Proteases and Their Inhibitors in Chronic Obstructive Pulmonary Disease




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
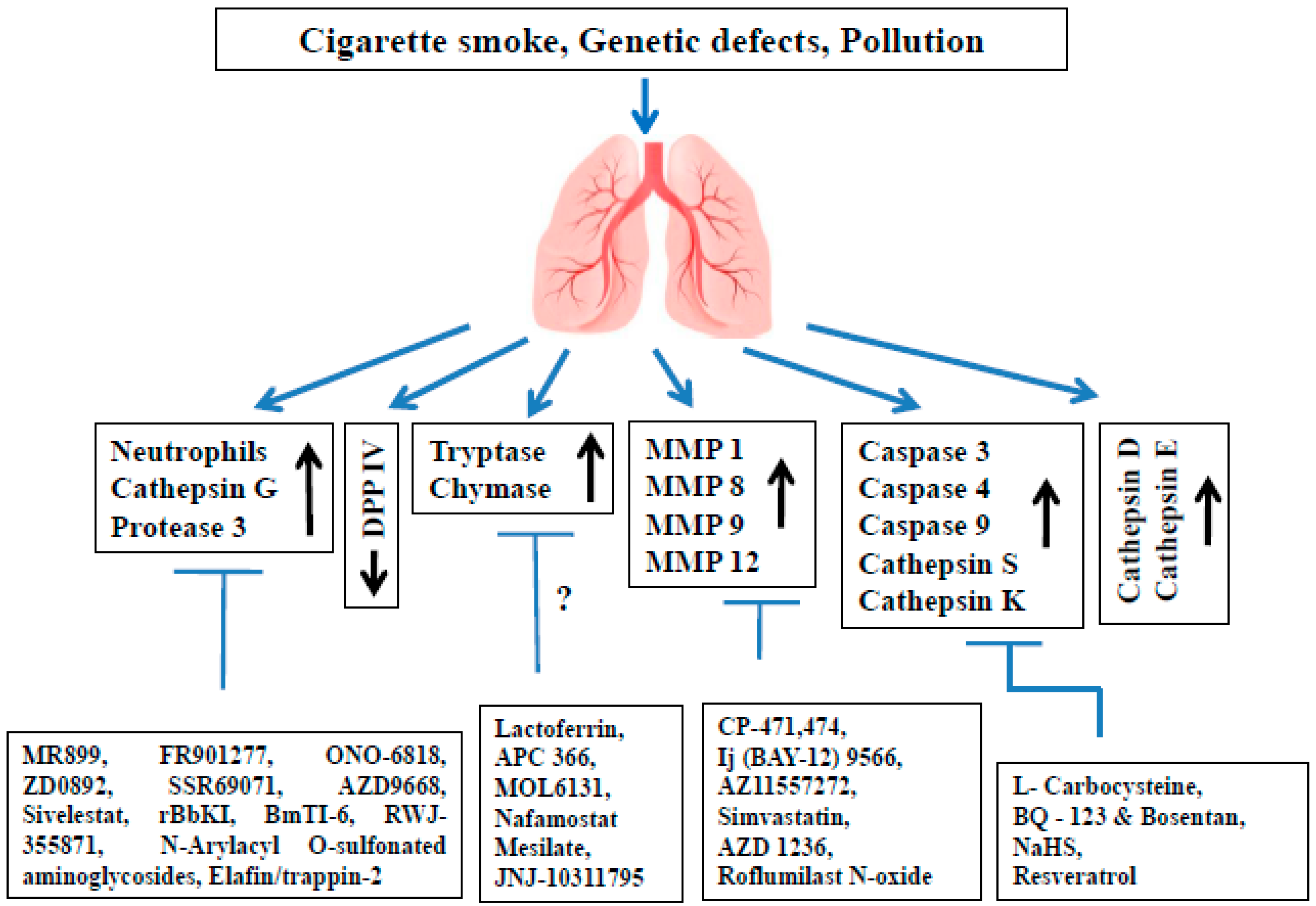
2. Proteases Involved in COPD Pathophysiology
3. The Role of Serine Proteases and Their Inhibitors in COPD
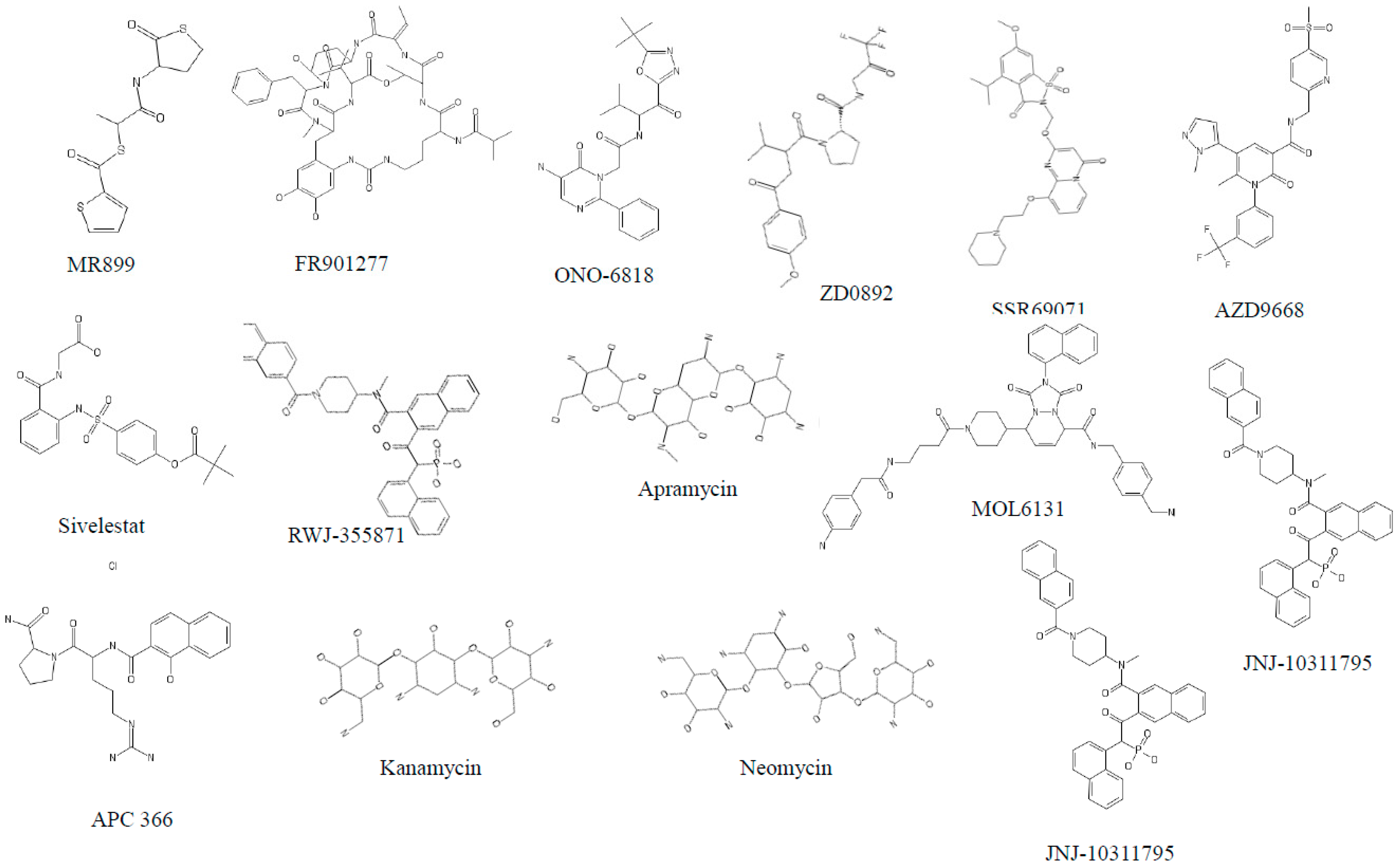
3.1. Neutrophil Elastase (NE)
3.2. Cathepsin G (cat G)
3.3. Proteinase 3 (PR3)
3.4. Dipeptidyl Peptidase IV (DPP IV)
3.5. Tryptases
3.6. Chymases
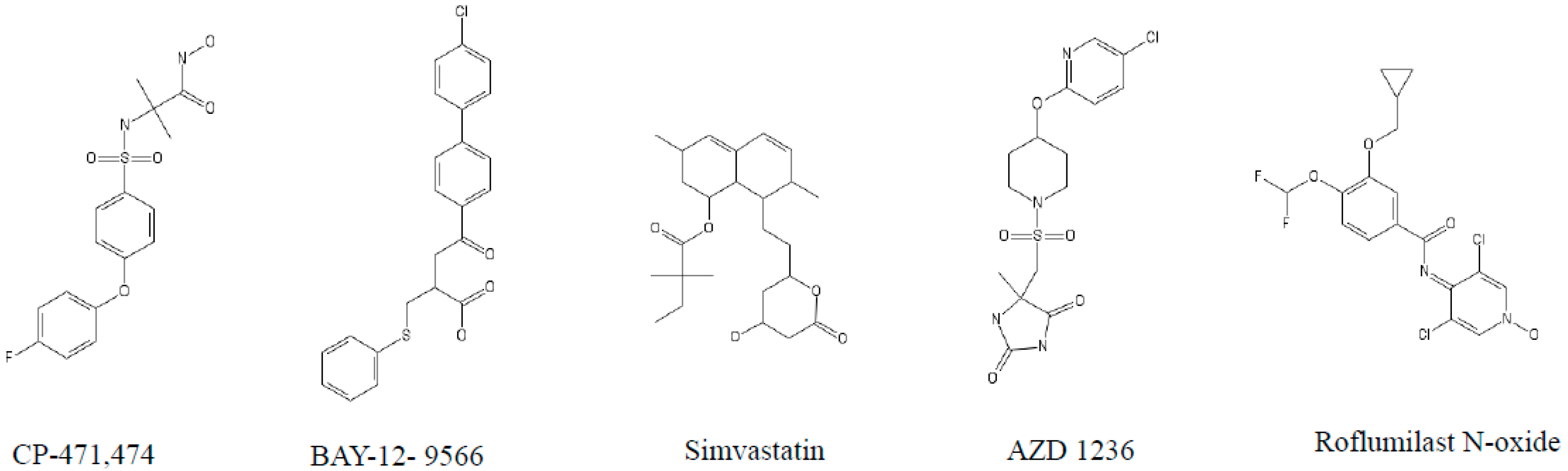
4. The Role of MMPs and Their Inhibitors in COPD
5. The Role of Cysteine Proteases and Their Inhibitors in COPD
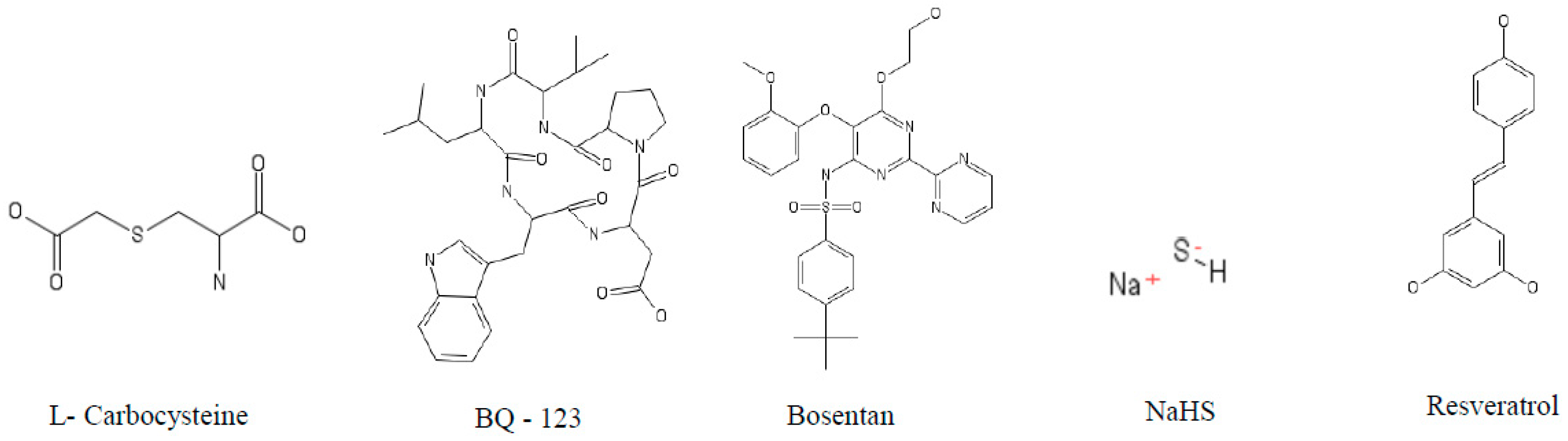
5.1. Caspases
5.2. Cathepsin S (cat S)
5.3. Cathepsin K (cat K)
6. The Role of Aspartic Proteases and Their Inhibitors in COPD
6.1. Cathepsin D (cat D)
6.2. Cathepsin E (cat E)
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A1AT | α-1 antitrypsin |
| Akt | Protein kinase B |
| Bad | Bcl-2-associated death promoter |
| BAL | Broncho alveolar lavage |
| Bax | BCL2-associated X protein |
| BbCI | Bauhinia bauhinioide scruzipain inhibitor |
| BmTI-6-D1 | Kunitz-type serine protease inhibitor 6 Recombinant Protein Domain 1 |
| CB | Chronic bronchitis |
| CE/CSE | Cigarette smoke extract |
| COPD | Chronic obstructive pulmonary disease |
| CXCL 12 | C-X-C motif chemokine 12 |
| CXCR1 | C-X-C chemokine receptor type 1 |
| DPP IV | Dipeptidyl peptidase IV |
| eNOS | Endothelial nitric oxide synthases |
| FcγRIIIb | Fcγ receptor IIIb |
| FEV1/VC | Forced expiratory volume (first second)/vital capacity. |
| H2O2 | Hydrogen peroxide |
| IFN-γ | Interferon gamma |
| IL-1 | Interleukin 1 |
| IL-18 | Interleukin 18 |
| IL-1β | Interleukin 1 beta |
| IL-8 | Interleukin 8 |
| iNOS | Inducible nitric oxide synthase |
| Lm | Mean linear intercept |
| LPS | Lipopolysaccharide |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MMP | Matrix metalloproteinase |
| NaHS | Sodium hydrosulfide |
| NE | Neutrophil elastase |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| P2X7 | P2X purinoceptor 7 |
| PAH | Pulmonary arterial hypertension |
| PF | Pulmonary fibrosis |
| PPE | Porcine pancreatic elastase |
| PR3 | Proteinase 3 |
| rBbKI | recombinant Bauhinia bauhinioides Kallikrein proteinase Inhibitor |
| RLV | Relative lung volumes |
| RVH | Right ventricular hypertrophy |
| SLPI | Secretory leukocyte protease inhibitor |
| Smad | Mothers against decapentaplegic homolog transcription factor |
| TGF-β | Transforming growth factor beta 1 |
| TNFα | Tumor necrosis factor-alpha |
| VL | Lung volumes |
| WT-elafin | Wild type elafin |
References
- Lopez, A.D.; Shibuya, K.; Rao, C.; Mathers, C.D.; Hansell, A.L.; Held, L.S.; Schmid, V.; Buist, S. Chronic obstructive pulmonary disease: Current burden and future projections. Eur. Respir. J. 2006, 27, 397–412. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Report: Burden of Chronic Respiratory Disease; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Dalal, A.A.; Christensen, L.; Liu, F.; Riedel, A.A. Direct costs of chronic obstructive pulmonary disease among managed care patients. Int. J. Chron. Obstr. Pulm. Dis. 2010, 5, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Bhome, A.B. COPD in India: Iceberg or volcano? J. Thorac. Dis. 2012, 4, 298–309. [Google Scholar] [PubMed]
- Chen, X.; Wang, N.; Chen, Y.; Xiao, T.; Fu, C.; Xu, B. Costs of chronic obstructive pulmonary disease in urban areas of China: A cross-sectional study in four cities. Int. J. Chron. Obstr. Pulm. Dis. 2016, 11, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Adeloye, D.; Chua, S.; Lee, C.; Basquill, C.; Papana, A.; Theodoratou, E.; Nair, H.; Gasevic, D.; Sridhar, D.; Campbell, H.; et al. Global and regional estimates of COPD prevalence: Systematic review and meta-analysis. J. Glob. Health 2015, 5, 020415. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Berger, K.; Lippert, B.; Kilgert, K.; Caeser, M.; Sandtmann, R. Epidemiology and health economics of COPD across Europe: A critical analysis. Treat. Respir. Med. 2005, 4, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Lahousse, L.; Seys, L.J.M.; Joos, G.F.; Franco, O.H.; Stricker, B.H.; Brusselle, G.G. Epidemiology and impact of chronic bronchitis in chronic obstructive pulmonary disease. Eur. Respir. J. 2017, 50, 1602470. [Google Scholar] [CrossRef] [PubMed]
- Sharafkhaneh, A.; Hanania, N.A.; Kim, V. Pathogenesis of emphysema: From the bench to the bedside. Proc. Am. Thorac. Soc. 2008, 5, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Kamal, R.; Srivastava, A.K.; Kesavachandran, C.N. Meta-analysis approach to study the prevalence of chronic obstructive pulmonary disease among current, former and non-smokers. Toxicol. Rep. 2015, 2, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Murray, R.P. Smoking cessation in chronic obstructive pulmonary disease. Respir. Med. 2009, 103, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Brode, S.K.; Ling, S.C.; Chapman, K.R. Alpha-1 antitrypsin deficiency: A commonly overlooked cause of lung disease. CMAJ 2012, 184, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- DeMeo, D.L.; Silverman, E.K. Alpha1-antitrypsin deficiency. 2: Genetic aspects of alpha(1)-antitrypsin deficiency: Phenotypes and genetic modifiers of emphysema risk. Thorax 2004, 59, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, Y. The aging lung and chronic obstructive pulmonary disease: Similarity and difference. Proc. Am. Thorac. Soc. 2009, 6, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Lowery, E.M.; Brubaker, A.L.; Kuhlmann, E.; Kovacs, E.J. The aging lung. Clin. Interv. Aging 2013, 8, 1489–1496. [Google Scholar] [PubMed]
- Montuschi, P. Pharmacological treatment of chronic obstructive pulmonary disease. Int. J. Chron. Obstr. Pulm. Dis. 2006, 1, 409–423. [Google Scholar] [CrossRef]
- Currie, G.P.; Lipworth, B.J. Inhaled treatment for chronic obstructive pulmonary disease: What’s new and how does it fit? QJM Int. J. Med. 2016, 109, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Ferguson, G.T. Combination bronchodilator therapy in the management of chronic obstructive pulmonary disease. Respir. Res. 2013, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Erkan, L.; Uzun, O.; Findik, S.; Katar, D.; Sanic, A.; Atici, A.G. Role of bacteria in acute exacerbations of chronic obstructive pulmonary disease. Int. J. Chron. Obstr. Pulm. Dis. 2008, 3, 463–467. [Google Scholar]
- Beasley, V.; Joshi, P.V.; Singanayagam, A.; Molyneaux, P.L.; Johnston, S.L.; Mallia, P. Lung microbiology and exacerbations in COPD. Int. J. Chron. Obstr. Pulm. Dis. 2012, 7, 555–569. [Google Scholar]
- Sethi, S. Bacteria in exacerbations of chronic obstructive pulmonary disease: Phenomenon or epiphenomenon? Proc. Am. Thorac. Soc. 2004, 1, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Kinases as novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Pharmacol. Rev. 2016, 68, 788–815. [Google Scholar] [CrossRef] [PubMed]
- Page, C.P. Phosphodiesterase inhibitors for the treatment of asthma and chronic obstructive pulmonary disease. Int. Arch. Allergy Immunol. 2014, 165, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Antoniu, S.A. Targeting 5-lipoxygenase-activating protein in asthma and chronic obstructive pulmonary disease. Expert Opin. Ther. Targets 2014, 18, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat. Rev. Drug Discov. 2013, 12, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Alamo, A.; Crisafulli, E.; Raciti, C.; Fisichella, A.; Polosa, R. Adenosine signaling pathways as potential therapeutic targets in respiratory disease. Expert Opin. Ther. Targets 2013, 17, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Drakatos, P.; Lykouras, D.; Sampsonas, F.; Karkoulias, K.; Spiropoulos, K. Targeting leukotrienes for the treatment of COPD? Inflamm. Allergy Drug Targets 2009, 8, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Mercer, B.A.; D’Armiento, J.M. Emerging role of map kinase pathways as therapeutic targets in COPD. Int. J. Chron. Obstr. Pulm. Dis. 2006, 1, 137–150. [Google Scholar] [CrossRef]
- Bals, R.; Hiemstra, P.S. Antimicrobial peptides in COPD—Basic biology and therapeutic applications. Curr. Drug Targets 2006, 7, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Macnee, W.; Rahman, I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 160, S58–S65. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R.T.; Vimalanathan, S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int. J. Tuberc. Lung Dis. 2008, 12, 361–367. [Google Scholar] [PubMed]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Kohnlein, T.; Welte, T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Bergin, D.A.; Reeves, E.P.; Meleady, P.; Henry, M.; McElvaney, O.J.; Carroll, T.P.; Condron, C.; Chotirmall, S.H.; Clynes, M.; O’Neill, S.J.; et al. Alpha-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J. Clin. Investig. 2010, 120, 4236–4250. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, P.; Rogan, M.P.; Greene, C.M.; Brantly, M.L.; O’Neill, S.J.; Taggart, C.C.; McElvaney, N.G. Alpha-1-antitrypsin aerosolised augmentation abrogates neutrophil elastase-induced expression of cathepsin B and matrix metalloprotease 2 in vivo and in vitro. Thorax 2008, 63, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Nadel, J.A. Role of neutrophil elastase in hypersecretion during COPD exacerbations, and proposed therapies. Chest 2000, 117, 386S–389S. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.Y.; Yang, Y.; Jia, X.Q.; Wang, Y.; Peng, L.N.; Ai, X.H.; Jiang, C.Y.; Guo, J.H.; Wu, T.T. Expression and clinical significance of serum dipeptidyl peptidase iv chronic obstructive pulmonary disease. Am. J. Med. Sci. 2016, 351, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Guyot, N.; Wartelle, J.; Malleret, L.; Todorov, A.A.; Devouassoux, G.; Pacheco, Y.; Jenne, D.E.; Belaaouaj, A. Unopposed cathepsin G, neutrophil elastase, and proteinase 3 cause severe lung damage and emphysema. Am. J. Pathol. 2014, 184, 2197–2210. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.A. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int. J. Chron. Obstr. Pulm. Dis. 2008, 3, 253–268. [Google Scholar] [CrossRef]
- Andersson, C.K.; Mori, M.; Bjermer, L.; Lofdahl, C.G.; Erjefalt, J.S. Alterations in lung mast cell populations in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Gosman, M.M.; Postma, D.S.; Vonk, J.M.; Rutgers, B.; Lodewijk, M.; Smith, M.; Luinge, M.A.; Ten Hacken, N.H.; Timens, W. Association of mast cells with lung function in chronic obstructive pulmonary disease. Respir. Res. 2008, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Baraldo, S.; Bazzan, E.; Zanin, M.E.; Turato, G.; Garbisa, S.; Maestrelli, P.; Papi, A.; Miniati, M.; Fabbri, L.M.; Zuin, R.; et al. Matrix metalloproteinase-2 protein in lung periphery is related to COPD progression. Chest 2007, 132, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Hunninghake, G.M.; Cho, M.H.; Tesfaigzi, Y.; Soto-Quiros, M.E.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melen, E.; Soderhall, C.; Hallberg, J.; et al. MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 2009, 361, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Molet, S.; Belleguic, C.; Lena, H.; Germain, N.; Bertrand, C.P.; Shapiro, S.D.; Planquois, J.M.; Delaval, P.; Lagente, V. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm. Res. 2005, 54, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Vernooy, J.H.; Lindeman, J.H.; Jacobs, J.A.; Hanemaaijer, R.; Wouters, E.F. Increased activity of matrix metalloproteinase-8 and matrix metalloproteinase-9 in induced sputum from patients with COPD. Chest 2004, 126, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; In, K.H.; Kim, J.H.; Lee, S.Y.; Shin, C.; Shim, J.J.; Kang, K.H.; Yoo, S.H.; Kim, C.H.; Kim, H.K.; et al. Proteomic analysis in lung tissue of smokers and COPD patients. Chest 2009, 135, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, E.; Karakiulakis, G.; Batzios, S.; Savic, S.; Roth, M.; Tamm, M.; Stolz, D. Acute exacerbations of COPD are associated with significant activation of matrix metalloproteinase 9 irrespectively of airway obstruction, emphysema and infection. Respir. Res. 2015, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Eltom, S.; Belvisi, M.G.; Stevenson, C.S.; Maher, S.A.; Dubuis, E.; Fitzgerald, K.A.; Birrell, M.A. Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: An insight into the pathogenesis of COPD. PLoS ONE 2014, 9, e112829. [Google Scholar] [CrossRef] [PubMed]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Lockett, A.D.; Van Demark, M.; Gu, Y.; Schweitzer, K.S.; Sigua, N.; Kamocki, K.; Fijalkowska, I.; Garrison, J.; Fisher, A.J.; Serban, K.; et al. Effect of cigarette smoke exposure and structural modifications on the alpha-1 antitrypsin interaction with caspases. Mol. Med. 2012, 18, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Podolin, P.L.; Foley, J.P.; Carpenter, D.C.; Bolognese, B.J.; Logan, G.A.; Long, E., III; Harrison, O.J.; Walsh, P.T. T cell depletion protects against alveolar destruction due to chronic cigarette smoke exposure in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L312–L323. [Google Scholar] [CrossRef] [PubMed]
- Gogebakan, B.; Bayraktar, R.; Ulasli, M.; Oztuzcu, S.; Tasdemir, D.; Bayram, H. The role of bronchial epithelial cell apoptosis in the pathogenesis of COPD. Mol. Biol. Rep. 2014, 41, 5321–5327. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, K.; Yoshimi, M.; Maeyama, T.; Hamada, N.; Yamada, M.; Nakanishi, Y. Apoptosis signaling pathways in lung diseases. Med. Chem. 2005, 1, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Homer, R.J.; Gallo, A.; Lee, C.G.; Crothers, K.A.; Cho, S.J.; Rochester, C.; Cain, H.; Chupp, G.; Yoon, H.J.; et al. IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J. Immunol. 2007, 178, 1948–1959. [Google Scholar] [CrossRef] [PubMed]
- Golovatch, P.; Mercer, B.A.; Lemaitre, V.; Wallace, A.; Foronjy, R.F.; D’Armiento, J. Role for cathepsin K in emphysema in smoke-exposed guinea pigs. Exp. Lung Res. 2009, 35, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Nakamura, H.; Owen, C.A.; Yoshida, S.; Tsuduki, K.; Chubachi, S.; Shirahata, T.; Mashimo, S.; Nakamura, M.; Takahashi, S.; et al. Plasma cathepsin S and cathepsin S/cystatin C ratios are potential biomarkers for COPD. Dis. Mark. 2016, 2016, 4093870. [Google Scholar] [CrossRef] [PubMed]
- Bracke, K.; Cataldo, D.; Maes, T.; Gueders, M.; Noel, A.; Foidart, J.M.; Brusselle, G.; Pauwels, R.A. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int. Arch. Allergy Immunol. 2005, 138, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Zhu, Z.; Wang, Z.; Homer, R.J.; Ma, B.; Riese, R.J., Jr.; Chapman, H.A., Jr.; Shapiro, S.D.; Elias, J.A. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J. Clin. Investig. 2000, 106, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.J.; Li, M.H.; Li, J.X.; Xu, X.; Ren, S.X.; Rajbanshi, B.; Xu, J.F. High expression of cathepsin E is associated with the severity of airflow limitation in patients with COPD. COPD 2016, 13, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shan, P.; Homer, R.; Zhang, Y.; Petrache, I.; Mannam, P.; Lee, P.J. Cathepsin E promotes pulmonary emphysema via mitochondrial fission. Am. J. Pathol. 2014, 184, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Stockley, R.A. The multiple facets of alpha-1-antitrypsin. Ann. Transl. Med. 2015, 3, 130. [Google Scholar] [PubMed]
- Tonelli, A.R.; Brantly, M.L. Augmentation therapy in alpha-1 antitrypsin deficiency: Advances and controversies. Ther. Adv. Respir. Dis. 2010, 4, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Higashimoto, Y.; Iwata, T.; Okada, M.; Satoh, H.; Fukuda, K.; Tohda, Y. Serum biomarkers as predictors of lung function decline in chronic obstructive pulmonary disease. Respir. Med. 2009, 103, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Belcher, J.; Thorpe, G.; Forsyth, N.R.; Spiteri, M.A. Measurement of c-reactive protein, procalcitonin and neutrophil elastase in saliva of COPD patients and healthy controls: Correlation to self-reported wellbeing parameters. Respir. Res. 2015, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Sng, J.J.; Prazakova, S.; Thomas, P.S.; Herbert, C. MMP-8, MMP-9 and neutrophil elastase in peripheral blood and exhaled breath condensate in COPD. COPD 2017, 14, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Luisetti, M.; Sturani, C.; Sella, D.; Madonini, E.; Galavotti, V.; Bruno, G.; Peona, V.; Kucich, U.; Dagnino, G.; Rosenbloom, J.; et al. MR889, a neutrophil elastase inhibitor, in patients with chronic obstructive pulmonary disease: A double-blind, randomized, placebo-controlled clinical trial. Eur. Respir. J. 1996, 9, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Kuraki, T.; Ishibashi, M.; Takayama, M.; Shiraishi, M.; Yoshida, M. A novel oral neutrophil elastase inhibitor (ONO-6818) inhibits human neutrophil elastase-induced emphysema in rats. Am. J. Respir. Crit. Care Med. 2002, 166, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Farmer, S.G.; Churg, A. Synthetic serine elastase inhibitor reduces cigarette smoke-induced emphysema in guinea pigs. Am. J. Respir. Crit. Care Med. 2002, 166, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Kapui, Z.; Varga, M.; Urban-Szabo, K.; Mikus, E.; Szabo, T.; Szeredi, J.; Batori, S.; Finance, O.; Aranyi, P. Biochemical and pharmacological characterization of 2-(9-(2-piperidinoethoxy)-4-oxo-4h-pyrido[1,2-a]pyrimidin-2-yloxymethyl)-4-(1-methylethyl)-6-methoxy-1,2-benzisothiazol-3(2h)-one-1,1-dioxide (SSR69071), a novel, orally active elastase inhibitor. J. Pharmacol. Exp. Ther. 2003, 305, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.; Ekholm, K.; Granse, M.; Lindahl, M.; Kozma, V.; Jungar, C.; Ottosson, T.; Falk-Hakansson, H.; Churg, A.; Wright, J.L.; et al. AZD9668: Pharmacological characterization of a novel oral inhibitor of neutrophil elastase. J. Pharmacol. Exp. Ther. 2011, 339, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kuna, P.; Jenkins, M.; O’Brien, C.D.; Fahy, W.A. AZD9668, a neutrophil elastase inhibitor, plus ongoing budesonide/formoterol in patients with COPD. Respir. Med. 2012, 106, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Vogelmeier, C.; Aquino, T.O.; O’Brien, C.D.; Perrett, J.; Gunawardena, K.A. A randomised, placebo-controlled, dose-finding study of AZD9668, an oral inhibitor of neutrophil elastase, in patients with chronic obstructive pulmonary disease treated with tiotropium. COPD 2012, 9, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Martins-Olivera, B.T.; Almeida-Reis, R.; Theodoro-Junior, O.A.; Oliva, L.V.; Neto Dos Santos Nunes, N.; Olivo, C.R.; Vilela de Brito, M.; Prado, C.M.; Leick, E.A.; Martins Mde, A.; et al. The plant-derived Bauhinia bauhinioides kallikrein proteinase inhibitor (rBbKI) attenuates elastase-induced emphysema in mice. Mediat. Inflamm. 2016, 2016, 5346574. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Reis, R.; Theodoro-Junior, O.A.; Oliveira, B.T.M.; Oliva, L.V.; Toledo-Arruda, A.C.; Bonturi, C.R.; Brito, M.V.; Lopes, F.; Prado, C.M.; Florencio, A.C.; et al. Plant proteinase inhibitor BbCI modulates lung inflammatory responses and mechanic and remodeling alterations induced by elastase in mice. Biomed. Res. Int. 2017, 2017, 8287125. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.F.A.; Neves, L.P.; da Silva, F.R.S.; Machado, G.C.; Ferreira, G.C.; Lourenco, J.D.; Tanaka, A.S.; Martins, M.A.; Lopes, F.; Sasaki, S.D. rBmTI-6 attenuates pathophysiological and inflammatory parameters of induced emphysema in mice. Int. J. Biol. Macromol. 2018, 111, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Shafer, W.M.; Pohl, J.; Onunka, V.C.; Bangalore, N.; Travis, J. Human lysosomal cathepsin G and granzyme b share a functionally conserved broad spectrum antibacterial peptide. J. Biol. Chem. 1991, 266, 112–116. [Google Scholar] [PubMed]
- Burster, T.; Macmillan, H.; Hou, T.; Boehm, B.O.; Mellins, E.D. Cathepsin G: Roles in antigen presentation and beyond. Mol. Immunol. 2010, 47, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Hahn, I.; Klaus, A.; Janze, A.K.; Steinwede, K.; Ding, N.; Bohling, J.; Brumshagen, C.; Serrano, H.; Gauthier, F.; Paton, J.C.; et al. Cathepsin G and neutrophil elastase play critical and nonredundant roles in lung-protective immunity against streptococcus pneumoniae in mice. Infect. Immun. 2011, 79, 4893–4901. [Google Scholar] [CrossRef] [PubMed]
- Almansa, R.; Socias, L.; Sanchez-Garcia, M.; Martin-Loeches, I.; del Olmo, M.; Andaluz-Ojeda, D.; Bobillo, F.; Rico, L.; Herrero, A.; Roig, V.; et al. Critical COPD respiratory illness is linked to increased transcriptomic activity of neutrophil proteases genes. BMC Res. Notes 2012, 5, 401. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Sommerhoff, C.P.; Nadel, J.A.; Basbaum, C.B.; Caughey, G.H. Neutrophil elastase and cathepsin G stimulate secretion from cultured bovine airway gland serous cells. J. Clin. Investig. 1990, 85, 682–689. [Google Scholar] [CrossRef] [PubMed]
- De Garavilla, L.; Greco, M.N.; Sukumar, N.; Chen, Z.W.; Pineda, A.O.; Mathews, F.S.; Di Cera, E.; Giardino, E.C.; Wells, G.I.; Haertlein, B.J.; et al. A novel, potent dual inhibitor of the leukocyte proteases cathepsin G and chymase: Molecular mechanisms and anti-inflammatory activity in vivo. J. Biol. Chem. 2005, 280, 18001–18007. [Google Scholar] [CrossRef] [PubMed]
- Maryanoff, B.E.; de Garavilla, L.; Greco, M.N.; Haertlein, B.J.; Wells, G.I.; Andrade-Gordon, P.; Abraham, W.M. Dual inhibition of cathepsin G and chymase is effective in animal models of pulmonary inflammation. Am. J. Respir. Crit. Care Med. 2010, 181, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Craciun, I.; Fenner, A.M.; Kerns, R.J. N-arylacyl o-sulfonated aminoglycosides as novel inhibitors of human neutrophil elastase, cathepsin G and proteinase 3. Glycobiology 2016, 26, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.J.; Campbell, M.A.; Owen, C.A. Bioactive proteinase 3 on the cell surface of human neutrophils: Quantification, catalytic activity, and susceptibility to inhibition. J. Immunol. 2000, 165, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Voswinkel, J.; Muller, A.; Lamprecht, P. Is PR3-anca formation initiated in wegener’s granulomatosis lesions? Granulomas as potential lymphoid tissue maintaining autoantibody production. Ann. N. Y. Acad. Sci. 2005, 1051, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, O.E.; Follin, P.; Johnsen, A.H.; Calafat, J.; Tjabringa, G.S.; Hiemstra, P.S.; Borregaard, N. Human cathelicidin, hcap-18, is processed to the antimicrobial peptide ll-37 by extracellular cleavage with proteinase 3. Blood 2001, 97, 3951–3959. [Google Scholar] [CrossRef] [PubMed]
- Coeshott, C.; Ohnemus, C.; Pilyavskaya, A.; Ross, S.; Wieczorek, M.; Kroona, H.; Leimer, A.H.; Cheronis, J. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc. Natl. Acad. Sci. USA 1999, 96, 6261–6266. [Google Scholar] [CrossRef] [PubMed]
- Sinden, N.J.; Stockley, R.A. Proteinase 3 activity in sputum from subjects with alpha-1-antitrypsin deficiency and COPD. Eur. Respir. J. 2013, 41, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Janelle, M.F.; Doucet, A.; Bouchard, D.; Bourbonnais, Y.; Tremblay, G.M. Increased local levels of granulocyte colony-stimulating factor are associated with the beneficial effect of pre-elafin (skalp/trappin-2/wap3) in experimental emphysema. Biol. Chem. 2006, 387, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Tanga, A.; Saidi, A.; Jourdan, M.L.; Dallet-Choisy, S.; Zani, M.L.; Moreau, T. Protection of lung epithelial cells from protease-mediated injury by trappin-2 a62l, an engineered inhibitor of neutrophil serine proteases. Biochem. Pharmacol. 2012, 83, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M.; Zani, M.L.; Quinn, D.J.; Dallet-Choisy, S.; Glasgow, A.M.; O’Kane, C.; McAuley, D.F.; McNally, P.; Weldon, S.; Moreau, T.; et al. A functional variant of elafin with improved anti-inflammatory activity for pulmonary inflammation. Mol. Ther. 2015, 23, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Doucet, A.; Bouchard, D.; Janelle, M.F.; Bellemare, A.; Gagne, S.; Tremblay, G.M.; Bourbonnais, Y. Characterization of human pre-elafin mutants: Full antipeptidase activity is essential to preserve lung tissue integrity in experimental emphysema. Biochem. J. 2007, 405, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Camerini, D.; Seed, B.; Torimoto, Y.; Dang, N.H.; Kameoka, J.; Dahlberg, H.N.; Schlossman, S.F.; Morimoto, C. Cloning and functional expression of the T cell activation antigen CD26. J. Immunol. 1992, 149, 481–486. [Google Scholar] [PubMed]
- Shubrook, J.; Colucci, R.; Guo, A.; Schwartz, F. Saxagliptin: A selective DPP-4 inhibitor for the treatment of type 2 diabetes mellitus. Clin. Med. Insights Endocrinol. Diabetes 2011, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meyerholz, D.K.; Lambertz, A.M.; McCray, P.B., Jr. Dipeptidyl peptidase 4 distribution in the human respiratory tract: Implications for the middle east respiratory syndrome. Am. J. Pathol. 2016, 186, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Somborac-Bacura, A.; Buljevic, S.; Rumora, L.; Culic, O.; Detel, D.; Pancirov, D.; Popovic-Grle, S.; Varljen, J.; Cepelak, I.; Zanic-Grubisic, T. Decreased soluble dipeptidyl peptidase IV activity as a potential serum biomarker for COPD. Clin. Biochem. 2012, 45, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Seys, L.J.M.; Widagdo, W.; Verhamme, F.M.; Kleinjan, A.; Janssens, W.; Joos, G.F.; Bracke, K.R.; Haagmans, B.L.; Brusselle, G.G. DPP4, the middle east respiratory syndrome coronavirus receptor, is upregulated in lungs of smokers and chronic obstructive pulmonary disease patients. Clin. Infect. Dis. 2018, 66, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Herlihy, S.E.; Pilling, D.; Maharjan, A.S.; Gomer, R.H. Dipeptidyl peptidase IV is a human and murine neutrophil chemorepellent. J. Immunol. 2013, 190, 6468–6477. [Google Scholar] [CrossRef] [PubMed]
- Busso, N.; Wagtmann, N.; Herling, C.; Chobaz-Peclat, V.; Bischof-Delaloye, A.; So, A.; Grouzmann, E. Circulating CD26 is negatively associated with inflammation in human and experimental arthritis. Am. J. Pathol. 2005, 166, 433–442. [Google Scholar] [CrossRef]
- Herlihy, S.E.; Brown, M.L.; Pilling, D.; Weeks, B.R.; Myers, L.K.; Gomer, R.H. Role of the neutrophil chemorepellent soluble dipeptidyl peptidase iv in decreasing inflammation in a murine model of arthritis. Arthritis Rheumatol. 2015, 67, 2634–2638. [Google Scholar] [CrossRef] [PubMed]
- Rohrborn, D.; Eckel, J.; Sell, H. Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up-regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Lett. 2014, 588, 3870–3877. [Google Scholar] [CrossRef] [PubMed]
- Payne, V.; Kam, P.C. Mast cell tryptase: A review of its physiology and clinical significance. Anaesthesia 2004, 59, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.B. Tryptase, a mediator of human mast cells. J. Allergy Clin. Immunol. 1990, 86, 594–598. [Google Scholar] [CrossRef]
- Peng, Q.; McEuen, A.R.; Benyon, R.C.; Walls, A.F. The heterogeneity of mast cell tryptase from human lung and skin. Eur. J. Biochem. 2003, 270, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Kalenderian, R.; Raju, L.; Roth, W.; Schwartz, L.B.; Gruber, B.; Janoff, A. Elevated histamine and tryptase levels in smokers’ bronchoalveolar lavage fluid. Do lung mast cells contribute to smokers’ emphysema? Chest 1988, 94, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Soltani, A.; Ewe, Y.P.; Lim, Z.S.; Sohal, S.S.; Reid, D.; Weston, S.; Wood-Baker, R.; Walters, E.H. Mast cells in COPD airways: Relationship to bronchodilator responsiveness and angiogenesis. Eur. Respir. J. 2012, 39, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Ballarin, A.; Bazzan, E.; Zenteno, R.H.; Turato, G.; Baraldo, S.; Zanovello, D.; Mutti, E.; Hogg, J.C.; Saetta, M.; Cosio, M.G. Mast cell infiltration discriminates between histopathological phenotypes of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 186, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, H.; Ma, W.; Wang, F.; Zeng, X.; Liu, C.; He, S. Tryptase enzyme activity is correlated with severity of chronic obstructive pulmonary disease. Tohoku J. Exp. Med. 2011, 224, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Elrod, K.C.; Moore, W.R.; Abraham, W.M.; Tanaka, R.D. Lactoferrin, a potent tryptase inhibitor, abolishes late-phase airway responses in allergic sheep. Am. J. Respir. Crit. Care Med. 1997, 156, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.T.; Chauhan, A.; Little, L.; Sampson, K.; Hawksworth, R.; Mant, T.; Djukanovic, R.; Lee, T.; Holgate, S. Inhibition of mast cell tryptase by inhaled APC 366 attenuates allergen-induced late-phase airway obstruction in asthma. J. Allergy Clin. Immunol. 2001, 107, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.W.; Pae, C.I.; Lee, D.K.; Jones, F.; Chiang, G.K.; Kim, H.O.; Moon, S.H.; Cao, B.; Ogbu, C.; Jeong, K.W.; et al. Tryptase inhibition blocks airway inflammation in a mouse asthma model. J. Immunol. 2002, 168, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, M.; Tanaka, H.; Kajiwara, D.; Toyohara, T.; Wakahara, K.; Inagaki, N.; Nagai, H. Nafamostatmesilate, a potent serine protease inhibitor, inhibits airway eosinophilic inflammation and airway epithelial remodeling in a murine model of allergic asthma. J. Pharmacol. Sci. 2008, 108, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Caughey, G.H. Mast cell tryptases and chymases in inflammation and host defense. Immunol. Rev. 2007, 217, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Han, S.X.; Zhang, S.F.; Ning, Y.Y.; Chen, L.; Chen, Y.J.; He, G.M.; Xu, D.; An, J.; Yang, T.; et al. Role of chymase in cigarette smoke-induced pulmonary artery remodeling and pulmonary hypertension in hamsters. Respir. Res. 2010, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Wypij, D.M.; Nichols, J.S.; Novak, P.J.; Stacy, D.L.; Berman, J.; Wiseman, J.S. Role of mast cell chymase in the extracellular processing of big-endothelin-1 to endothelin-1 in the perfused rat lung. Biochem. Pharmacol. 1992, 43, 845–853. [Google Scholar] [CrossRef]
- Tchougounova, E.; Lundequist, A.; Fajardo, I.; Winberg, J.O.; Abrink, M.; Pejler, G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J. Biol. Chem. 2005, 280, 9291–9296. [Google Scholar] [CrossRef] [PubMed]
- Karlson, U.; Pejler, G.; Froman, G.; Hellman, L. Rat mast cell protease 4 is a beta-chymase with unusually stringent substrate recognition profile. J. Biol. Chem. 2002, 277, 18579–18585. [Google Scholar] [CrossRef] [PubMed]
- Amin, K. The role of mast cells in allergic inflammation. Respir. Med. 2012, 106, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kosanovic, D.; Dahal, B.K.; Peters, D.M.; Seimetz, M.; Wygrecka, M.; Hoffmann, K.; Antel, J.; Reiss, I.; Ghofrani, H.A.; Weissmann, N.; et al. Histological characterization of mast cell chymase in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Pulm. Circ. 2014, 4, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Kosanovic, D.; Luitel, H.; Dahal, B.K.; Cornitescu, T.; Janssen, W.; Danser, A.H.; Garrelds, I.M.; De Mey, J.G.; Fazzi, G.; Schiffers, P.; et al. Chymase: A multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur. Respir. J. 2015, 46, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- He, S.H.; Zheng, J. Stimulation of mucin secretion from human bronchial epithelial cells by mast cell chymase. Acta Pharmacol. Sin. 2004, 25, 827–832. [Google Scholar] [PubMed]
- Tinel, H.; Zubov, D.; Zimmermann, K.; Tersteegen, A.; Boerngen, K.; Joerissen, H.; Ackerstaff, J.; Fuerstner, C. Abstract 13624: A novel chymase inhibitor bay 1142524 reduces fibrosis and improves cardiac function after myocardial infarction in hamster. Circulation 2017, 136, A13624. [Google Scholar]
- Bot, I.; Bot, M.; van Heiningen, S.H.; van Santbrink, P.J.; Lankhuizen, I.M.; Hartman, P.; Gruener, S.; Hilpert, H.; van Berkel, T.J.; Fingerle, J.; et al. Mast cell chymase inhibition reduces atherosclerotic plaque progression and improves plaque stability in ApoE−/− mice. Cardiovasc. Res. 2011, 89, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Jin, D.; Tashiro, K.; Komeda, K.; Masubuchi, S.; Hirokawa, F.; Hayashi, M.; Takai, S.; Uchiyama, K. Chymase inhibitor prevents the development and progression of non-alcoholic steatohepatitis in rats fed a high-fat and high-cholesterol diet. J. Pharmacol. Sci. 2017, 134, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted role of matrix metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Gueders, M.M.; Foidart, J.M.; Noel, A.; Cataldo, D.D. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: Potential implications in asthma and other lung diseases. Eur. J. Pharmacol. 2006, 533, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Elkington, P.T.; Friedland, J.S. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006, 61, 259–266. [Google Scholar] [CrossRef] [PubMed]
- D’Armiento, J.; Dalal, S.S.; Okada, Y.; Berg, R.A.; Chada, K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 1992, 71, 955–961. [Google Scholar] [CrossRef]
- Mercer, P.F.; Shute, J.K.; Bhowmik, A.; Donaldson, G.C.; Wedzicha, J.A.; Warner, J.A. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir. Res. 2005, 6, 151. [Google Scholar] [CrossRef] [PubMed]
- Ilumets, H.; Rytila, P.; Demedts, I.; Brusselle, G.G.; Sovijarvi, A.; Myllarniemi, M.; Sorsa, T.; Kinnula, V.L. Matrix metalloproteinases-8, -9 and -12 in smokers and patients with stage 0 COPD. Int. J. Chron. Obstr. Pulm. Dis. 2007, 2, 369–379. [Google Scholar]
- Brajer, B.; Batura-Gabryel, H.; Nowicka, A.; Kuznar-Kaminska, B.; Szczepanik, A. Concentration of matrix metalloproteinase-9 in serum of patients with chronic obstructive pulmonary disease and a degree of airway obstruction and disease progression. J. Physiol. Pharmacol. 2008, 59 (Suppl. 6), 145–152. [Google Scholar] [PubMed]
- Selman, M.; Cisneros-Lira, J.; Gaxiola, M.; Ramirez, R.; Kudlacz, E.M.; Mitchell, P.G.; Pardo, A. Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in guinea pigs. Chest 2003, 123, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, P.A.; Cantwell, J.S.; Kim, K.M.; Sundin, D.J.; Kobayashi, D.; Fink, J.B.; Shapiro, S.D.; Barr, P.J. An inhaled matrix metalloprotease inhibitor prevents cigarette smoke-induced emphysema in the mouse. COPD 2005, 2, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Jiang, Y.; Chen, F.; Gong, L.K.; Ding, K.; Xu, Y.; Wang, R.; Ge, A.; Ren, J.; Li, J.; et al. Selective inhibition of matrix metalloproteinase isozymes and in vivo protection against emphysema by substituted gamma-keto carboxylic acids. J. Med. Chem. 2006, 49, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Wang, R.; Wang, X.; Onnervik, P.O.; Thim, K.; Wright, J.L. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 2007, 62, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Dahl, R.; Titlestad, I.; Lindqvist, A.; Wielders, P.; Wray, H.; Wang, M.; Samuelsson, V.; Mo, J.; Holt, A. Effects of an oral MMP-9 and -12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: A randomised controlled trial. Pulm. Pharmacol. Ther. 2012, 25, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Thanh Thuy, T.T.; Lee, J.H.; Ro, J.Y.; Bae, Y.A.; Kong, Y.; Ahn, J.Y.; Lee, D.S.; Oh, Y.M.; Lee, S.D.; et al. Simvastatin inhibits induction of matrix metalloproteinase-9 in rat alveolar macrophages exposed to cigarette smoke extract. Exp. Mol. Med. 2009, 41, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Bao, J.; Shi, Y.; Zhang, B.; Yuan, L.; Li, J.; Zhang, L.; Sun, M.; Sun, W. Effect of simvastatin on MMPs and TIMPs in cigarette smoke-induced rat COPD model. Int. J. Chron. Obstr. Pulm. Dis. 2017, 12, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Perng, D.W.; Tao, C.W.; Su, K.C.; Tsai, C.C.; Liu, L.Y.; Lee, Y.C. Anti-inflammatory effects of salmeterol/fluticasone, tiotropium/fluticasone or tiotropium in COPD. Eur. Respir. J. 2009, 33, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Investig. 2000, 106, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Aoshiba, K.; Yokohori, N.; Nagai, A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am. J. Respir. Cell. Mol. Biol. 2003, 28, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Yokohori, N.; Aoshiba, K.; Nagai, A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 2004, 125, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Mercer, B.A.; Schulman, L.L.; Sonett, J.R.; D’Armiento, J.M. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur. Respir. J. 2005, 25, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Holmes, M.; Reynolds, P.N. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur. Respir. J. 2005, 25, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Couillin, I.; Vasseur, V.; Charron, S.; Gasse, P.; Tavernier, M.; Guillet, J.; Lagente, V.; Fick, L.; Jacobs, M.; Coelho, F.R.; et al. IL-1R1/MyD88 signaling is critical for elastase-induced lung inflammation and emphysema. J. Immunol. 2009, 183, 8195–8202. [Google Scholar] [CrossRef] [PubMed]
- Eltom, S.; Stevenson, C.S.; Rastrick, J.; Dale, N.; Raemdonck, K.; Wong, S.; Catley, M.C.; Belvisi, M.G.; Birrell, M.A. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS ONE 2011, 6, e24097. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hanaoka, M.; Droma, Y.; Chen, P.; Voelkel, N.F.; Kubo, K. Endothelin-1 receptor antagonists prevent the development of pulmonary emphysema in rats. Eur. Respir. J. 2010, 35, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Nakayama, K.; Yasuda, H.; Kubo, H.; Kuwano, K.; Arai, H.; Yamaya, M. Carbocisteine inhibits oxidant-induced apoptosis in cultured human airway epithelial cells. Respirology 2009, 14, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, M.; Droma, Y.; Chen, Y.; Agatsuma, T.; Kitaguchi, Y.; Voelkel, N.F.; Kubo, K. Carbocisteine protects against emphysema induced by cigarette smoke extract in rats. Chest 2011, 139, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Dong, Z.; Dimitropoulou, C.; Su, Y. Hydrogen sulfide ameliorates tobacco smoke-induced oxidative stress and emphysema in mice. Antioxid. Redox Signal. 2011, 15, 2121–2134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, X.; Xie, W.; Li, Y.; Ma, M.; Yuan, T.; Luo, B. Resveratrol exerts an anti-apoptotic effect on human bronchial epithelial cells undergoing cigarette smoke exposure. Mol. Med. Rep. 2015, 11, 1752–1758. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Kang, M.J.; Crothers, K.; Zhu, Z.; Liu, W.; Lee, C.G.; Rabach, L.A.; Chapman, H.A.; Homer, R.J.; Aldous, D.; et al. Role of cathepsin S-dependent epithelial cell apoptosis in IFN-gamma-induced alveolar remodeling and pulmonary emphysema. J. Immunol. 2005, 174, 8106–8115. [Google Scholar] [CrossRef] [PubMed]
- Saetta, M.; Di Stefano, A.; Turato, G.; Facchini, F.M.; Corbino, L.; Mapp, C.E.; Maestrelli, P.; Ciaccia, A.; Fabbri, L.M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 157, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Lams, B.E.; Sousa, A.R.; Rees, P.J.; Lee, T.H. Subepithelial immunopathology of the large airways in smokers with and without chronic obstructive pulmonary disease. Eur. Respir. J. 2000, 15, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Grumelli, S.; Corry, D.B.; Song, L.Z.; Song, L.; Green, L.; Huh, J.; Hacken, J.; Espada, R.; Bag, R.; Lewis, D.E.; et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004, 1, e8. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, T.C.; Ansari, T.W.; Barnes, N.C.; Jeffery, P.K. Inflammation in bronchial biopsies of subjects with chronic bronchitis: Inverse relationship of CD8+ T lymphocytes with FEV1. Am. J. Respir. Crit. Care Med. 1997, 155, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zheng, T.; Zhu, Z.; Homer, R.J.; Riese, R.J.; Chapman, H.A., Jr.; Shapiro, S.D.; Elias, J.A. Interferon gamma induction of pulmonary emphysema in the adult murine lung. J. Exp. Med. 2000, 192, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, P.; Greene, C.M.; O’Mahony, M.; O’Neill, S.J.; Taggart, C.C.; McElvaney, N.G. Secretory leucocyte protease inhibitor inhibits interferon-gamma-induced cathepsin S expression. J. Biol. Chem. 2007, 282, 33389–33395. [Google Scholar] [CrossRef] [PubMed]
- Buhling, F.; Gerber, A.; Hackel, C.; Kruger, S.; Kohnlein, T.; Bromme, D.; Reinhold, D.; Ansorge, S.; Welte, T. Expression of cathepsin K in lung epithelial cells. Am. J. Respir. Cell. Mol. Biol. 1999, 20, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J. Cathepsin D. Purification of isoenzymes from human and chicken liver. Biochem. J. 1970, 117, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Mizuochi, T.; Yee, S.T.; Kasai, M.; Kakiuchi, T.; Muno, D.; Kominami, E. Both cathepsin B and cathepsin D are necessary for processing of ovalbumin as well as for degradation of class II mhc invariant chain. Immunol. Lett. 1994, 43, 189–193. [Google Scholar] [CrossRef]
- Diment, S.; Martin, K.J.; Stahl, P.D. Cleavage of parathyroid hormone in macrophage endosomes illustrates a novel pathway for intracellular processing of proteins. J. Biol. Chem. 1989, 264, 13403–13406. [Google Scholar] [PubMed]
- Khalkhali-Ellis, Z.; Abbott, D.E.; Bailey, C.M.; Goossens, W.; Margaryan, N.V.; Gluck, S.L.; Reuveni, M.; Hendrix, M.J. IFN-gamma regulation of vacuolarph, cathepsin D processing and autophagy in mammary epithelial cells. J. Cell. Biochem. 2008, 105, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Kalbacher, H. Cathepsin E: A mini review. Biochem. Biophys. Res. Commun. 2008, 367, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Keliher, E.J.; Reiner, T.; Earley, S.; Klubnick, J.; Tassa, C.; Lee, A.J.; Ramaswamy, S.; Bardeesy, N.; Hanahan, D.; Depinho, R.A.; et al. Targeting cathepsin E in pancreatic cancer by a small molecule allows in vivo detection. Neoplasia 2013, 15, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Kawakubo, T.; Yasukochi, A.; Toyama, T.; Takahashi, S.; Okamoto, K.; Tsukuba, T.; Nakamura, S.; Ozaki, Y.; Nishigaki, K.; Yamashita, H.; et al. Repression of cathepsin E expression increases the risk of mammary carcinogenesis and links to poor prognosis in breast cancer. Carcinogenesis 2014, 35, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Konno-Shimizu, M.; Yamamichi, N.; Inada, K.; Kageyama-Yahara, N.; Shiogama, K.; Takahashi, Y.; Asada-Hirayama, I.; Yamamichi-Nishina, M.; Nakayama, C.; Ono, S.; et al. Cathepsin E is a marker of gastric differentiation and signet-ring cell carcinoma of stomach: A novel suggestion on gastric tumorigenesis. PLoS ONE 2013, 8, e56766. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, T.; Kalita, J.; Weldon, S.; Taggart, C.C. Proteases and Their Inhibitors in Chronic Obstructive Pulmonary Disease. J. Clin. Med. 2018, 7, 244. https://doi.org/10.3390/jcm7090244
Dey T, Kalita J, Weldon S, Taggart CC. Proteases and Their Inhibitors in Chronic Obstructive Pulmonary Disease. Journal of Clinical Medicine. 2018; 7(9):244. https://doi.org/10.3390/jcm7090244
Chicago/Turabian StyleDey, Tapan, Jatin Kalita, Sinéad Weldon, and Clifford C. Taggart. 2018. "Proteases and Their Inhibitors in Chronic Obstructive Pulmonary Disease" Journal of Clinical Medicine 7, no. 9: 244. https://doi.org/10.3390/jcm7090244
APA StyleDey, T., Kalita, J., Weldon, S., & Taggart, C. C. (2018). Proteases and Their Inhibitors in Chronic Obstructive Pulmonary Disease. Journal of Clinical Medicine, 7(9), 244. https://doi.org/10.3390/jcm7090244