Main Oral Manifestations in Immune-Mediated and Inflammatory Rheumatic Diseases




Abstract
1. Introduction
2. Oral Manifestations in Rheumatic Diseases
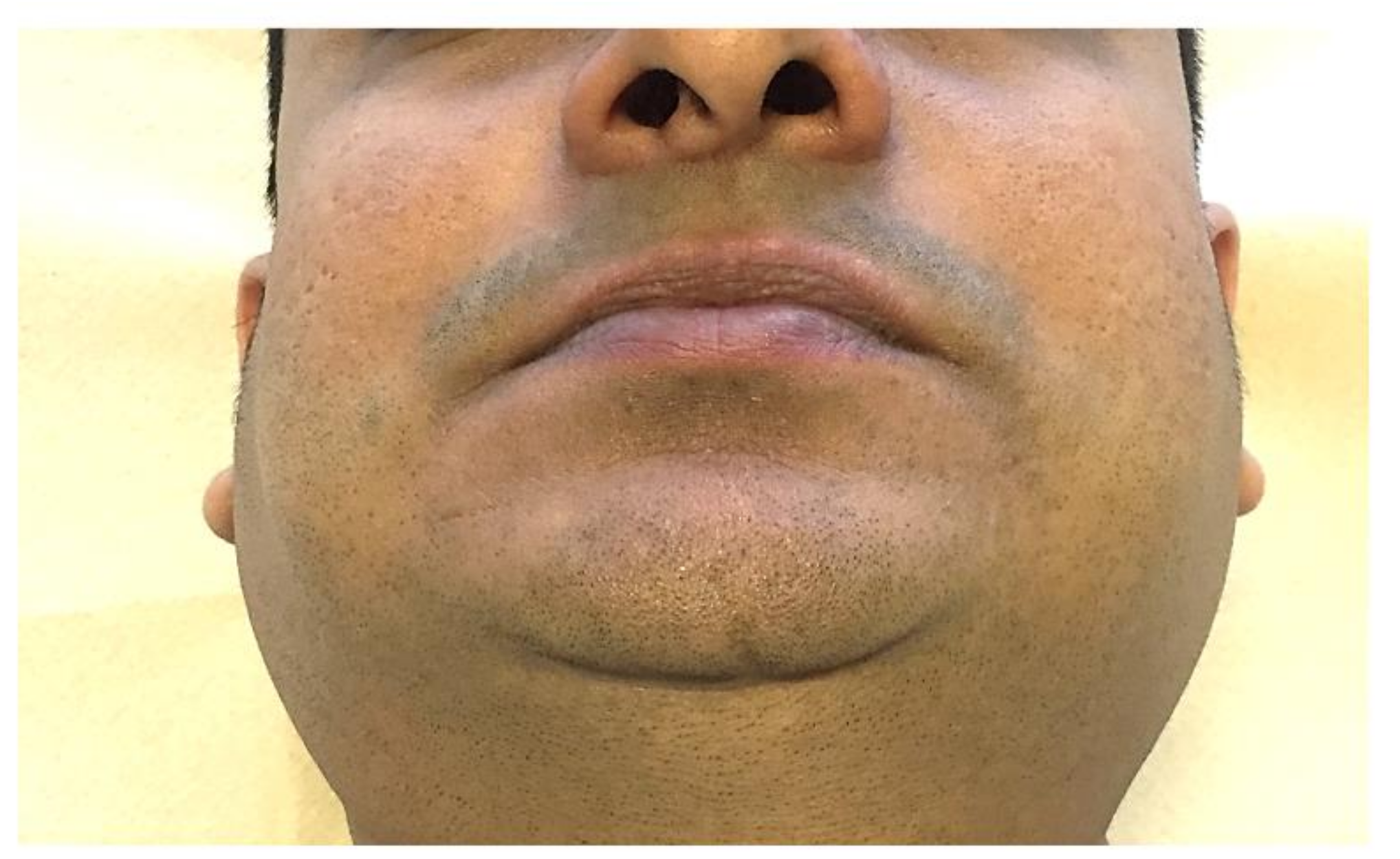
2.1. Microstomia/Microcheilia
2.2. Dysphagia
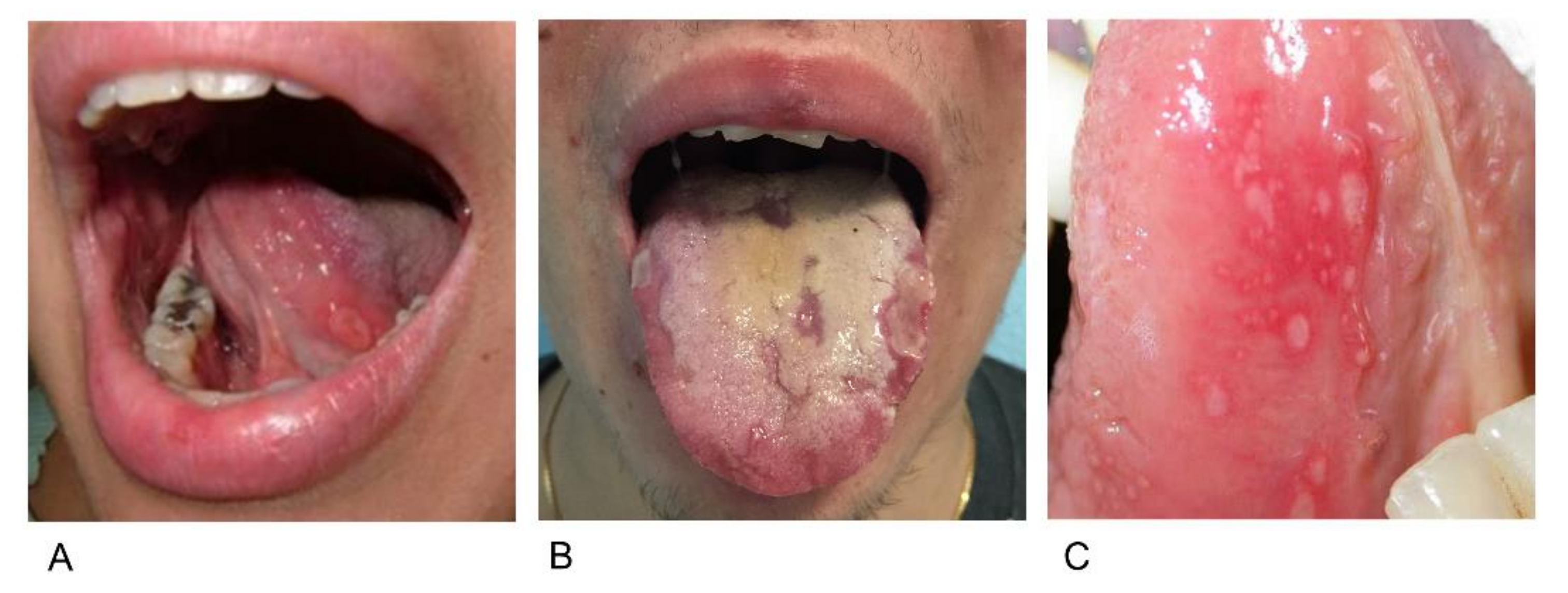
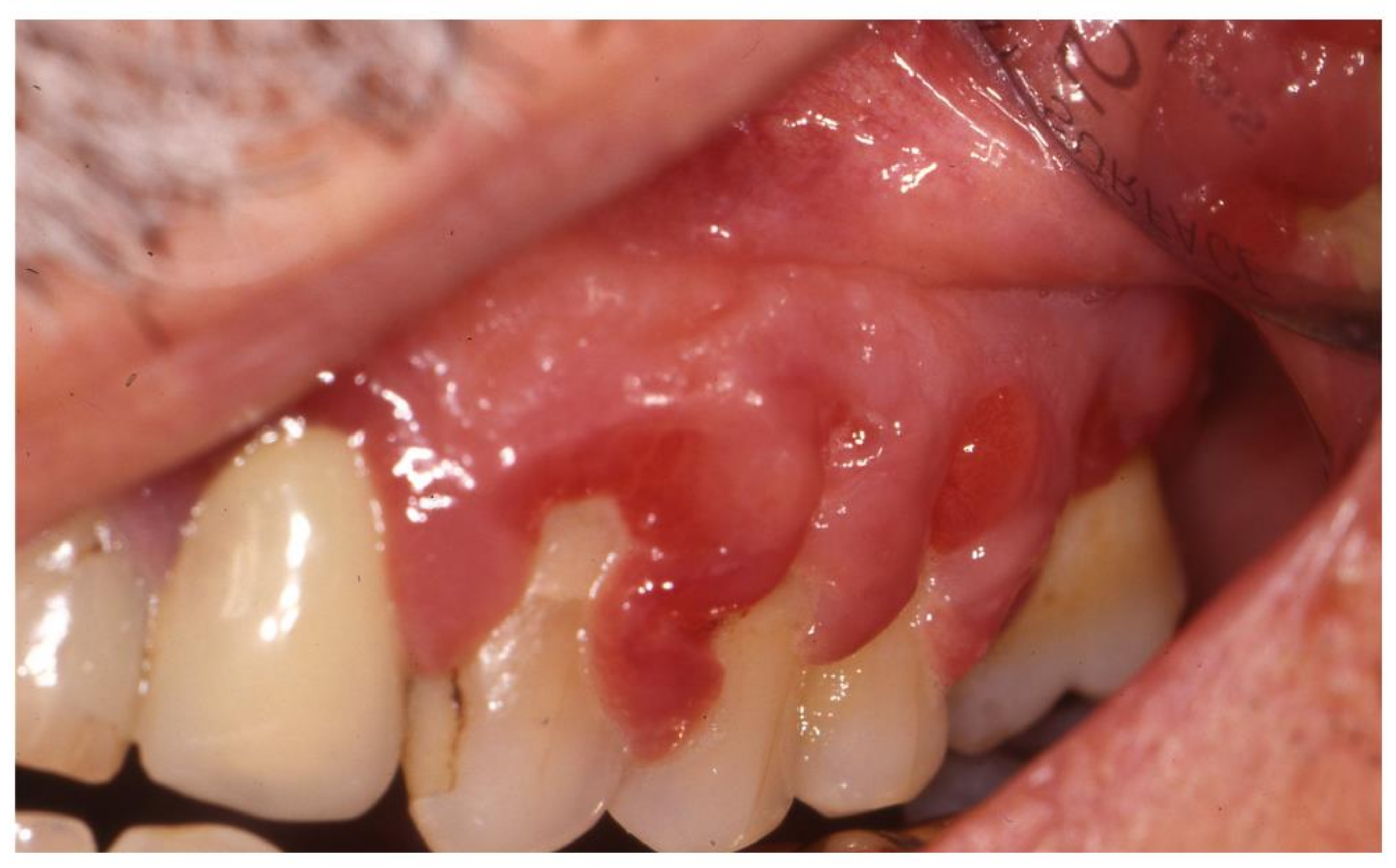
2.3. Aphthosis
2.4. Xerostomia
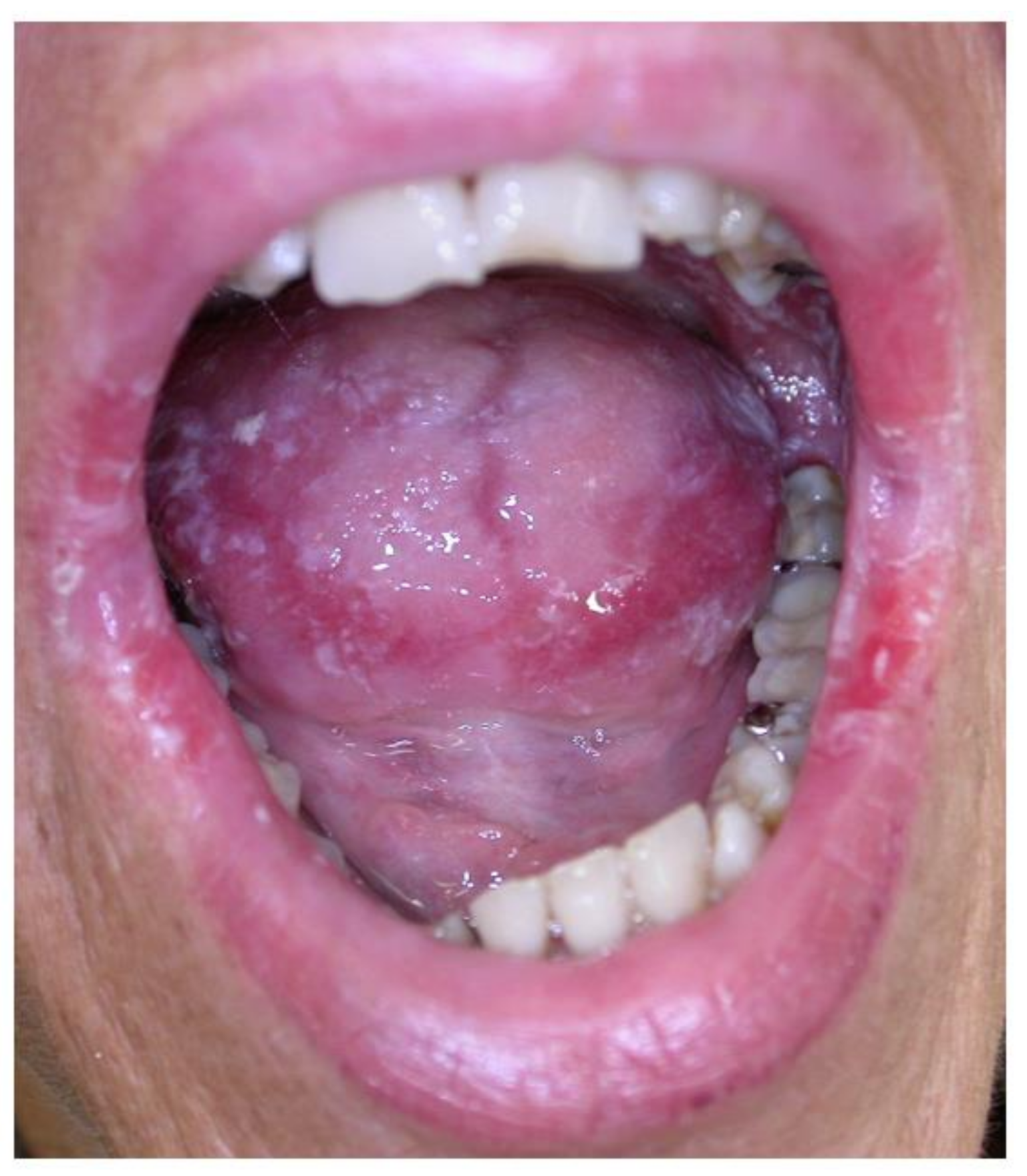
2.5. Gingival Overgrowth
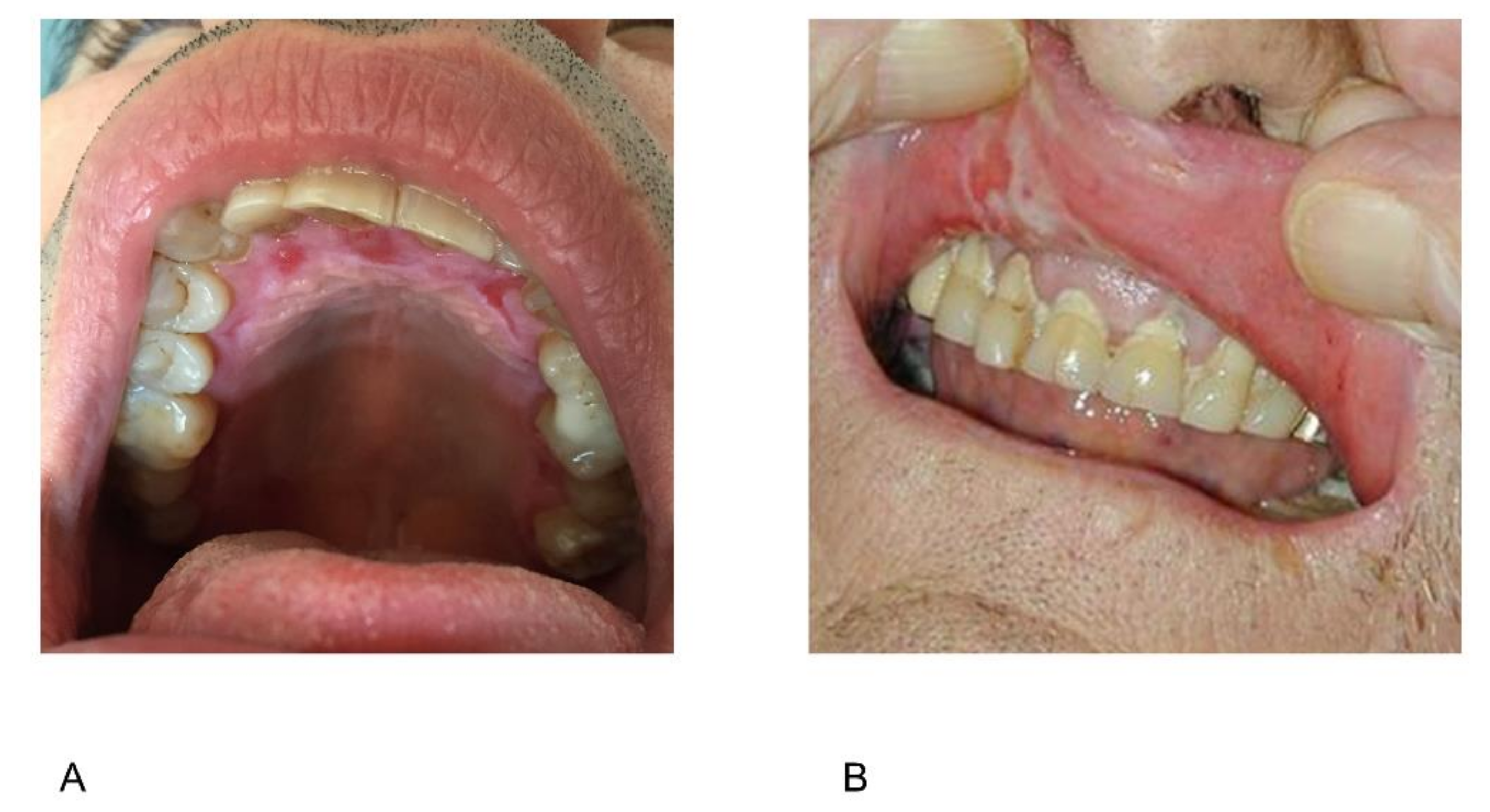
2.6. Periodontal Disease
2.7. Jaw Claudication
2.8. Temporomandibular Joint Involvement
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States- Part I. Arthritis Rheum. 2008, 58, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Khatibi, M.; Shakoorpour, A.H.; Jahromi, Z.M.; Ahmadzadeh, A. The prevalence of oral mucosal lesions and related factors in 188 patients with systemic lupus erythematosus. Lupus 2012, 21, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Kleinheinz, J.; Handschel, J.; Kruse-Losler, B.; Weingart, D.; Joos, U. Oral findings in three different groups of immunocompromised patients. J. Oral Pathol. Med. 2000, 29, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Mayes, M.D.; Lacey, J.V., Jr.; Beebe-Dimmer, J.; Gillespie, B.W.; Cooper, B.; Laing, T.J.; Schottenfeld, D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003, 48, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Nikpour, M.; Stevens, W.M.; Herrick, A.L.; Proudman, S.M. Epidemiology of systemic sclerosis. Best Pract. Res. Clin. Rheumatol. 2010, 24, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P. Systemic sclerosis: From pathogenesis to targeted therapy. Clin. Exp. Rheumatol. 2015, 33, S3–S7. [Google Scholar] [PubMed]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic sclerosis. Nat. Rev. Dis. Primers 2015, 23, 15002. [Google Scholar] [CrossRef]
- Preliminary Criteria for the Classification of Systemic Sclerosis (Scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980, 23, 581–590. [Google Scholar]
- Leader, D.; Papas, A.; Finkelman, M. A survey of dentists’ knowledge and attitudes with respect to the treatment of scleroderma patients. J. Clin. Rheumatol. 2014, 20, 189–194. [Google Scholar] [CrossRef]
- Yuen, H.K.; Marlow, N.M.; Reed, S.G.; Mahoney, S.; Summerlin, L.M.; Leite, R.; Slate, E.; Silver, R.M. Effect of orofacial exercises on oral aperture in adults with systemic sclerosis. Disabil. Rehabil. 2012, 34, 84–89. [Google Scholar] [CrossRef]
- Pischon, N.; Pischon, T.; Kroger, J.; Gulmez, E.; Kleber, B.M.; Bernimoulin, J.P.; Landau, H.; Brinkmann, P.G.; Schlattmann, P.; Zernicke, J.; et al. Association among rheumatoid arthritis, oral hygiene, and periodontitis. J. Periodontol. 2008, 79, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Torkzaban, P.; Hjiabadi, T.; Basiri, Z.; Poorolajal, J. Effect of rheumatoid arthritis on periodontitis: A historical cohort study. J. Periodontal Implant Sci. 2012, 42, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Scrivani, S.J.; Keith, D.A.; Kaban, L.B. Temporomandibular disorders. N. Engl. J. Med. 2008, 359, 2693–2705. [Google Scholar] [CrossRef] [PubMed]
- Goupille, P.; Fouquet, B.; Cotty, P.; Goga, D.; Mateu, J.; Valat, J.P. The temporomandibular joint in rheumatoid arthritis. Correlations between clinical and computed tomography features. J. Rheumatol. 1990, 17, 1285–1291. [Google Scholar] [PubMed]
- Arabshahi, B.; Cron, R.Q. Temporomandibular joint arthritis in juvenile idiopathic arthritis: The forgotten joint. Curr. Opin. Rheumatol. 2006, 18, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Twilt, M.; Arends, L.R.; Cate, R.T.; van Suijlekom-Smit, L.W. Incidence of temporomandibular involvement in juvenile idiopathic arthritis. Scand. J. Rheumatol. 2007, 36, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, S.V.; de Carvalho, F.R.; Boggio, P.; Sotto, M.N.; Vilela, M.A.; Rivitti, E.A.; Nico, M.M. Lupus erythematosus: Clinical and histopathological study of oral manifestations and immunohistochemical profile of the inflammatory infiltrate. J. Cutan. Pathol. 2007, 34, 558–564. [Google Scholar] [CrossRef]
- Correa, J.D.; Calderaro, D.C.; Ferreira, G.A.; Mendonca, S.M.; Fernandes, G.R.; Xiao, E.; Teixeira, A.L.; Leys, E.J.; Graves, D.T.; Silva, T.A. Subgingival microbiota dysbiosis in systemic lupus erythematosus: Association with periodontal status. Microbiome 2017, 5, 34. [Google Scholar] [CrossRef]
- Rutter-Locher, Z.; Smith, T.O.; Giles, I.; Sofat, N. Association between Systemic Lupus Erythematosus and Periodontitis: A Systematic Review and Meta-analysis. Front. Immunol. 2017, 8, 1295. [Google Scholar] [CrossRef]
- Brito-Zeron, P.; Baldini, C.; Bootsma, H.; Bowman, S.J.; Jonsson, R.; Mariette, X.; Sivils, K.; Theander, E.; Tzioufas, A.; Ramos-Casals, M. Sjögren syndrome. Nat. Rev. Dis. Primers 2016, 2, 16047. [Google Scholar] [CrossRef]
- Thoua, N.M.; Bunce, C.; Brough, G.; Forbes, A.; Emmanuel, A.V.; Denton, C.P. Assessment of gastrointestinal symptoms in patients with systemic sclerosis in a UK tertiary referral centre. Rheumatology 2010, 49, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Veale, B.J.; Jablonski, R.Y.; Frech, T.M.; Pauling, J.D. Orofacial manifestations of systemic sclerosis. Br. Dent. J. 2016, 221, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Gadiparthi, C.; Hans, A.; Potts, K.; Ismail, M.K. Gastrointestinal and Hepatic Disease in the Inflammatory Myopathies. Rheum. Dis. Clin. N. Am. 2018, 44, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Al-Otaibi, L.M.; Porter, S.R.; Poate, T.W. Behcet’s disease: A review. J. Dent. Res. 2005, 84, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; De Virgilio, A.; Ralli, M.; Ciofalo, A.; Mancini, P.; Attanasio, G.; de Vincentiis, M.; Lambiase, A. Behcet’s disease: New insights into pathophysiology, clinical features and treatment options. Autoimmun. Rev. 2018, 17, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Paraskevas, K.I.; Boumpas, D.T.; Vrentzos, G.E.; Mikhailidis, D.P. Oral and ocular/orbital manifestations of temporal arteritis: A disease with deceptive clinical symptoms and devastating consequences. Clin. Rheumatol. 2007, 26, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, M. Strawberry Gingivitis in Granulomatosis with Polyangiitis. N. Engl. J. Med. 2017, 377, 2073. [Google Scholar] [CrossRef]
- Hernandez, G.; Serrano, C.; Porras, L.; Lopez-Pintor, R.; Rubio, L.; Yanes, J. Strawberry-like gingival tumor as the first clinical sign of Wegener’s granulomatosis. J. Periodontol. 2008, 79, 1297–1303. [Google Scholar] [CrossRef]
- Alantar, A.; Cabane, J.; Hachulla, E.; Princ, G.; Ginisty, D.; Hassin, M.; Sorel, M.; Maman, L.; Pilat, A.; Mouthon, L. Recommendations for the care of oral involvement in patients with systemic sclerosis. Arthritis Care Res. 2011, 63, 1126–1133. [Google Scholar] [CrossRef]
- Derk, C.T.; Rasheed, M.; Spiegel, J.R.; Jimenez, S.A. Increased incidence of carcinoma of the tongue in patients with systemic sclerosis. J. Rheumatol. 2005, 32, 637–641. [Google Scholar]
- Katada, Y.; Tanaka, T. Images in clinical medicine. Lingual Raynaud’s phenomenon. N. Engl. J. Med. 2012, 366, e12. [Google Scholar] [CrossRef] [PubMed]
- Launay, D.; Sitbon, O.; Hachulla, E.; Mouthon, L.; Gressin, V.; Rottat, L.; Clerson, P.; Cordier, J.F.; Simonneau, G.; Humbert, M. Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era. Ann. Rheum. Dis. 2013, 72, 1940–1946. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger, J.P.; Khanna, D.; Gerry Coghlan, J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur. Respir. Rev. 2013, 22, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Del Papa, N.; Caviggioli, F.; Sambataro, D.; Zaccara, E.; Vinci, V.; Di Luca, G.; Parafioriti, A.; Armiraglio, E.; Maglione, W.; Polosa, R.; et al. Autologous fat grafting in the treatment of fibrotic perioral changes in patients with systemic sclerosis. Cell Transpl. 2015, 24, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Maria, A.T.; Maumus, M.; Le Quellec, A.; Jorgensen, C.; Noel, D.; Guilpain, P. Adipose-Derived Mesenchymal Stem Cells in Autoimmune Disorders: State of the Art and Perspectives for Systemic Sclerosis. Clin. Rev. Allergy Immunol. 2017, 52, 234–259. [Google Scholar] [CrossRef] [PubMed]
- Desbois, A.C.; Cacoub, P. Systemic sclerosis: An update in 2016. Autoimmun. Rev. 2016, 15, 417–426. [Google Scholar] [CrossRef]
- De Bleecker, J.L.; De Paepe, B.; Aronica, E.; de Visser, M.; Group, E.M.M.B.S.; Amato, A.; Aronica, E.; Benveniste, O.; De Bleecker, J.; de Boer, O.; et al. 205th ENMC International Workshop: Pathology diagnosis of idiopathic inflammatory myopathies part II 28–30 March 2014, Naarden, The Netherlands. Neuromuscul. Disord. 2015, 25, 268–272. [Google Scholar] [CrossRef]
- Milone, M. Diagnosis and Management of Immune-Mediated Myopathies. Mayo Clin. Proc. 2017, 92, 826–837. [Google Scholar] [CrossRef]
- Lundberg, I.E.; de Visser, M.; Werth, V.P. Classification of myositis. Nat. Rev. Rheumatol. 2018, 14, 269–278. [Google Scholar] [CrossRef]
- Iaccarino, L.; Ghirardello, A.; Bettio, S.; Zen, M.; Gatto, M.; Punzi, L.; Doria, A. The clinical features, diagnosis and classification of dermatomyositis. J. Autoimmun. 2014, 48–49, 122–127. [Google Scholar] [CrossRef]
- Ertekin, C.; Secil, Y.; Yuceyar, N.; Aydogdu, I. Oropharyngeal dysphagia in polymyositis/dermatomyositis. Clin. Neurol. Neurosurg. 2004, 107, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Mugii, N.; Hasegawa, M.; Matsushita, T.; Hamaguchi, Y.; Oohata, S.; Okita, H.; Yahata, T.; Someya, F.; Inoue, K.; Murono, S.; et al. Oropharyngeal Dysphagia in Dermatomyositis: Associations with Clinical and Laboratory Features Including Autoantibodies. PLoS ONE 2016, 11, e0154746. [Google Scholar] [CrossRef]
- Yazici, Y.; Kagen, L.J. Clinical presentation of the idiopathic inflammatory myopathies. Rheum. Dis. Clin. N. Am. 2002, 28, 823–832. [Google Scholar] [CrossRef]
- Lundberg, I.E.; Dastmalchi, M. Possible pathogenic mechanisms in inflammatory myopathies. Rheum. Dis. Clin. N. Am. 2002, 28, 799–822. [Google Scholar] [CrossRef]
- Oddis, C.V.; Reed, A.M.; Aggarwal, R.; Rider, L.G.; Ascherman, D.P.; Levesque, M.C.; Barohn, R.J.; Feldman, B.M.; Harris-Love, M.O.; Koontz, D.C.; et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheum. 2013, 65, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.H.; Rubin, A.D. Dysphagia due to inclusion body myositis: Case presentation and review of the literature. Ann. Otol. Rhinol. Laryngol. 2014, 123, 605–608. [Google Scholar] [CrossRef]
- Petri, M.; Orbai, A.M.; Alarcon, G.S.; Gordon, C.; Merrill, J.T.; Fortin, P.R.; Bruce, I.N.; Isenberg, D.; Wallace, D.J.; Nived, O.; et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2677–2686. [Google Scholar] [CrossRef]
- Hochberg, M.C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997, 40, 1725. [Google Scholar] [CrossRef]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Lopez-Labady, J.; Villarroel-Dorrego, M.; Gonzalez, N.; Perez, R.; Mata de Henning, M. Oral manifestations of systemic and cutaneous lupus erythematosus in a Venezuelan population. J. Oral. Pathol. Med. 2007, 36, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Schiodt, M. Oral manifestations of lupus erythematosus. Int. J. Oral Surg. 1984, 13, 101–147. [Google Scholar] [CrossRef]
- Gul, A. Behcet’s disease as an autoinflammatory disorder. Curr. Drug Targets Inflamm. Allergy 2005, 4, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, C.; Lopalco, G.; Iannone, F.; Vitale, A.; Talarico, R.; Galeazzi, M.; Lapadula, G.; Cantarini, L. Mucocutaneous Involvement in Behcet’s Disease: How Systemic Treatment Has Changed in the Last Decades and Future Perspectives. Mediat. Inflamm. 2015, 2015, 451675. [Google Scholar] [CrossRef] [PubMed]
- Hatemi, G.; Christensen, R.; Bang, D.; Bodaghi, B.; Celik, A.F.; Fortune, F.; Gaudric, J.; Gul, A.; Kotter, I.; Leccese, P.; et al. 2018 update of the EULAR recommendations for the management of Behcet’s syndrome. Ann. Rheum. Dis. 2018, 77, 808–818. [Google Scholar] [CrossRef]
- Lankarani, K.B.; Sivandzadeh, G.R.; Hassanpour, S. Oral manifestation in inflammatory bowel disease: A review. World J. Gastroenterol. 2013, 19, 8571–8579. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Zarkadas, M.; Anca, A.; Limeback, H. Oral manifestations of celiac disease: A clinical guide for dentists. J. Can. Dent. Assoc. 2011, 77, b39. [Google Scholar] [PubMed]
- Scully, C.; Hodgson, T.; Lachmann, H. Auto-inflammatory syndromes and oral health. Oral Dis. 2008, 14, 690–699. [Google Scholar] [CrossRef]
- Schmidt, E.; Zillikens, D. Pemphigoid diseases. Lancet 2013, 381, 320–332. [Google Scholar] [CrossRef]
- Kalantzis, A.; Marshman, Z.; Falconer, D.T.; Morgan, P.R.; Odell, E.W. Oral effects of low-dose methotrexate treatment. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2005, 100, 52–62. [Google Scholar] [CrossRef]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Ince, A.; Yazici, Y.; Hamuryudan, V.; Yazici, H. The frequency and clinical characteristics of methotrexate (MTX) oral toxicity in rheumatoid arthritis (RA): A masked and controlled study. Clin. Rheumatol. 1996, 15, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Visser, K.; van der Heijde, D. Optimal dosage and route of administration of methotrexate in rheumatoid arthritis: A systematic review of the literature. Ann. Rheum. Dis. 2009, 68, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Arnet, I.; Bernhardt, V.; Hersberger, K.E. Methotrexate intoxication: The Pharmaceutical Care process reveals a critical error. J. Clin. Pharm. Ther. 2012, 37, 242–244. [Google Scholar] [CrossRef]
- Shiroky, J.B.; Neville, C.; Esdaile, J.M.; Choquette, D.; Zummer, M.; Hazeltine, M.; Bykerk, V.; Kanji, M.; St-Pierre, A.; Robidoux, L.; et al. Low-dose methotrexate with leucovorin (folinic acid) in the management of rheumatoid arthritis. Results of a multicenter randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 1993, 36, 795–803. [Google Scholar] [CrossRef]
- Shea, B.; Swinden, M.V.; Ghogomu, E.T.; Ortiz, Z.; Katchamart, W.; Rader, T.; Bombardier, C.; Wells, G.A.; Tugwell, P. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. J. Rheumatol. 2014, 41, 1049–1060. [Google Scholar] [CrossRef]
- Konings, A.W.; Coppes, R.P.; Vissink, A. On the mechanism of salivary gland radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1187–1194. [Google Scholar] [CrossRef]
- Bowman, S.J.; Ibrahim, G.H.; Holmes, G.; Hamburger, J.; Ainsworth, J.R. Estimating the prevalence among Caucasian women of primary Sjögren’s syndrome in two general practices in Birmingham, UK. Scand. J. Rheumatol. 2004, 33, 39–43. [Google Scholar] [CrossRef]
- Reksten, T.R.; Jonsson, M.V. Sjögren’s syndrome: An update on epidemiology and current insights on pathophysiology. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26, 1–12. [Google Scholar] [CrossRef]
- Voulgarelis, M.; Ziakas, P.D.; Papageorgiou, A.; Baimpa, E.; Tzioufas, A.G.; Moutsopoulos, H.M. Prognosis and outcome of non-Hodgkin lymphoma in primary Sjögren syndrome. Medicine 2012, 91, 1–9. [Google Scholar] [CrossRef]
- Vitali, C.; Bombardieri, S.; Jonsson, R.; Moutsopoulos, H.M.; Alexander, E.L.; Carsons, S.E.; Daniels, T.E.; Fox, P.C.; Fox, R.I.; Kassan, S.S.; et al. Classification criteria for Sjögren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 2002, 61, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol. 2017, 69, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Burger-Calderon, R.; Madden, V.; Hallett, R.A.; Gingerich, A.D.; Nickeleit, V.; Webster-Cyriaque, J. Replication of oral BK virus in human salivary gland cells. J. Virol. 2014, 88, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, V.S.; Mattoo, H.; Deshpande, V.; Pillai, S.S.; Stone, J.H. IgG4-related disease. Annu. Rev. Pathol. 2014, 9, 315–347. [Google Scholar] [CrossRef] [PubMed]
- Umehara, H.; Okazaki, K.; Masaki, Y.; Kawano, M.; Yamamoto, M.; Saeki, T.; Matsui, S.; Yoshino, T.; Nakamura, S.; Kawa, S.; et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod. Rheumatol. 2012, 22, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Price, E.J.; Rauz, S.; Tappuni, A.R.; Sutcliffe, N.; Hackett, K.L.; Barone, F.; Granata, G.; Ng, W.F.; Fisher, B.A.; Bombardieri, M.; et al. The British Society for Rheumatology guideline for the management of adults with primary Sjögren’s Syndrome. Rheumatology 2017, 56, 1828. [Google Scholar] [CrossRef] [PubMed]
- Gawron, K.; Łazarz-Bartyzel, K.; Potempa, J.; Chomyszyn-Gajewska, M. Gingival fibromatosis: Clinical, molecular and therapeutic issues. Orphanet J. Rare Dis. 2016, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.J.; Jacobsson, L.T.H.; Westman, K.W.A.; Sturfelt, G.; Segelmark, M. Incidence and survival rates in Wegener’s granulomatosis, microscopic polyangiitis, Churg–Strauss syndrome and polyarteritis nodosa. Rheumatology 2009, 48, 1560–1565. [Google Scholar] [CrossRef]
- Pagnoux, C. Updates in ANCA-associated vasculitis. Eur. J. Rheumatol. 2016, 3, 122–133. [Google Scholar] [CrossRef]
- Bachmeyer, C.; Petitjean, B.; Testart, F.; Richecoeur, J.; Ammouri, W.; Blum, L. Lingual necrosis as the presenting sign of Wegener’s granulomatosis. Clin. Exp. Dermatol. 2006, 31, 321–322. [Google Scholar] [CrossRef]
- Guillevin, L.; Pagnoux, C.; Karras, A.; Khouatra, C.; Aumaître, O.; Cohen, P.; Maurier, F.; Decaux, O.; Ninet, J.; Gobert, P.; et al. Rituximab versus Azathioprine for Maintenance in ANCA-Associated Vasculitis. N. Engl. J. Med. 2014, 371, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; St Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Dreizen, S.; McCredie, K.B.; Keating, M.J.; Luna, M.A. Malignant gingival and skin “infiltrates” in adult leukemia. Oral Surg. Oral Med. Oral Pathol. 1983, 55, 572–579. [Google Scholar] [CrossRef]
- Paixao, C.G.; Sekiguchi, R.T.; Saraiva, L.; Pannuti, C.M.; Silva, H.T.; Medina-Pestana, J.; Romito, G.A. Gingival overgrowth among patients medicated with cyclosporin A and tacrolimus undergoing renal transplantation: A prospective study. J. Periodontol. 2011, 82, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, V.L.; Ramalho, H.J.; Cipullo, J.P.; Azoubel, R.; Burdmann, E.A. Comparison of azithromycin and oral hygiene program in the treatment of cyclosporine-induced gingival hyperplasia. Ren. Fail. 2007, 29, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ilgenli, T.; Atilla, G.; Baylas, H. Effectiveness of periodontal therapy in patients with drug-induced gingival overgrowth. Long-term results. J. Periodontol. 1999, 70, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Primers 2017, 3, 17038. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.M.; Vissink, A.; de Smit, M.J.; Westra, J.; Brouwer, E. Lessons to be learned from periodontitis. Curr. Opin. Rheumatol. 2013, 25, 241–247. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid factors: Clinical applications. Dis. Markers 2013, 35, 727–734. [Google Scholar] [CrossRef]
- Ortiz, P.; Bissada, N.F.; Palomo, L.; Han, Y.W.; Al-Zahrani, M.S.; Panneerselvam, A.; Askari, A. Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J. Periodontol. 2009, 80, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Erciyas, K.; Sezer, U.; Ustun, K.; Pehlivan, Y.; Kisacik, B.; Senyurt, S.Z.; Tarakcioglu, M.; Onat, A.M. Effects of periodontal therapy on disease activity and systemic inflammation in rheumatoid arthritis patients. Oral Dis. 2013, 19, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Savioli, C.; Ribeiro, A.C.; Fabri, G.M.; Calich, A.L.; Carvalho, J.; Silva, C.A.; Viana, V.S.; Bonfa, E.; Siqueira, J.T. Persistent periodontal disease hampers anti-tumor necrosis factor treatment response in rheumatoid arthritis. J. Clin. Rheumatol. 2012, 18, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, M.; MacDonald, D.; Baron, M.; Hudson, M.; Tatibouet, S.; Steele, R.; Gravel, S.; Mohit, S.; El Sayegh, T.; Pope, J.; et al. The Canadian Systemic Sclerosis Oral Health Study IV: Oral radiographic manifestations in systemic sclerosis compared with the general population. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2015, 120, 104–111. [Google Scholar] [CrossRef]
- Ozcelik, O.; Haytac, M.C.; Ergin, M.; Antmen, B.; Seydaoglu, G. The immunohistochemical analysis of vascular endothelial growth factors A and C and microvessel density in gingival tissues of systemic sclerosis patients: Their possible effects on gingival inflammation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2008, 105, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Hudson, M.; Dagenais, M.; Macdonald, D.; Gyger, G.; El Sayegh, T.; Pope, J.; Fontaine, A.; Masetto, A.; Matthews, D.; et al. Relationship Between Disease Characteristics and Oral Radiologic Findings in Systemic Sclerosis: Results From a Canadian Oral Health Study. Arthritis Care Res. 2016, 68, 673–680. [Google Scholar] [CrossRef]
- Isola, G.; Williams, R.C.; Lo Gullo, A.; Ramaglia, L.; Matarese, M.; Iorio-Siciliano, V.; Cosio, C.; Matarese, G. Risk association between scleroderma disease characteristics, periodontitis, and tooth loss. Clin. Rheumatol. 2017, 36, 2733–2741. [Google Scholar] [CrossRef]
- Scardina, G.A.; Ruggieri, A.; Messina, P. Periodontal disease and Sjögren syndrome: A possible correlation? Angiology 2010, 61, 289–293. [Google Scholar] [CrossRef]
- Schiodt, M.; Christensen, L.B.; Petersen, P.E.; Thorn, J.J. Periodontal disease in primary Sjögren’s syndrome. Oral Dis. 2001, 7, 106–108. [Google Scholar] [PubMed]
- Celenligil, H.; Eratalay, K.; Kansu, E.; Ebersole, J.L. Periodontal status and serum antibody responses to oral microorganisms in Sjögren’s syndrome. J. Periodontol. 1998, 69, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Ozcaka, O.; Alpoz, E.; Nalbantsoy, A.; Karabulut, G.; Kabasakal, Y. Clinical periodontal status and inflammatory cytokines in primary sjögren syndrome and rheumatoid artritis. J. Periodontol. 2018, 89, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Garcia-Porrua, C. Epidemiology of the vasculitides. Rheum. Dis. Clin. N. Am. 2001, 27, 729–749. [Google Scholar] [CrossRef]
- Salvarani, C.; Pipitone, N.; Versari, A.; Hunder, G.G. Clinical features of polymyalgia rheumatica and giant cell arteritis. Nat. Rev. Rheumatol. 2012, 8, 509–521. [Google Scholar] [CrossRef]
- Sheldon, C.A.; White, V.A.; Holland, S.P. Giant cell arteritis presenting with bilateral loss of vision and jaw pain: Reminder of a potentially devastating condition. J. Can. Dent. Assoc. 2011, 77, b55. [Google Scholar] [PubMed]
- Villiger, P.M.; Adler, S.; Kuchen, S.; Wermelinger, F.; Dan, D.; Fiege, V.; Butikofer, L.; Seitz, M.; Reichenbach, S. Tocilizumab for induction and maintenance of remission in giant cell arteritis: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1921–1927. [Google Scholar] [CrossRef]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar] [PubMed]
- Martini, A. It is time to rethink juvenile idiopathic arthritis classification and nomenclature. Ann. Rheum. Dis. 2012, 71, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Martini, A. Juvenile idiopathic arthritis. Lancet 2007, 369, 767–778. [Google Scholar] [CrossRef]
- Tugal-Tutkun, I.; Quartier, P.; Bodaghi, B. Disease of the year: Juvenile idiopathic arthritis-associated uveitis--classification and diagnostic approach. Ocul. Immunol. Inflamm. 2014, 22, 56–63. [Google Scholar] [CrossRef]
- Prakken, B.; Albani, S.; Martini, A. Juvenile idiopathic arthritis. Lancet 2011, 377, 2138–2149. [Google Scholar] [CrossRef]
- Hugle, B.; Spiegel, L.; Hotte, J.; Wiens, S.; Herlin, T.; Cron, R.Q.; Stoll, M.L.; Vinod, S.; Stoustrup, P.; Pedersen, T.K.; et al. Isolated Arthritis of the Temporomandibular Joint as the Initial Manifestation of Juvenile Idiopathic Arthritis. J. Rheumatol. 2017, 44, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Piancino, M.G.; Cannavale, R.; Dalmasso, P.; Tonni, I.; Garagiola, U.; Perillo, L.; Olivieri, A.N. Cranial structure and condylar asymmetry of patients with juvenile idiopathic arthritis: A risky growth pattern. Clin. Rheumatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S.M. The temporomandibular joint in rheumatoid arthritis. Acta Radiol. Diagn. 1985, 26, 235–243. [Google Scholar] [CrossRef]
- Kallenberg, A.; Wenneberg, B.; Carlsson, G.E.; Ahlmen, M. Reported symptoms from the masticatory system and general well-being in rheumatoid arthritis. J. Oral Rehabil. 1997, 24, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, D.; Calderazzi, A.; Maddali Bongi, S.; Cristofani, R.; Bazzichi, L.; Eligi, C.; Maresca, M.; Ciompi, M. A comparison of ultrasonography and magnetic resonance imaging in the evaluation of temporomandibular joint involvement in rheumatoid arthritis and psoriatic arthritis. Rheumatology 2003, 42, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Scarfe, W.C.; Farman, A.G.; Sukovic, P. Clinical applications of cone-beam computed tomography in dental practice. J. Can. Dent. Assoc. 2006, 72, 75–80. [Google Scholar]
- Carrasco, R. Juvenile idiopathic arthritis overview and involvement of the temporomandibular joint: Prevalence, systemic therapy. Oral Maxillofac. Surg. Clin. N. Am. 2015, 27, 1–10. [Google Scholar] [CrossRef]
- Hammer, M.R.; Kanaan, Y. Imaging of the Pediatric Temporomandibular Joint. Oral Maxillofac. Surg. Clin. N. Am. 2018, 30, 25–34. [Google Scholar] [CrossRef]
- Tamimi, D.; Jalali, E.; Hatcher, D. Temporomandibular Joint Imaging. Radiol. Clin. N. Am. 2018, 56, 157–175. [Google Scholar] [CrossRef]
- Wright, E.F.; North, S.L. Management and treatment of temporomandibular disorders: A clinical perspective. J. Man. Manip. Ther. 2009, 17, 247–254. [Google Scholar] [CrossRef]
- Stoustrup, P.; Twilt, M. Therapy. Intra-articular steroids for TMJ arthritis--caution needed. Nat. Rev. Rheumatol. 2015, 11, 566–567. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Definition | Hallmark Biomarkers | Main Oral Manifestation | Ref. |
|---|---|---|---|---|
| Rheumatoid arthritis | A chronic inflammatory disease marked by symmetrical, peripheral polyarthritis | RF, ACPA | Periodontitis, TMJ involvement | [11,12] [13,14] |
| Juvenile idiopathic arthritis | A clinically heterogeneous group of arthritides that appear before the age of 16 | ANA, RF, HLA-B27 | TMJ involvement | [13,15,16] |
| Systemic lupus erythematosus | A chronic autoimmune disease potentially targeting any organ | ANA, anti-dsDNA, anti-Sm | Oral aphthous ulcers, periodontitis | [2,3,17] [18,19] |
| Sjögren’s syndrome | A chronic autoimmune inflammatory disease primarily targeting exocrine glands | ANA, anti-SSA/Ro, anti-SSB/La | Xerostomia | [20] |
| Systemic sclerosis | A connective tissue disease characterized by multi-system involvement (skin, lungs, cardiovascular and gastro-intestinal systems) | Anti-Scl70-topoisomerase I, anti-CENPA/B | Dysphagia, microstomia, periodontitis | [10,21,22] |
| Immune-mediated myopathies | A group of acquired heterogeneous autoimmune disorders characterized by muscle weakness | ANA, myositis-specific antibodies | Dysphagia | [23] |
| Behçet’s disease | A multi-systemic disorder characterized by oral aphthous ulcers, genital ulcers and ocular involvement | HLA-B51 | Oral aphthous ulcers | [24,25] |
| Giant cell arteritis | Vasculitis of medium-sized and large vessels typically involving branches of the carotid artery such as the temporal artery | None | Jaw claudication | [26] |
| Granulomatosis with polyangiitis | Granulomatous vasculitis involving small arterial and venous vessels | cANCA (anti-PR3) | Strawberry-like gingivitis | [27,28] |
| Classification | Frequency | Onset | F:M |
|---|---|---|---|
| Oligoarticular Extended Persistent | (27% to 56%) | Early childhood, peaks at 2–4 years | F > M |
| Polyarticular RF negative RF positive | (11% to 28%) (2% to 7%) | Late childhood or adolescence Peaks at 2–4 and 6–12 years | F > M |
| Systemic arthritis | (4% to 17%) | Throughout childhood | F = M |
| Enthesitis-related arthritis | (3% to 11%) | Late childhood or adolescence | M > F |
| Psoriatic arthritis | (2% to 11%) | Peaks at 2–4 and 9–11 years | F > M |
| Undifferentiated arthritis | (11% to 21%) | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gualtierotti, R.; Marzano, A.V.; Spadari, F.; Cugno, M. Main Oral Manifestations in Immune-Mediated and Inflammatory Rheumatic Diseases. J. Clin. Med. 2019, 8, 21. https://doi.org/10.3390/jcm8010021
Gualtierotti R, Marzano AV, Spadari F, Cugno M. Main Oral Manifestations in Immune-Mediated and Inflammatory Rheumatic Diseases. Journal of Clinical Medicine. 2019; 8(1):21. https://doi.org/10.3390/jcm8010021
Chicago/Turabian StyleGualtierotti, Roberta, Angelo Valerio Marzano, Francesco Spadari, and Massimo Cugno. 2019. "Main Oral Manifestations in Immune-Mediated and Inflammatory Rheumatic Diseases" Journal of Clinical Medicine 8, no. 1: 21. https://doi.org/10.3390/jcm8010021
APA StyleGualtierotti, R., Marzano, A. V., Spadari, F., & Cugno, M. (2019). Main Oral Manifestations in Immune-Mediated and Inflammatory Rheumatic Diseases. Journal of Clinical Medicine, 8(1), 21. https://doi.org/10.3390/jcm8010021