de Graaf, B.M.; Verhagen, J.M.; Hoedemaekers, Y.M.; Willemsen, R.; Severijnen, L.A.; Venselaar, H.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121–126. [Google Scholar] [CrossRef]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; Verhagen, J.M.A.; de Graaf, B.M.; van de Beek, G.; Gallo, E.; Kruithof, B.P.T.; Venselaar, H.; Myers, L.A.; et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, I.M.; van der Linde, D.; Oei, E.H.; Bos, P.K.; Bessems, J.H.; Bierma-Zeinstra, S.M.; van Meer, B.L.; Pals, G.; Oldenburg, R.A.; Bekkers, J.A.; et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J. Med. Genet. 2012, 49, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Kuechler, A.; Altmuller, J.; Nurnberg, P.; Kotthoff, S.; Kubisch, C.; Borck, G. Exome sequencing identifies a novel heterozygous TGFB3 mutation in a disorder overlapping with Marfan and Loeys-Dietz syndrome. Mol. Cell Probes. 2015, 29, 330–334. [Google Scholar] [CrossRef]
- Schepers, D.; Tortora, G.; Morisaki, H.; MacCarrick, G.; Lindsay, M.; Liang, D.; Mehta, S.G.; Hague, J.; Verhagen, J.; van de Laar, I.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621–634. [Google Scholar] [CrossRef]
- Engelfriet, P.M.; Boersma, E.; Tijssen, J.G.; Bouma, B.J.; Mulder, B.J. Beyond the root: Dilatation of the distal aorta in Marfan’s syndrome. Heart 2006, 92, 1238–1243. [Google Scholar] [CrossRef]
- Attias, D.; Stheneur, C.; Roy, C.; Collod-Beroud, G.; Detaint, D.; Faivre, L.; Delrue, M.A.; Cohen, L.; Francannet, C.; Beroud, C.; et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation 2009, 120, 2541–2549. [Google Scholar] [CrossRef]
- Girdauskas, E.; Schulz, S.; Borger, M.A.; Mierzwa, M.; Kuntze, T. Transforming Growth Factor-Beta Receptor Type II Mutation in a patient with bicuspid aortic valve disease and intraoperative aortic dissection. Ann. Thorac. Surg. 2011, 91, e70–e71. [Google Scholar] [CrossRef]
- Iba, Y.; Minatoya, K.; Matsuda, H.; Sasaki, H.; Tanaka, H.; Morisaki, H.; Morisaki, T.; Kobayashi, J.; Ogino, H. Surgical experience with aggressive aortic pathologic process in Loeys-Dietz syndrome. Ann. Thorac. Surg. 2012, 94, 1413–1417. [Google Scholar] [CrossRef]
- Kari, F.A.; Russe, M.F.; Peter, P.; Blanke, P.; Rylski, B.; Euringer, W.; Beyersdorf, F.; Siepe, M. Late complications and distal growth rates of Marfan aortas after proximal aortic repair. Eur. J. Cardiothorac. Surg. 2013, 44, 163–171. [Google Scholar] [CrossRef]
- Williams, J.A.; Hanna, J.M.; Shah, A.A.; Andersen, N.D.; McDonald, M.T.; Jiang, Y.H.; Wechsler, S.B.; Zomorodi, A.; McCann, R.L.; Hughes, G.C. Adult surgical experience with Loeys-Dietz syndrome. Ann. Thorac. Surg. 2015, 99, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Jondeau, G.; Ropers, J.; Regalado, E.; Braverman, A.; Evangelista, A.; Teixedo, G.; De Backer, J.; Muino-Mosquera, L.; Naudion, S.; Zordan, C.; et al. International registry of patients carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ. Cardiovasc. Genet. 2016, 9, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Krohg-Sorensen, K.; Lingaas, P.S.; Lundblad, R.; Seem, E.; Paus, B.; Geiran, O.R. Cardiovascular surgery in Loeys-Dietz syndrome types 1-4. Eur. J. Cardiothorac. Surg. 2017. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.D.; Crawford, T.; Magruder, J.T.; Alejo, D.E.; Hibino, N.; Black, J.; Dietz, H.C.; Vricella, L.A.; Cameron, D.E. Cardiovascular operations for Loeys-Dietz syndrome: Intermediate-term results. J. Thorac. Cardiovasc. Surg. 2017, 153, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Aftab, M.; Cikach, F.S.; Zhu, Y.; Idrees, J.J.; Rigelsky, C.M.; Kalahasti, V.; Roselli, E.E.; Svensson, L.G. Loeys-Dietz syndrome: Intermediate-term outcomes of medically and surgically managed patients. J. Thorac. Cardiovasc. Surg. 2019, 157, 439–450. [Google Scholar] [CrossRef]
- Hostetler, E.M.; Regalado, E.S.; Guo, D.C.; Hanna, N.; Arnaud, P.; Muino-Mosquera, L.; Callewaert, B.L.; Lee, K.; Leal, S.M.; Wallace, S.E.; et al. SMAD3 pathogenic variants: Risk for thoracic aortic disease and associated complications from the montalcino aortic consortium. J. Med. Genet. 2019. [Google Scholar] [CrossRef]
- von Elm, E.; Altman, D.G.; Egger, M.; Pocock, S.J.; Gotzsche, P.C.; Vandenbroucke, J.P. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement: Guidelines for reporting observational studies. Int. J. Surg. 2014, 12, 1495–1499. [Google Scholar] [CrossRef]
- Kühne, K.; Keyser, B.; Groene, E.F.; Sheikhzadeh, S.; Detter, C.; Lorenzen, V.; Hillebrand, M.; Bernhardt, A.M.J.; Hoffmann, B.; Mir, T.S.; et al. FBN1 gene mutation characteristics and clinical features for the prediction of mitral valve disease progression. Int. J. Cardiol. 2013, 168, 953–959. [Google Scholar] [CrossRef]
- Weinrich, J.M.; Lenz, A.; Girdauskas, E.; Adam, G.; von Kodolitsch, Y.; Bannas, P. Current and emerging imaging techniques in patients with genetic aortic syndromes. RoFo 2019. [Google Scholar] [CrossRef]
- Sheikhzadeh, S.; Brockstaedt, L.; Habermann, C.R.; Sondermann, C.; Bannas, P.; Mir, T.S.; Staebler, A.; Seidel, H.; Keyser, B.; Arslan-Kirchner, M.; et al. Dural ectasia in Loeys-Dietz syndrome: Comprehensive study of 30 patients with a TGFBR1 or TGFBR2 mutation. Clin. Genet. 2014, 86, 545–551. [Google Scholar] [CrossRef]
- Rybczynski, M.; Bernhardt, A.M.; Rehder, U.; Fuisting, B.; Meiss, L.; Voss, U.; Habermann, C.; Detter, C.; Robinson, P.N.; Arslan-Kirchner, M.; et al. The spectrum of syndromes and manifestations in individuals screened for suspected Marfan syndrome. Am. J. Med. Genet. A 2008, 146, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Rybczynski, M.; Mir, T.S.; Sheikhzadeh, S.; Bernhardt, A.M.; Schad, C.; Treede, H.; Veldhoen, S.; Groene, E.F.; Kuhne, K.; Koschyk, D.; et al. Frequency and age-related course of mitral valve dysfunction in the Marfan syndrome. Am. J. Cardiol. 2010, 106, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Freed, L.A.; Levy, D.; Levine, R.A.; Larson, M.G.; Evans, J.C.; Fuller, D.L.; Lehman, B.; Benjamin, E.J. Prevalence and clinical outcome of mitral-valve prolapse. N. Engl. J. Med. 1999, 341, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Detaint, D.; Fermanian, C.; Aegerter, P.; Delorme, G.; Arnoult, F.; Milleron, O.; Raoux, F.; Stheneur, C.; Boileau, C.; et al. Nomograms for aortic root diameters in children using two-dimensional echocardiography. Am. J. Cardiol. 2010, 105, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Devereux, R.B.; de Simone, G.; Arnett, D.K.; Best, L.G.; Boerwinkle, E.; Howard, B.V.; Kitzman, D.; Lee, E.T.; Mosley, T.H., Jr.; Weder, A.; et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons ≥15 years of age. Am. J. Cardiol. 2012, 110, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Renner, S.; Schuler, H.; Alawi, M.; Kolbe, V.; Rybczynski, M.; Woitschach, R.; Sheikhzadeh, S.; Stark, V.C.; Olfe, J.; Roser, E.; et al. Next-generation sequencing of 32 genes associated with hereditary aortopathies and related disorders of connective tissue in a cohort of 199 patients. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015. [Google Scholar] [CrossRef]
- Erbel, R.; Aboyans, V.; Boileau, C.; Bossone, E.; Bartolomeo, R.D.; Eggebrecht, H.; Evangelista, A.; Falk, V.; Frank, H.; Gaemperli, O.; et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases. Eur. Heart J. 2014, 35, 2873–2926. [Google Scholar]
- Bernhardt, A.M.; Treede, H.; Rybczynski, M.; Sheikzadeh, S.; Kersten, J.F.; Meinertz, T.; von Kodolitsch, Y.; Reichenspurner, H. Comparison of aortic root replacement in patients with Marfan syndrome. Eur. J. Cardiothorac. Surg. 2011, 40, 1052–1057. [Google Scholar] [CrossRef]
- Detter, C.; Demal, T.J.; Bax, L.; Tsilimparis, N.; Koelbel, T.; von Kodolitsch, Y.; Vettorazzi, E.; Reichenspurner, H.; Brickwedel, J. Simplified frozen elephant trunk technique for combined open and endovascular treatment of extensive aortic diseases. Eur. J. Cardiothorac. Surg. 2019. [Google Scholar] [CrossRef]
- Kolbel, T.; Tsilimparis, N.; Mani, K.; Rohlffs, F.; Wipper, S.; Debus, E.S.; Kodolitsch, Y.V.; Wanhainen, A. Physician-modified thoracic stent-graft with low distal radial force to prevent distal stent-graft-induced new entry tears in patients with genetic aortic syndromes and aortic dissection. J. Endovasc. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, A.M.; Treede, H.; Detter, C.; Rybczynski, M.; Sheikhzadeh, S.; Wagner, F.M.; Von Kodolitsch, Y.; Reichenspurner, H. Results of modern mitral valve repair in patients with Marfan syndrome. Thorac. Cardiovasc. Surg. 2014, 62, 35–41. [Google Scholar] [PubMed]
- Nistri, S.; Porciani, M.C.; Attanasio, M.; Abbate, R.; Gensini, G.F.; Pepe, G. Association of Marfan syndrome and bicuspid aortic valve: Frequency and outcome. Int. J. Cardiol. 2012, 155, 324–325. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.W.; Silberbach, G.M.; Liliana, P.; Maslen, C.L.; Dietz, H.C.; Ravekes, W.J.; LeMaire, S.A.; Milewicz, D.M.; Devereux, R.; Roman, M.J.; et al. Abstract 13178: Bicuspid aortic valve and marfan syndrome: Two strikes? Circulation 2019, 128, A13178. [Google Scholar]
- Karur, G.R.; Pagano, J.J.; Bradley, T.; Lam, C.Z.; Seed, M.; Yoo, S.J.; Grosse-Wortmann, L. Diffuse myocardial fibrosis in children and adolescents with marfan syndrome and loeys-dietz syndrome. J. Am. Coll. Cardiol. 2018, 72, 2279–2281. [Google Scholar] [CrossRef] [PubMed]
- Minatoya, K.; Inoue, Y.; Seike, Y.; Omura, A.; Uehara, K.; Sasaki, H.; Matsuda, H.; Kobayashi, J. Thoracoabdominal aortic replacement in patients aged 50 and younger. Gen. Thorac. Cardiovasc. Surg. 2019, 67, 53–58. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Indication for Corroborative Genetic Testing | ||
|---|---|---|---|
| Clinical Suspicion | Cascade Screening | p | |
| Total number of individuals | 45 | 38 | |
| Cardiovascular features | <0.001 | ||
| No cardiovascular features | 5 (11%) | 16 (42%) | |
| Isolated aortic features | 6 (13%) | 1 (3%) | |
| Isolated mitral valve features | 3 (7%) | 6 (16%) | |
| Isolated malformation (ASD, BAV, or PDA, or combinations) | 3 (7%) | 3 (8%) | |
| Stroke | 0 | 2 (5%) | |
| Combination of these cardiovascular features | 28 (62%) | 10 (26%) | |
| Systemic features | 0.128 | ||
| No formal systemic features | 5 (11%) | 11 (29%) | |
| Isolated systemic features (according to Ghent score) | 25 (56%) | 18 (47%) | |
| Isolated craniofacial features (according to Loeys et at.) | 15 (33%) | 9 (24%) | |
| Cardiovascular plus systemic features | <0.001 | ||
| No formally defined clinical features 1 | 0 | 8 (21%) | |
| Isolated systemic features (according to Ghent score) | 4 (9%) | 0 | |
| Isolated craniofacial features (according to Loeys et at) | 5 (11%) | 8 (21%) | |
| Cardiovascular and systemic features | 36 (80%) | 22 (58%) | |
| Variable | Syndrome Group | Loeys-Dietz Group by Causative Genes | |||||
|---|---|---|---|---|---|---|---|
| Loeys-Dietz | Marfan | p | TGFBR1 | TGFBR2 | SMAD3 | p | |
| Total number of individuals | 83 | 83 | 26 | 40 | 17 | ||
| Age at initial contact (years) | 34 ± 18 | 34 ± 18 | 0.873 | 34 ± 17 | 34 ± 18 | 35 ± 22 | 0.983 |
| Age at final contact (years) | 38 ± 18 | 40 ± 18 | 0.523 | 39 ± 17 | 38 ± 18 | 37 ± 21 | 0.898 |
| Male sex | 43 (52%) | 43 (52%) | 1.000 | 12 (46%) | 20 (50%) | 11 (65%) | 0.446 |
| Previous ischemic neurologic event | 6/80 (8%) | 4 (5%) | 0.538 | 2 (8%) | 3/39 (8%) | 1/15 (7%) | 1.000 |
| Atrial septal defect | 4 (5%) | 1/82 (1%) | 0.367 | 1 (4%) | 3 (8%) | 0 | 0.812 |
| History of patent ductus arteriosus | 7 (8%) | 0 | 0.014 | 1 (4%) | 6 (15%) | 0 | 0.178 |
| Bicuspid aortic valve | 5 (6%) | 0 | 0.059 | 1 (4%) | 2 (5%) | 2 (12%) | 0.591 |
| Systemic score (points) | 3.5 ± 3.5 | 6.6 ± 3.2 | <0.001 | 3.4 ± 3.8 | 4.2 ± 3.7 | 2.1 ± 2.1 | 0.176 |
| Craniofacial severity index (points) | 1.3 ± 1.8 | 0 | <0.001 | 0.7 ± 1.4 | 1.6 ± 2 | 1.3 ± 1.7 | 0.227 |
| LV ejection fraction (%) | 62 ± 11 | 57 ± 12 | 0.019 | 62 ± 10 | 60 ± 12 | 68 ± 8 | 0.103 |
| Indexed LVESD (mm/m2) | 19 ± 6 | 19 ± 6 | 0.392 | 19 ± 6 | 19 ± 6 | 16 ± 3 | 0.345 |
| Indexed LVEDD (mm/m2) | 29 ± 8 | 29 ± 8 | 0.867 | 29 ± 7 | 30 ± 10 | 27 ± 3 | 0.754 |
| Indexed left atrial diameter (mm/m2) | 19 ± 5 | 19 ± 6 | 0.663 | 18 ± 5 | 20 ± 6 | 19 ± 5 | 0.493 |
| Aortic sinus dimensions at initial presentation | |||||||
| Diameter (cm) 1 | 3.6 ± 0.8 | 3.6 ± 0.8 | 0.701 | 3.3 ± 0.5 | 3.8 ± 0.7 | 3.4±1.3 | 0.102 |
| Z-score 1 | 2.2 ± 3.2 | 2.4 ± 2.9 | 0.761 | 1.8 ± 1.9 | 2.2 ± 3.6 | 2.5±3.6 | 0.675 |
| Aortic sinus dimensions at aortic surgery | |||||||
| Diameter (cm) | 4.8 ± 0.9 | 5.1 ± 0.9 | 0.208 | 4.7 ± 0.3 | 4.8 ± 0.8 | 5.0 | 0.688 |
| Z-score | 5.0 ± 3.2 | 6.7 ± 3.1 | 0.091 | 5.3 ± 1.5 | 4.8 ± 3.1 | 0.751 | |
| Moderate degree of MVR at baseline | 7/78 (9%) | 11/82 (13%) | 0.457 | 2/25 (8%) | 4/39 (10%) | 1/14 (7%) | 1.000 |
| MV prolapse | 28 (34%) | 48 (58%) | 0.003 | 5 (19%) | 17 (43%) | 6 (35%) | 0.152 |
| MV leaflet prolapse location (N) | 19 | 38 | 0.063 | 5 | 11 | 3 | 0.162 |
| Isolated anterior | 10 (53%) | 11 (29%) | 4 (80%) | 6 (55%) | 0 | ||
| Isolated posterior | 2 (11%) | 1 (3%) | 0 | 2 (18%) | 0 | ||
| Combined anterior and posterior | 7 (37%) | 26 (68%) | 1 (20%) | 3 (27%) | 3 (100%) | ||
| Tricuspid valve prolapse | 5/79 (6%) | 33/82 (40%) | <0.001 | 1/25 (4%) | 4 (10%) | 0/14 | 0.570 |
| Outcome Variables | Syndrome Group | Loeys-Dietz Group by Causative Genes | |||||
|---|---|---|---|---|---|---|---|
| Loeys-Dietz | Marfan | p | TGFBR1 | TGFBR2 | SMAD3 | p | |
| Number of individuals | 83 | 83 | 26 | 40 | 17 | ||
| Deaths of any cause | 8 (10%) | 6 (7%) | 0.781 | 2 (8%) | 4 (10%) | 2 (12%) | 1.000 |
| Deaths of any cause by age (years) 1 | 47 ± 22 | 50 ± 26 | 0.755 | 27–32 | 52 ± 20 | 26–81 | 0.570 |
| Deaths by cause | 0.867 | 0.829 | |||||
| Unknown | 4 (50%) | 2 (33%) | 1 (50%) | 2 (50%) | 1 (50%) | ||
| Aorta-related | 3 (38%) | 2 (33%) | 1 (50%) | 2 (50%) | 0 | ||
| Heart-valve related heart failure | 1 (13%) | 1 (17%) | 0 | 0 | 1 (50%) | ||
| Heart failure of unknown cause | 0 | 1 (17%) | 0 | 0 | 0 | ||
| Proximal aortic surgery | 33 (40%) | 37 (45%) | 0.637 | 12 (46%) | 18 (45%) | 3 (18%) | 0.119 |
| Proximal aortic surgery by age (years) 1 | 34 ± 14 | 35 ± 16 | 0.864 | 31 ± 10 | 36 ± 17 | 39 ± 7 | 0.512 |
| Proximal aortic surgery by indication/location | |||||||
| Urgent surgery (rupture/dissection) | 11 (30%) | 11 (30%) | 0.800 | 5 (42%) | 5 (28%) | 1 (33%) | 0.749 |
| Surgery involving arch 2 | 2 (6%) | 5 (14%) | 0.434 | 2 (17%) | 0 | 0 | 0.301 |
| Proximal aortic surgery by technique | 0.637 | 0.153 | |||||
| Aortic root reconstruction | 23 (70%) | 19 (51%) | 8 (67%) | 14 (78%) | 1 (33%) | ||
| Aortic root replacement (biological valve) | 3 (9%) | 3 (8%) | 0 | 2 (11%) | 1 (33%) | ||
| Aortic root replacement (mechanical valve) | 7 (21%) | 15 (41%) | 4 (33%) | 2 (11%) | 1 (33%) | ||
| Distal aortic repair | 8 (10%) | 7 (8%) | 1.000 | 1 (4%) | 7 (18%) | 0 | 0.085 |
| Distal aortic repair by age (years) 1 | 41 ± 15 | 38 ± 11 | 0.536 | 23 | 43 ± 14 | 0.272 | |
| Distal aortic repair by indication/technique | |||||||
| Urgent surgery (rupture/dissection) | 2 (25%) | 2 (29%) | 1.000 | 0 | 2 (29%) | 0 | 1.000 |
| Elective surgery (true or false lumen expansion) | 6 (75%) | 5 (71%) | 1 (100%) | 5 (71%) | 0 | ||
| Endovascular (versus surgical) | 0 | 2 (29%) | 0.200 | 0 | 0 | 0 | |
| Distal aortic repair by location | 0.322 | 0.500 | |||||
| Isolated thoracic aortic repair 3 | 4 (50%) | 4 (57%) | 0.793 | 0 | 4 (57%) | 0 | |
| Thoracoabdominal repair | 1 (13%) | 2 (29%) | 0 | 1 (14%) | 0 | ||
| Isolated abdominal aortic repair | 3 (38%) | 1 (14%) | 1 (100%) | 2 (29%) | 0 | ||
| Mitral valve surgery | 6 (7%) | 14 (17%) | 0.093 | 0 | 3 (8%) | 3 (18%) | 0.063 |
| Mitral valve surgery by age (years) 1 | 47 ± 15 | 40 ± 14 | 0.239 | 21-50 | 50–65 | 0.077 | |
| Mitral valve surgery by indication | |||||||
| Urgent surgery | 0 | 1 (7%) | 1.000 | 0 | 0 | 0 | |
| Mitral valve surgery by technique | 0.482 | 1.000 | |||||
| Reconstruction | 3 (50%) | 9 (64%) | 0 | 1 (33%) | 2 (67%) | ||
| Biological valve prosthesis | 1 (17%) | 0 | 0 | 1 (33%) | 0 | ||
| Mechanical valve prosthesis | 2 (33%) | 5 (36%) | 0 | 1 (33%) | 1 (33%) | ||
| Number of events/individual | 0.449 | 0.952 | |||||
| None | 44 (53%) | 41 (49%) | 14 (54%) | 20 (50%) | 10 (59%) | ||
| One | 28 (34%) | 24 (29%) | 10 (39%) | 12 (30%) | 6 (35%) | ||
| Two | 7 (8%) | 13 (16%) | 1 (4%) | 5 (13%) | 1 (6%) | ||
| Three | 3 (4%) | 5 (6%) | 1 (4%) | 2 (5%) | 0 | ||
| Four | 1 (1%) | 0 | 0 | 1 (3%) | 0 | ||
| First event in all individuals | 39 (47%) | 42 (51%) | 0.756 | 12 (46%) | 20 (50%) | 7 (41%) | 0.845 |
| First event by age (years) 1 | 36 ± 14 | 36 ± 16 | 0.977 | 31 ± 10 | 37 ± 16 | 45 ± 13 | 0.141 |
| First event by type | 0.872 | 0.020 | |||||
| Death | 2 (5%) | 2 (5%) | 0 | 1 (5%) | 1 (14%) | ||
| Proximal aortic surgery | 31 (80%) | 36 (86%) | 12 (100%) | 16 (80%) | 3 (43%) | ||
| Distal aortic intervention | 2 (5%) | 1 (2%) | 0 | 2 (10%) | 0 | ||
| Mitral valve surgery | 4 (10%) | 3 (7%) | 0 | 1 (5%) | 3 (43%) | ||
| First procedure by | 37 | 40 | 12 | 19 | 6 | ||
| Combined (versus isolated) procedure | 0 | 4 (10%) | 0.116 | 0 | 0 | 0 | |
| Second event after non-lethal first event | 11/37 (30%) | 18/40 (45%) | 0.239 | 2/12 (17%) | 8/19 (42%) | 1/6 (17%) | 0.299 |
| Second event by age (years) 1 | 46 ± 19 | 40 ± 15 | 0.387 | 23–32 | 47 ± 15 | 81 | 0.103 |
| Second event by type | 0.006 | 0.106 | |||||
| Death | 3 (27%) | 1 (6%) | 1 (50%) | 1 (13%) | 1 (100%) | ||
| Proximal aortic surgery | 2 (18%) | 1 (6%) | 0 | 2 (25%) | 0 | ||
| Distal aortic repair | 6 (55%) | 6 (33%) | 1 (50%) | 5 (63%) | 0 | ||
| Mitral valve surgery | 0 | 10 (56%) | 0 | 0 | 0 | ||
| Distal aortic repair by age (years) | 41 ± 15 | 38 ± 11 | 0.536 | 23 | 43 ± 14 | 41 ± 15 | 0.272 |
| Distal aortic repair as second event by indication | 0.318 | 1.000 | |||||
| True aneurysm | 3 (50%) | 0 | 1 (100%) | 2 (40%) | |||
| False lumen expansion after type A dissection | 2 (33%) | 4 (67%) | 0 | 2 (40%) | |||
| False lumen expansion of type-B dissection | 1 (17%) | 2 (33%) | 0 | 1 (20%) | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mühlstädt, K.; De Backer, J.; von Kodolitsch, Y.; Kutsche, K.; Muiño Mosquera, L.; Brickwedel, J.; Girdauskas, E.; Mir, T.S.; Mahlmann, A.; Tsilimparis, N.; et al. Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome. J. Clin. Med. 2019, 8, 2079. https://doi.org/10.3390/jcm8122079
Mühlstädt K, De Backer J, von Kodolitsch Y, Kutsche K, Muiño Mosquera L, Brickwedel J, Girdauskas E, Mir TS, Mahlmann A, Tsilimparis N, et al. Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome. Journal of Clinical Medicine. 2019; 8(12):2079. https://doi.org/10.3390/jcm8122079
Chicago/Turabian StyleMühlstädt, Kristina, Julie De Backer, Yskert von Kodolitsch, Kerstin Kutsche, Laura Muiño Mosquera, Jens Brickwedel, Evaldas Girdauskas, Thomas S. Mir, Adrian Mahlmann, Nikolaos Tsilimparis, and et al. 2019. "Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome" Journal of Clinical Medicine 8, no. 12: 2079. https://doi.org/10.3390/jcm8122079
APA StyleMühlstädt, K., De Backer, J., von Kodolitsch, Y., Kutsche, K., Muiño Mosquera, L., Brickwedel, J., Girdauskas, E., Mir, T. S., Mahlmann, A., Tsilimparis, N., Staebler, A., Schoof, L., Seidel, H., Berger, J., Bernhardt, A. M., Blankenberg, S., Kölbel, T., Detter, C., Szöcs, K., & Kaemmerer, H. (2019). Case-matched Comparison of Cardiovascular Outcome in Loeys-Dietz Syndrome versus Marfan Syndrome. Journal of Clinical Medicine, 8(12), 2079. https://doi.org/10.3390/jcm8122079