Baseline High-Resolution CT Findings Predict Acute Exacerbation of Idiopathic Pulmonary Fibrosis: German and Japanese Cohort Study

 , , , , and
, , , , and
Abstract
1. Introduction
2. Methods
2.1. Subjects
2.2. Definition of AE-IPF
2.3. Pulmonary Function Tests
2.4. Radiographic Assessments
2.5. Statistical Analysis
3. Results
3.1. Patients’ Characteristics
3.2. Inter-Observer Agreement
3.3. Correlation between HRCT Findings
3.4. HRCT Findings and Pulmonary Function
3.5. HRCT According to Ethnicity
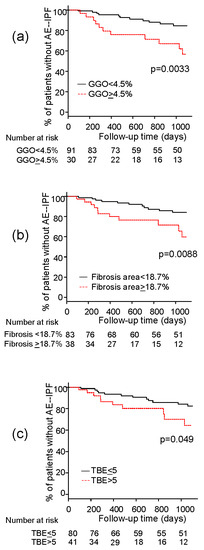
3.6. Association between HRCT Findings and Risk of AE-IPF
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AE-IPF | acute exacerbation of idiopathic pulmonary fibrosis |
| BAL | bronchoalveolar lavage |
| CPFE | combined pulmonary fibrosis and emphysema |
| DLco | diffusing capacity of carbon monoxide |
| FVC | forced vital capacity |
| GGO | ground glass opacity |
| HRCT | high-resolution computed tomography |
| IPF | idiopathic pulmonary fibrosis |
| ROC | receiver-operating characteristic |
| VC | vital capacity |
References
- Kim, D.S.; Park, J.H.; Park, B.K.; Lee, J.S.; Nicholson, A.G.; Colby, T. Acute exacerbation of idiopathic pulmonary fibrosis: Frequency and clinical features. Eur. Respir. J. 2006, 27, 143–150. [Google Scholar] [CrossRef]
- Akira, M.; Kozuka, T.; Yamamoto, S.; Sakatani, M. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 372–378. [Google Scholar] [CrossRef]
- Song, J.W.; Hong, S.B.; Lim, C.M.; Koh, Y.; Kim, D.S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 2011, 37, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Porretta, M.A.; Bargagli, E.; Sergiacomi, G.; Zompatori, M.; Sverzellati, N.; Taglieri, A.; Mezzasalma, F.; Rottoli, P.; Saltini, C.; et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: A 3-year prospective study. Eur. Respir. J. 2012, 40, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Richeldi, L.; Kim, D.S.; Taniguchi, H.; Tschoepe, I.; Luisetti, M.; Roman, J.; Tino, G.; Schlenker-Herceg, R.; Hallmann, C.; et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1601339. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Taniguchi, H.; Katsuta, T.; Kataoka, K.; Kimura, T.; Nishiyama, O.; Sakamoto, K.; Johkoh, T.; Nishimura, M.; Ono, K.; et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2010, 27, 103–110. [Google Scholar]
- Kondoh, Y.; Taniguchi, H.; Ebina, M.; Azuma, A.; Ogura, T.; Taguchi, Y.; Suga, M.; Takahashi, H.; Nakata, K.; Sugiyama, Y.; et al. Risk factors for acute exacerbation of idiopathic pulmonary fibrosis-extended analysis of pirfenidone trial in Japan. Respir. Investig. 2015, 53, 271–278. [Google Scholar] [CrossRef]
- Reichmann, W.M.; Yu, Y.F.; Macaulay, D.; Wu, E.Q.; Nathan, S.D. Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm. Med. 2015, 15, 167. [Google Scholar] [CrossRef]
- Judge, E.P.; Fabre, A.; Adamali, H.I.; Egan, J.J. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 40, 93–100. [Google Scholar] [CrossRef]
- Rev, E.; Med, R.; Campo, I.; Zorzetto, M.; Bonella, F. Facts and promises on lung biomarkers in interstitial lung diseases. Expert Rev. Respir. Med. 2015, 9, 437–457. [Google Scholar]
- Lynch, D.A.; Godwin, J.D.; Safrin, S.; Starko, K.M.; Hormel, P.; Brown, K.K.; Raghu, G.; King, T.E., Jr.; Bradford, W.Z.; Schwartz, D.A.; et al. High-resolution computed tomography in idiopathic pulmonary fibrosis: Diagnosis and prognosis. Am. J. Respir. Crit. Care Med. 2005, 172, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Sumikawa, H.; Johkoh, T.; Colby, T.V.; Ichikado, K.; Suga, M.; Taniguchi, H.; Kondoh, Y.; Ogura, T.; Arakawa, H.; Fujimoto, K.; et al. Computed tomography findings in pathological usual interstitial pneumonia: Relationship to survival. Am. J. Respir. Crit. Care Med. 2008, 177, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Best, A.C.; Meng, J.; Lynch, A.M.; Bozic, C.M.; Miller, D.; Grunwald, G.K.; Lynch, D.A. Idiopathic pulmonary fibrosis: Physiologic tests, quantitative CT indexes, and CT visual scores as predictors of mortality. Radiology 2008, 246, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Natsuizaka, M.; Chiba, H.; Kuronuma, K.; Otsuka, M.; Kudo, K.; Mori, M.; Bando, M.; Sugiyama, Y.; Takahashi, H. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am. J. Respir. Crit. Care Med. 2014, 190, 773–779. [Google Scholar] [CrossRef]
- Taniguchi, H.; Xu, Z.; Azuma, A.; Inoue, Y.; Li, H.; Fujimoto, T.; Bailes, Z.; Schlenker-Herceg, R.; Kim, D.S. Subgroup analysis of Asian patients in the INPULSIS® trials of nintedanib in idiopathic pulmonary fibrosis. Respirology 2016, 2016, 1–6. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar] [CrossRef]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E., Jr.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Idiopathic Pulmonary Fibrosis Clinical Research Network Investigators. Acute Exacerbations of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef]
- Renzetti, A.D., Jr. Standardization of Spirometry, 1994 Update. American Thoracic Society. Am. J. Respir. Crit. Care Med. 1995, 152, 1107–1136. [Google Scholar]
- Hansell, D.M.; Bankier, A.A.; MacMahon, H.; McLoud, T.C.; Müller, N.L.; Remy, J. Fleischner Society: Glossary of terms for thoracic imaging. Radiology 2008, 246, 697–722. [Google Scholar] [CrossRef]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.A.; Müller, N.L.; Flint, J.; Wright, J.L.; Churg, A. Idiopathic pulmonary fibrosis: Spectrum of high-resolution CT findings. Am. J. Roentgenol. 2005, 185, 1531–1539. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society. Diagnosis of Idiopathic Pulmonary Fibrosis An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Med. Crit. Care 2018, 198. [Google Scholar] [CrossRef]
- Mai, C.; Verleden, S.E.; McDonough, J.E.; Willems, S.; De Wever, W.; Coolen, J.; Dubbeldam, A.; Van Raemdonck, D.E.; Verbeken, E.K.; Verleden, G.M.; et al. Thin-Section CT Features of Idiopathic Pulmonary Fibrosis Correlated with Micro-CT and Histologic Analysis. Radiology 2017, 283, 252–263. [Google Scholar] [CrossRef]
- Suzuki, H.; Sekine, Y.; Yoshida, S.; Suzuki, M.; Shibuya, K.; Yonemori, Y.; Hiroshima, K.; Nakatani, Y.; Mizuno, S.; Takiguchi, Y.; et al. Risk of acute exacerbation of interstitial pneumonia after pulmonary resection for lung cancer in patients with idiopathic pulmonary fibrosis based on preoperative high-resolution computed tomography. Surg. Today 2011, 41, 914–921. [Google Scholar] [CrossRef]
- Cottin, V.; Nunes, H.; Brillet, P.Y.; Delaval, P.; Devouassoux, G.; Tillie-Leblond, I.; Israel-Biet, D.; Court-Fortune, I.; Valeyre, D.; Cordier, J.F. Groupe d’Etude et de Recherche sur les Maladies Orphelines Pulmonaires (GERM O P). Combined pulmonary fibrosis and emphysema: A distinct underrecognised entity. Eur. Respir. J. 2005, 26, 586–593. [Google Scholar] [CrossRef]
- Sugino, K.; Nakamura, Y.; Ito, T.; Isshiki, T.; Sakamoto, S.; Homma, S. Comparison of clinical characteristics and outcomes between combined pulmonary fibrosis and emphysema associated with usual interstitial pneumonia pattern and non-usual interstitial pneumonia. Sarcoidosis Vasc. Diffus. Lung Dis. 2015, 32, 129–137. [Google Scholar]
- Fernández Pérez, E.R.; Daniels, C.E.; Schroeder, D.R.; St Sauver, J.; Hartman, T.E.; Bartholmai, B.J.; Yi, E.S.; Ryu, J.H. Incidence, Prevalence, and Clinical Course of Idiopathic Pulmonary Fibrosis. Chest 2010, 137, 129–137. [Google Scholar] [CrossRef]
- Peljto, A.L.; Selman, M.; Kim, D.S.; Murphy, E.; Tucker, L.; Pardo, A.; Lee, J.S.; Ji, W.; Schwarz, M.I.; Yang, I.V.; et al. The MUC5B Promoter Polymorphism Is Associated With Idiopathic Pulmonary Fibrosis in a Mexican Cohort but Is Rare Among Asian Ancestries. Chest. 2015, 147, 460. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.N.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Horimasu, Y.A.; Ohshimo, S.H.; Bonella, F.R.; Tanaka, S.O.; Ishikawa, N.O.; Hattori, N.O.; Kohno, N.; Guzman, J.; Costabel, U. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology 2015, 2014, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Costabel, U.; Inoue, Y.; Richeldi, L.; Collard, H.R.; Tschoepe, I.; Stowasser, S.; Azuma, A. Efficacy of Nintedanib in Idiopathic Pulmonary Fibrosis across Prespecified Subgroups in INPULSIS. Am. J. Respir. Crit. Care Med. 2016, 193, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Johannson, K.A.; Vittinghoff, E.; Lee, K.; Balmes, J.R.; Ji, W.; Kaplan, G.G.; Kim, D.S.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur. Respir. J. 2014, 43, 1124–1131. [Google Scholar] [CrossRef]
- Rosas, I.O.; Yao, J.; Avila, N.A.; Chow, C.K.; Gahl, W.A.; Gochuico, B.R. Automated quantification of high-resolution CT scan findings in individuals at risk for pulmonary fibrosis. Chest 2011, 140, 1590–1597. [Google Scholar] [CrossRef]
- Collard, H.R.; Yow, E.; Richeldi, L.; Anstrom, K.J.; Glazer, C.; Schwarz, M.; IPFnet investigators. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir. Res. 2013, 14, 1. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| All | German | Japanese | p Value | Missing Values | |
|---|---|---|---|---|---|
| Number of the subjects | 121 | 54 | 67 | ||
| Age (years) | 68.5 ±7.6 | 68.1 ± 7.6 | 68.9 ± 7.5 | NS | 0 |
| Gender (male/female) | 98/23 | 41/13 | 57/10 | NS | 0 |
| Smoking (Cu or Ex/Non) | 86/30 | 33/16 | 53/14 | NS | 5(4.1%) |
| VC (percent predicted) | 75.0 ± 16.5 | 74.7 ±14.9 | 75.3 ± 18.1 | NS | 0 |
| DLCO (percent predicted) | 45.5 ± 15.5 | 45.8 ±17.3 | 45.4 ± 14.2 | NS | 6(5.0%) |
| Use of corticosteroid (yes/no) | 57/64 | 41/13 | 16/51 | <0.01 | 0 |
| Use of immunosuppressive agent (yes/no) | 30/91 | 27/27 | 3/64 | <0.01 | 0 |
| HRCT-Findings | German Patients | Japanese Patients | p Value |
|---|---|---|---|
| GGO (%) | 2.92 ± 4.11 | 3.85 ± 5.12 | 0.015 |
| Fibrosis (%) | 16.60 ± 7.99 | 12.80 ± 6.68 | <0.01 |
| Emphysema (%) | 1.72 ± 3.63 | 4.02 ± 8.62 | 0.010 |
| Zones with traction bronchiectasis (n) | 5.19 ± 1.13 | 4.27 ± 1.61 | <0.01 |
| Variables | HR | 95%CI | p Value |
|---|---|---|---|
| Age (continuous) | 0.99 | 0.94–1.05 | 0.73 |
| Male sex | 0.87 | 0.29–2.57 | 0.80 |
| Positive smoking history | 0.97 | 0.36–2.64 | 0.96 |
| Pack-year | 0.99 | 0.97–1.01 | 0.19 |
| Japanese Ethnicity | 3.17 | 1.07–9.37 | 0.037 |
| %VC ≥ 74% (median) | 0.37 | 0.15–0.92 | 0.031 |
| %DLco ≥ 44% (median) | 0.51 | 0.21–1.23 | 0.13 |
| Use of corticosteroid | 1.76 | 0.76–4.08 | 0.19 |
| Use of immunosuppressive agent | 1.03 | 0.35–3.06 | 0.96 |
| Use of pirfenidone | 0.52 | 0.16–1.77 | 0.30 |
| GGO > 4.5% | 3.27 | 1.42–7.55 | 0.0055 |
| Fibrosis > 18.7% | 2.93 | 1.26–6.79 | 0.012 |
| Emphysema > 8.3% | 1.67 | 0.56–4.92 | 0.36 |
| Zones with traction bronchiectasis >5 (n) | 2.29 | 0.98–5.34 | 0.055 |
| Variables | HR | 95%CI | p Value |
|---|---|---|---|
| Age (continuous) | 0.97 | 0.91–1.03 | 0.29 |
| Male sex | 0.59 | 0.18–1.89 | 0.37 |
| Japanese Ethnicity | 4.59 | 1.50–13.98 | 0.0074 |
| %VC > 74% (median) | 0.55 | 0.21–1.43 | 0.22 |
| GGO > 4.5% | 2.81 | 1.13–6.96 | 0.026 |
| Fibrosis area > 18.7% | 2.68 | 1.06–6.76 | 0.037 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirano, C.; Ohshimo, S.; Horimasu, Y.; Iwamoto, H.; Fujitaka, K.; Hamada, H.; Kohno, N.; Komoto, D.; Awai, K.; Shime, N.; et al. Baseline High-Resolution CT Findings Predict Acute Exacerbation of Idiopathic Pulmonary Fibrosis: German and Japanese Cohort Study. J. Clin. Med. 2019, 8, 2069. https://doi.org/10.3390/jcm8122069
Hirano C, Ohshimo S, Horimasu Y, Iwamoto H, Fujitaka K, Hamada H, Kohno N, Komoto D, Awai K, Shime N, et al. Baseline High-Resolution CT Findings Predict Acute Exacerbation of Idiopathic Pulmonary Fibrosis: German and Japanese Cohort Study. Journal of Clinical Medicine. 2019; 8(12):2069. https://doi.org/10.3390/jcm8122069
Chicago/Turabian StyleHirano, Chihiro, Shinichiro Ohshimo, Yasushi Horimasu, Hiroshi Iwamoto, Kazunori Fujitaka, Hironobu Hamada, Nobuoki Kohno, Daisuke Komoto, Kazuo Awai, Nobuaki Shime, and et al. 2019. "Baseline High-Resolution CT Findings Predict Acute Exacerbation of Idiopathic Pulmonary Fibrosis: German and Japanese Cohort Study" Journal of Clinical Medicine 8, no. 12: 2069. https://doi.org/10.3390/jcm8122069
APA StyleHirano, C., Ohshimo, S., Horimasu, Y., Iwamoto, H., Fujitaka, K., Hamada, H., Kohno, N., Komoto, D., Awai, K., Shime, N., Bonella, F., Guzman, J., Kühl, H., Costabel, U., & Hattori, N. (2019). Baseline High-Resolution CT Findings Predict Acute Exacerbation of Idiopathic Pulmonary Fibrosis: German and Japanese Cohort Study. Journal of Clinical Medicine, 8(12), 2069. https://doi.org/10.3390/jcm8122069