Idiopathic Pulmonary Fibrosis (IPF): An Overview



Abstract
1. Introduction
2. Epidemiology
3. Diagnosis
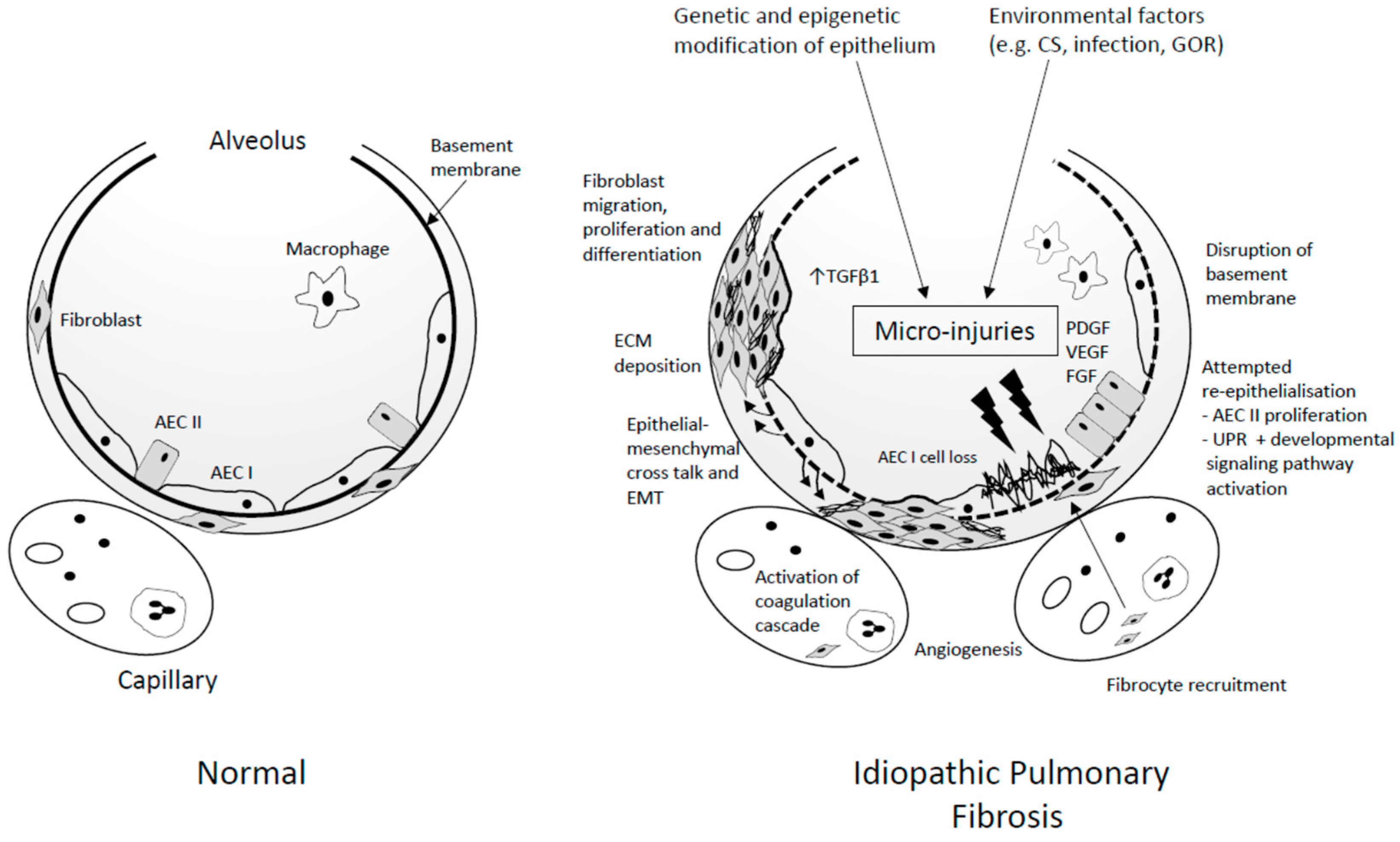
4. Pathogenesis
4.1. Initiation
4.1.1. Genetic Basis for IPF
4.1.2. Surfactant Protein-Related Genes
4.1.3. MUC5B Promoter Polymorphism
4.1.4. Other Gene Variants
4.1.5. Telomeres
4.2. Environmental Factors
4.3. Propagation
5. Treatment
6. Future Perspectives
6.1. Personalised Medicine
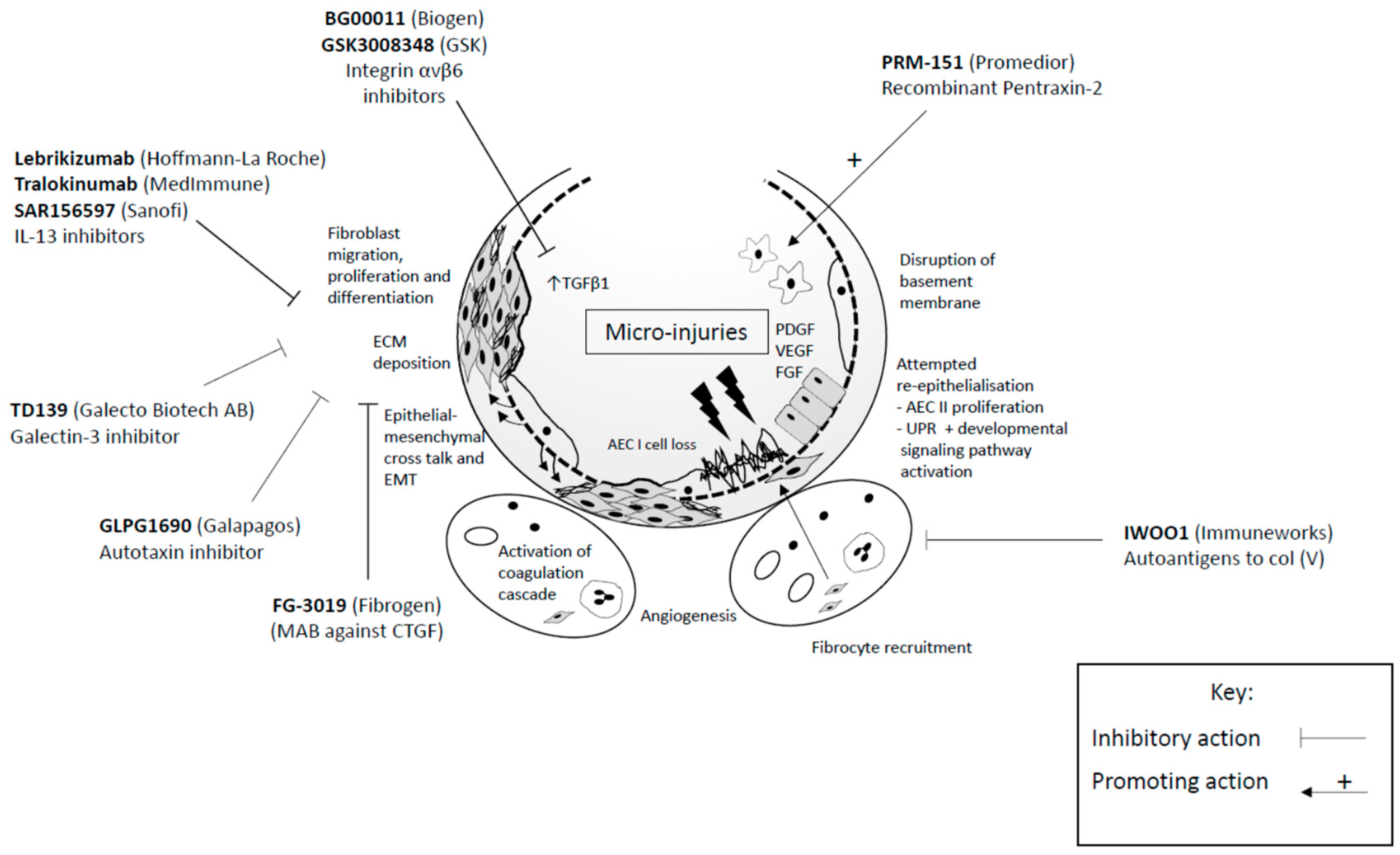
6.2. Novel Therapeutic Targets
6.2.1. Growth Factors and Associated Signalling Pathways
6.2.2. Cytokines
6.2.3. Alveolar Monocytes
6.2.4. Developing Tolerance to Autoantigens
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.; Jenkins, R.G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Chen, S.Y.; Yeh, W.S.; Maroni, B.; Li, Q.; Lee, Y.C.; Collard, H.R. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001–11. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Hopkins, R.B.; Burke, N.; Fell, C.; Dion, G.; Kolb, M. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur. Respir. J. 2016, 48, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Strongman, H.; Kausar, I.; Maher, T.M. Incidence, Prevalence, and Survival of Patients with Idiopathic Pulmonary Fibrosis in the UK. Adv. Ther. 2018, 35, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Gribbin, J.; Hubbard, R.B.; le Jeune, I.; Smith, C.J.; West, J.; Tata, L.J. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006, 61, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulos, A.; Wright, E.; Vlahopoulou, K.; Cornic, L.; Schoof, N.; Maher, T.M. The Burden of Illness of Idiopathic Pulmonary Fibrosis: A Comprehensive Evidence Review. Pharmacoeconomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Kaunisto, J.; Kelloniemi, K.; Sutinen, E.; Hodgson, U.; Piilonen, A.; Kaarteenaho, R.; Makitaro, R.; Purokivi, M.; Lappi-Blanco, E.; Saarelainen, S.; et al. Re-evaluation of diagnostic parameters is crucial for obtaining accurate data on idiopathic pulmonary fibrosis. BMC Pulm. Med. 2015, 15, 92. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.C.; Salciccioli, J.D.; Shea, B.S.; Akuthota, P. Trends in mortality from idiopathic pulmonary fibrosis in the European Union: An observational study of the WHO mortality database from 2001–2013. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Algranti, E.; Saito, C.A.; Silva, D.; Carneiro, A.P.S.; Bussacos, M.A. Mortality from idiopathic pulmonary fibrosis: A temporal trend analysis in Brazil, 1979–2014. J. Bras. Pneumol. 2017, 43, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Costabel, U.; Albera, C.; Lancaster, L.H.; Lin, C.Y.; Hormel, P.; Hulter, H.N.; Noble, P.W. An Open-Label Study of the Long-Term Safety of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (RECAP). Respiration 2017, 94, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Tooze, J.A.; Schwarz, M.I.; Brown, K.R.; Cherniack, R.M. Predicting survival in idiopathic pulmonary fibrosis: Scoring system and survival model. Am. J. Respir. Crit. Care Med. 2001, 164, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Ryerson, C.J.; Vittinghoff, E.; Ryu, J.H.; Tomassetti, S.; Lee, J.S.; Poletti, V.; Buccioli, M.; Elicker, B.M.; Jones, K.D.; et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann. Intern. Med. 2012, 156, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.P.; McKeever, T.M.; Fogarty, A.W.; Navaratnam, V.; Hubbard, R.B. Surgical lung biopsy for the diagnosis of interstitial lung disease in England: 1997–2008. Eur. Respir. J. 2016, 48, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Lynch, D.; Godwin, J.D.; Webb, R.; Colby, T.V.; Leslie, K.O.; Behr, J.; Brown, K.K.; Egan, J.J.; Flaherty, K.R.; et al. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: Secondary analysis of a randomised, controlled trial. Lancet Respir. Med. 2014, 2, 277–284. [Google Scholar] [CrossRef]
- Chung, J.H.; Chawla, A.; Peljto, A.L.; Cool, C.D.; Groshong, S.D.; Talbert, J.L.; McKean, D.F.; Brown, K.K.; Fingerlin, T.E.; Schwarz, M.I.; et al. CT scan findings of probable usual interstitial pneumonitis have a high predictive value for histologic usual interstitial pneumonitis. Chest 2015, 147, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.U. Any fool can make a rule and any fool will mind it. BMC Med. 2016, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V. Lung biopsy in interstitial lung disease: Balancing the risk of surgery and diagnostic uncertainty. Eur. Respir. J. 2016, 48, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Cosgrove, P.; Bianchi, P.; Danese, S.; Lederer, D.J. Barriers to timely diagnosis of interstitial lung disease in the real world: The INTENSITY survey. BMC Pulm. Med. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, S.; Wells, A.U.; Costabel, U.; Cavazza, A.; Colby, T.V.; Rossi, G.; Sverzellati, N.; Carloni, A.; Carretta, E.; Buccioli, M.; et al. Bronchoscopic Lung Cryobiopsy Increases Diagnostic Confidence in the Multidisciplinary Diagnosis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, H.; Nakagawa, H.; Niimi, A. Computer-based quantitative computed tomography image analysis in idiopathic pulmonary fibrosis: A mini review. Respir. Investig. 2018, 56, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.L.; Calandriello, L.; Sverzellati, N.; Wells, A.U.; Hansell, D.M. Interobserver agreement for the ATS/ERS/JRS/ALAT criteria for a UIP pattern on CT. Thorax 2016, 71, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Rabeyrin, M.; Thivolet, F.; Ferretti, G.R.; Chalabreysse, L.; Jankowski, A.; Cottin, V.; Pison, C.; Cordier, J.F.; Lantuejoul, S. Usual interstitial pneumonia end-stage features from explants with radiologic and pathological correlations. Ann. Diagn. Pathol. 2015, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Nogee, L.M.; Dunbar, A.E.; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.Q.; Lane, K.; Phillips, J.; Prince, M.; Markin, C.; Speer, M.; Schwartz, D.A.; Gaddipati, R.; Marney, A.; Johnson, J.; et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 2002, 165, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- van Moorsel, C.H.; van Oosterhout, M.F.; Barlo, N.P.; de Jong, P.A.; van der Vis, J.J.; Ruven, H.J.; van Es, H.W.; van den Bosch, J.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Usinger, W.; Nichols, B.; Gray, J.; Xu, L.; Seeley, T.W.; Brenner, M.; Guo, G.; Zhang, W.; Oliver, N.; et al. Cooperative interaction of CTGF and TGF-beta in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Crossno, P.F.; Polosukhin, V.V.; Blackwell, T.S.; Johnson, J.E.; Markin, C.; Moore, P.E.; Worrell, J.A.; Stahlman, M.T.; Phillips, J.A.; Loyd, J.E.; et al. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest 2010, 137, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Grant, S.W.; Ambrosini, V.; Womble, K.E.; Dawson, E.P.; Lane, K.B.; Markin, C.; Renzoni, E.; Lympany, P.; Thomas, A.Q.; et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 2004, 59, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Blackwell, T.S.; Lawson, W.E. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L721–L729. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; JMaguire, A.; Newitt, J.L.; Russo, S.J.; Kotorashvili, A.; Beers, M.F. Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L720–L729. [Google Scholar] [CrossRef] [PubMed]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Cheng, D.S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; AWise, L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Noth, I.; Garcia, J.G.; Kaminski, N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1576–1577. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.J.; Sato, H.; Fonseca, C.; Banya, W.A.; Molyneaux, P.L.; Adamali, H.; Russell, A.M.; Denton, C.P.; Abraham, D.J.; Hansell, D.M.; et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax 2013, 68, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Fingerlin, T.E.; Evans, C.M.; Schwarz, M.I.; Schwartz, D.A. MUC5B and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2015. [Google Scholar] [CrossRef]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Hong, A.; Solomon, M.J.; Lee, C.S. The role of telomeres and telomerase in the pathology of human cancer and aging. Pathology 2006, 38, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Shammas, M.A. Telomeres, lifestyle, cancer, and aging. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M. Telomeres and age-related disease: How telomere biology informs clinical paradigms. J. Clin. Invest. 2013, 123, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, K.D.; JCronkhite, T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Mitchell, D.B.; Markin, C.; Polosukhin, V.V.; Choi, L.; Johnson, J.E.; Lawson, W.E.; Phillips, J.A.; Cogan, J.D.; Blackwell, T.S.; et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest 2014, 146, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Stanley, S.E.; Wagner, C.L.; Hamilton, M.; Hanumanthu, V.S.; Armanios, M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 2015, 147, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Kannengiesser, C.; Borie, R.; Ménard, C.; Réocreux, M.; Nitschké, P.; Gazal, S.; Mal, H.; Taillé, C.; Cadranel, J.; Nunes, H.; et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur. Respir. J. 2015, 46, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.A.; Batra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.S.; Aravena, C.; Meyer, K.; Raghu, G.; Collard, H.R.; Garcia, C.K. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur. Respir. J. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.P.; Speer, M.C.; Loyd, J.E.; Brown, K.K.; Herron, A.; Slifer, S.H.; Burch, L.H.; Wahidi, M.M.; Phillips, J.A.; Sporn, T.A.; et al. Clinical and pathologic features of familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2005, 172, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Borie, R.; Kannengiesser, C.; Debray, M.P.; Crestani, B. The Genetic Diagnosis of Interstitial Lung Disease: A Need for an International Consensus. Am. J. Respir. Crit. Care Med. 2017, 195, 1538–1539. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Lee, J.S.; Kozlitina, J.; Noth, I.; Devine, M.S.; Glazer, C.S.; Torres, F.; Kaza, V.; Girod, C.E.; Jones, K.D.; et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: An observational cohort study with independent validation. Lancet Respir. Med. 2014, 2, 557–565. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, D.; Xu, Y.; Zhang, J.; Wang, Y.; Chen, M.; Lin, S.; Huang, L.; Chung, E.J.; Citrin, D.E.; et al. Inhibition of Bcl-2/xl with ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Manguan-Garcia, C.; Pintado-Berninches, L.; Carrillo, J.; Machado-Pinilla, R.; Sastre, L.; Pérez-Quilis, C.; Esmoris, I.; Gimeno, A.; García-Giménez, J.L.; Pallardó, F.V.; et al. Expression of the genetic suppressor element 24.2 (GSE24.2) decreases DNA damage and oxidative stress in X-linked dyskeratosis congenita cells. PLoS ONE 2014, 9, e101424. [Google Scholar] [CrossRef] [PubMed]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.; Beane, J.; Shah, V.; Liu, G.; Schembri, F.; Yang, X.; Palma, J.; Brody, J.S. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc. Natl. Acad. Sci. USA 2004, 101, 10143–10148. [Google Scholar] [CrossRef] [PubMed]
- Tennis, M.A.; Vanscoyk, M.M.; Wilson, L.A.; Kelley, N.; Winn, R.A. Methylation of Wnt7a is modulated by DNMT1 and cigarette smoke condensate in non-small cell lung cancer. PLoS ONE 2012, 7, e32921. [Google Scholar] [CrossRef] [PubMed]
- Buro-Auriemma, L.J.; Salit, J.; Hackett, N.R.; Walters, M.S.; Strulovici-Barel, Y.; Staudt, M.R.; Fuller, J.; Mahmoud, M.; Stevenson, C.S.; Hilton, H.; et al. Cigarette smoking induces small airway epithelial epigenetic changes with corresponding modulation of gene expression. Hum. Mol. Genet. 2013, 22, 4726–4738. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Killian, J.K.; Yang, M.; Walker, R.L.; Hong, J.A.; Zhang, M.; Davis, S.; Zhang, Y.; Hussain, M.; Xi, S.; et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene 2010, 29, 3650–3664. [Google Scholar] [CrossRef] [PubMed]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A.M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Bowers, J.; Vaziri, C.; et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Calin, G.A.; Steele, V.E.; Croce, C.M.; de Flora, S. Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J. 2009, 23, 3243–3250. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.W.; Johnson, J.E.; Browning, P.J.; Cruz-Gervis, R.A.; Davis, A.; Graham, B.S.; Brigham, K.L.; Oates, J.A.; Loyd, J.E.; Stecenko, A.A. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Microbiol. 2003, 41, 2633–2640. [Google Scholar] [CrossRef] [PubMed]
- Vergnon, J.M.; Vincent, M.; de Thé, G.; Mornex, J.F.; Weynants, P.; Brune, J. Cryptogenic fibrosing alveolitis and Epstein-Barr virus: An association? Lancet 1984, 2, 768–771. [Google Scholar] [CrossRef]
- Manika, K.; Alexiou-Daniel, S.; Papakosta, D.; Papa, A.; Kontakiotis, T.; Patakas, D.; Antoniadis, A. Epstein-Barr virus DNA in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2007, 24, 134–140. [Google Scholar] [PubMed]
- Stewart, J.P.; Egan, J.J.; Ross, A.J.; Kelly, B.G.; Lok, S.S.; Hasleton, P.S.; Woodcock, A.A. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1999, 159, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.G.; Lok, S.S.; Hasleton, P.S.; Egan, J.J.; Stewart, J.P. A rearranged form of Epstein-Barr virus DNA is associated with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2002, 166, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Lawson, W.E.; Blackwell, T.S. Right place, right time: The evolving role of herpesvirus infection as a “second hit” in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L441–L444. [Google Scholar] [CrossRef] [PubMed]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Shulgina, L.; Cahn, A.P.; Chilvers, E.R.; Parfrey, H.; Clark, A.B.; Wilson, E.C.; Twentyman, O.P.; Davison, A.G.; Curtin, J.J.; Crawford, M.B.; et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: A randomised controlled trial. Thorax 2013, 68, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.; Clark, A.B.; Cahn, A.P.; Chilvers, E.R.; Fraser, W.D.; Livermore, D.M.; Maher, T.M.; Parfrey, H.; Swart, A.M.; Stirling, S.; et al. The Efficacy and Mechanism Evaluation of Treating Idiopathic Pulmonary fibrosis with the Addition of Co-trimoxazole (EME-TIPAC): Study protocol for a randomised controlled trial. Trials 2018, 19, 89. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.; Crooks, M.; Hart, S.P. Gastroesophageal reflux and idiopathic pulmonary fibrosis: A review. Pulm. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Appel, J.Z.; Lee, S.M.; Hartwig, M.G.; Li, B.; Hsieh, C.C.; Cantu, E.; Yoon, Y.; Lin, S.S.; Parker, W.; Davis, R.D. Characterization of the innate immune response to chronic aspiration in a novel rodent model. Respir. Res. 2007, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Song, J.W.; Wolters, P.J.; Elicker, B.M.; King, T.E.; Kim, D.S.; Collard, H.R. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Fidler, L.; Sitzer, N.; Shapera, S.; Shah, P.S. Treatment of Gastroesophageal Reflux in Patients with Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Chest 2018. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Collard, H.R.; Raghu, G.; Sweet, M.P.; Hays, S.R.; Campos, G.M.; Golden, J.A.; King, T.E. Does chronic microaspiration cause idiopathic pulmonary fibrosis? Am. J. Med. 2010, 123, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Gnanapandithan, K.; Popkin, J.H.; Devadoss, R.; Martin, K. Gastroesophageal reflux and idiopathic pulmonary fibrosis: A long term relationship. Respir. Med. Case Rep. 2016, 17, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef] [PubMed]
- Basset, F.; Ferrans, V.J.; Soler, P.; Takemura, T.; Fukuda, Y.; Crystal, R.G. Intraluminal fibrosis in interstitial lung disorders. Am. J. Pathol. 1986, 122, 443–461. [Google Scholar] [PubMed]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Borensztajn, K.; Spek, C.A. Targeting coagulation factor receptors—Protease-activated receptors in idiopathic pulmonary fibrosis. J. Thromb. Haemost. 2017, 15, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, G.P.; Brown, K.K.; Schiemann, W.P.; Serls, A.E.; Parr, J.E.; Geraci, M.W.; Schwarz, M.I.; Cool, C.D.; Worthen, G.S. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: A role in aberrant angiogenesis. Am. J. Respir. Crit. Care Med. 2004, 170, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Ebina, M.; Shimizukawa, M.; Shibata, N.; Kimura, Y.; Suzuki, T.; Endo, M.; Sasano, H.; Kondo, T.; Nukiwa, T. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2004, 169, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Mechanisms of fibrogenesis. Exp. Biol. Med. 2008, 233, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [PubMed]
- Zavadil, J.; Böttinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell. Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu. Rev. Physiol. 2011, 73, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.L.; Katzenstein, A.L. Epithelial necrosis and alveolar collapse in the pathogenesis of usual interstitial pneumonia. Chest 1988, 94, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet. 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-Dose Acetylcysteine in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242. [Google Scholar] [CrossRef] [PubMed]
- The Idiopathic Pulmonary Fibrosis Clinical Research Network. Prednisone, Azathioprine, and N-Acetylcysteine for Pulmonary Fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; de Andrade, J.A.; Anstrom, K.J.; King, T.E.; Raghu, G.; Idiopathic Pulmonary Fibrosis Clinical Research Network. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Inoue, H.; Nakazawa, R.; Azuma, N.; Suzuki, M.; Yamauchi, S.; Margolin, S.B.; Tsubota, K.; Saito, I. Pirfenidone induces intercellular adhesion molecule-1 (ICAM-1) down-regulation on cultured human synovial fibroblasts. Clin. Exp. Immunol. 1998, 113, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Azuma, A.; Nukiwa, T.; Tsuboi, E.; Suga, M.; Abe, S.; Nakata, K.; Taguchi, Y.; Nagai, S.; Itoh, H.; Ohi, M.; et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010, 35, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Bois, R.M.d.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Lancaster, L.; et al. Pirfenidone for idiopathic pulmonary fibrosis: Analysis of pooled data from three multinational phase 3 trials. Eur. Respir. J. 2016, 47, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Albera, C.; Costabel, U.; Fagan, E.A.; Glassberg, M.K.; Gorina, E.; Lancaster, L.; Lederer, D.J.; Nathan, S.D.; Spirig, D.; Swigris, J.J. Efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis with more preserved lung function. Eur. Respir. J. 2016, 48, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Cottin, V.; Bois, R.M.d.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS((R)) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Richeldi, L.; Kim, D.S.; Taniguchi, H.; Tschoepe, I.; Luisetti, M.; Roman, J.; Tino, G.; Schlenker-Herceg, R.; Hallmann, C.; et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [PubMed]
- Costabel, U.; Inoue, Y.; Richeldi, L.; Collard, H.R.; Tschoepe, I.; Stowasser, S.; Azuma, A. Efficacy of Nintedanib in Idiopathic Pulmonary Fibrosis across Prespecified Subgroups in INPULSIS. Am. J. Respir. Crit. Care Med. 2016, 193, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Richeldi, L.; Behr, J.; Maher, T.M.; Tang, W.; Stowasser, S.; Hallmann, C.; du Bois, R.M. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax 2017, 72, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Wells, A.U.; Nicholson, A.G.; Richeldi, L.; Flaherty, K.R.; le Maulf, F.; Stowasser, S.; Schlenker-Herceg, R.; Hansell, D.M. Effect of Nintedanib in Subgroups of Idiopathic Pulmonary Fibrosis by Diagnostic Criteria. Am. J. Respir. Crit. Care Med. 2017, 195, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Canestaro, W.J.; Forrester, S.H.; Raghu, G.; Ho, L.; Devine, B.E. Drug Treatment of Idiopathic Pulmonary Fibrosis: Systematic Review and Network Meta-Analysis. Chest 2016, 149, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Loveman, E.; Copley, V.R.; Scott, D.A.; Colquitt, J.L.; Clegg, A.J.; O’Reilly, K.M. Comparing new treatments for idiopathic pulmonary fibrosis-a network meta-analysis. BMC Pulm. Med. 2015, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Hayton, C.; Chaudhuri, N. Managing Idiopathic Pulmonary Fibrosis: Which Drug for Which Patient? Drugs Aging 2017, 34, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.; Toellner, H.; Morris, H.; Leonard, C.; Chaudhuri, N. Real World Experiences: Pirfenidone and Nintedanib are Effective and Well Tolerated Treatments for Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Brunnemer, E.; Walscher, J.; Tenenbaum, S.; Hausmanns, J.; Schulze, K.; Seiter, M.; Heussel, C.P.; Warth, A.; Herth, F.J.F.; Kreuter, M. Real-World Experience with Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Respiration 2018, 95, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Karampitsakos, T.; Ntolios, P.; Tzilas, V.; Bouros, E.; Markozannes, E.; Malliou, I.; Anagnostopoulos, A.; Granitsas, A.; Steiropoulos, P.; et al. Longitudinal “Real-World” Outcomes of Pirfenidone in Idiopathic Pulmonary Fibrosis in Greece. Front. Med. 2017, 4, 213. [Google Scholar] [CrossRef] [PubMed]
- Galli, J.A.; Pandya, A.; Vega-Olivo, M.; Dass, C.; Zhao, H.; Criner, G.J. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: Tolerability and adverse drug reactions. Respirology 2017, 22, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Bonella, F.; Kreuter, M.; Hagmeyer, L.; Neurohr, C.; Keller, C.; Kohlhaeufl, M.J.; Muller-Quernheim, J.; Milger, K.; Prasse, A. Consortium German Nintedanib Compassionate Use. Insights from the German Compassionate Use Program of Nintedanib for the Treatment of Idiopathic Pulmonary Fibrosis. Respiration 2016, 92, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Karampitsakos, T.; Kontou, M.; Granitsas, A.; Malliou, I.; Anagnostopoulos, A.; Ntolios, P.; Tzilas, V.; Bouros, E.; Steiropoulos, P.; et al. Safety and efficacy of nintedanib in idiopathic pulmonary fibrosis: A real-life observational study in Greece. Pulm. Pharmacol. Ther. 2018, 49, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Marshall, T.; Morris, H.; Hayton, C.; Chaudhuri, N. Idiopathic pulmonary fibrosis: A holistic approach to disease management in the antifibrotic age. J. Thorac. Dis. 2017, 9, 4700–4707. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Taniguchi, H.; Azuma, A.; Inoue, Y.; Kondoh, Y.; Hasegawa, Y.; Bando, M.; Abe, S.; Mochizuki, Y.; Chida, K.; et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Kreuter, M.; Richeldi, L.; Ryerson, C.J.; Valeyre, D.; Grutters, J.C.; Wiebe, S.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Nintedanib with Add-on Pirfenidone in Idiopathic Pulmonary Fibrosis. Results of the INJOURNEY Trial. Am. J. Respir. Crit. Care Med. 2018, 197, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Brownell, R.; Kaminski, N.; Woodruff, P.G.; Bradford, W.Z.; Richeldi, L.; Martinez, F.J.; Collard, H.R. Precision Medicine: The New Frontier in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.A. Personalized medicine: A personal view. Clin. Pharmacol. Ther. 2012, 91, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Brown, K.K.; Collard, H.R. Molecular biomarkers in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L681–L691. [Google Scholar] [CrossRef] [PubMed]
- Guiot, J.; Moermans, C.; Henket, M.; Corhay, J.L.; Louis, R. Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Lung 2017, 195, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Oikonomidi, S.; Kostikas, K.; Tsilioni, I.; Tanou, K.; Gourgoulianis, K.I.; Kiropoulos, T.S. Matrix metalloproteinases in respiratory diseases: From pathogenesis to potential clinical implications. Curr. Med. Chem. 2009, 16, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E.; Lindell, K.O.; Cisneros, J.; Macdonald, S.D.; Pardo, A.; et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008, 5, e93. [Google Scholar] [CrossRef] [PubMed]
- White, E.S.; Xia, M.; Murray, S.; Dyal, R.; Flaherty, C.M.; Flaherty, K.R.; Moore, B.B.; Cheng, L.; Doyle, T.J.; Villalba, J.; et al. Plasma Surfactant Protein-D, Matrix Metalloproteinase-7, and Osteopontin Index Distinguishes Idiopathic Pulmonary Fibrosis from Other Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2016, 194, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Sun, J.; Molyneaux, P.L.; Li, Q.; Villalba, J.A.; Tzouvelekis, A.; Lynn, H.; Juan-Guardela, B.M.; Risquez, C.; Osorio, J.C.; et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: An international, multicentre, cohort study. Lancet Respir. Med. 2017, 5, 857–868. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Herazo-Maya, J.D.; Slade, M.; Chu, J.H.; Deiuliis, G.; Ryu, C.; Li, Q.; Sakamoto, K.; Ibarra, G.; Pan, H.; et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology 2017, 22, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M. PROFILEing idiopathic pulmonary fibrosis: Rethinking biomarker discovery. Eur. Respir. Rev. 2013, 22, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.G.; Simpson, J.K.; Saini, G.; Bentley, J.H.; Russell, A.M.; Braybrooke, R.; Molyneaux, P.L.; McKeever, T.M.; Wells, A.U.; Flynn, A.; et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: An analysis from the prospective, multicentre PROFILE study. Lancet Respir. Med. 2015, 3, 462–472. [Google Scholar] [CrossRef]
- Ishikawa, N.; Hattori, N.; Yokoyama, A.; Kohno, N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir. Investig. 2012, 50, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Yokoyama, A.; Kondo, K.; Hamada, H.; Abe, M.; Nishimura, K.; Hiwada, K.; Kohno, N. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am. J. Respir. Crit. Care Med. 2002, 165, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Greene, K.E.; TKing, E.; Kuroki, Y.; Bucher-Bartelson, B.; Hunninghake, G.W.; Newman, L.S.; Nagae, H.; Mason, R.J. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur. Respir. J. 2002, 19, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Mukae, H.; Kadota, J.; Kaida, H.; Nagata, T.; Abe, K.; Matsukura, S.; Kohno, S. High serum concentrations of surfactant protein A in usual interstitial pneumonia compared with non-specific interstitial pneumonia. Thorax 2003, 58, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Do, K.H.; Jang, S.J.; Colby, T.V.; Han, S.; Kim, D.S. Blood biomarkers MMP-7 and SP-A: Predictors of outcome in idiopathic pulmonary fibrosis. Chest 2013, 143, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Guiot, J.; Bondue, B.; Henket, M.; Corhay, J.L.; Louis, R. Raised serum levels of IGFBP-1 and IGFBP-2 in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2016, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Herazo-Maya, J.D.; Noth, I.; Duncan, S.R.; Kim, S.; Ma, S.F.; Tseng, G.C.; Feingold, E.; Juan-Guardela, B.M.; Richards, T.J.; Lussier, Y.; et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2013, 5, 205ra136. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M.; Agusti, A.; Crestani, B.; Schwartz, D.A.; Königshoff, M.; Chambers, R.C.; Maher, T.M.; Faner, R.; Mora, A.L.; Rojas, M.; et al. Towards a global initiative for fibrosis treatment (GIFT). ERJ Open Res. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Scholand, M.B.; de Andrade, J.; Lancaster, L.; Mageto, Y.; Goldin, J.; Brown, K.K.; Flaherty, K.R.; Wencel, M.; Wanger, J.; et al. FG-3019 anti-connective tissue growth factor monoclonal antibody: Results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 47, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Tatler, A.L.; Jenkins, G. TGF-beta activation and lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Maden, C.H.; Fairman, D.; Chalker, M.; Costa, M.J.; Fahy, W.A.; Garman, N.; Lukey, P.T.; Mant, T.; Parry, S.; Simpson, J.K.; et al. Safety, tolerability and pharmacokinetics of GSK3008348, a novel integrin alphavbeta6 inhibitor, in healthy participants. Eur. J. Clin. Pharmacol. 2018, 74, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M. Autotaxin emerges as a therapeutic target for idiopathic pulmonary fibrosis: Limiting fibrosis by limiting lysophosphatidic acid synthesis. Am. J. Respir. Cell. Mol. Biol. 2012, 47, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; van der Aar, E.M.; van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018. [Google Scholar] [CrossRef]
- Zhu, Z.; Homer, R.J.; Wang, Z.; Chen, Q.; Geba, G.P.; Wang, J.; Zhang, Y.; Elias, J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Investig. 1999, 103, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Belperio, J.A.; Dy, M.; Burdick, M.D.; Xue, Y.Y.; Li, K.; Elias, J.A.; Keane, M.P. Interaction of IL-13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2002, 27, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Zhang, H.; Oak, S.R.; Coelho, A.L.; Herath, A.; Flaherty, K.R.; Lee, J.; Bell, M.; Knight, D.A.; Martinez, F.J.; et al. Targeting interleukin-13 with tralokinumab attenuates lung fibrosis and epithelial damage in a humanized SCID idiopathic pulmonary fibrosis model. Am. J. Respir. Cell. Mol. Biol. 2014, 50, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Castaño, A.P.; Lin, S.L.; Surowy, T.; Nowlin, B.T.; Turlapati, S.A.; Patel, T.; Singh, A.; Li, S.; Lupher, M.L.; Duffield, J.S. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci. Transl. Med. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Dillingh, M.R.; van den Blink, B.; Moerland, M.; van Dongen, M.G.; Levi, M.; Kleinjan, A.; Wijsenbeek, M.S.; Lupher, M.L.; Harper, D.M.; Getsy, J.A.; et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm. Pharmacol. Ther. 2013, 26, 672–676. [Google Scholar] [CrossRef] [PubMed]
- van den Blink, B.; Dillingh, M.R.; Ginns, L.C.; Morrison, L.D.; Moerland, M.; Wijsenbeek, M.; Trehu, E.G.; Bartholmai, B.J.; Burggraaf, J. Recombinant human pentraxin-2 therapy in patients with idiopathic pulmonary fibrosis: Safety, pharmacokinetics and exploratory efficacy. Eur. Respir. J. 2016, 47, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.E.; Gao, W.; Levy, D.; Santhanakrishnan, R.; Araki, T.; Rosas, I.O.; Hatabu, H.; Latourelle, J.C.; Nishino, M.; Dupuis, J.; et al. Galectin-3 Is Associated with Restrictive Lung Disease and Interstitial Lung Abnormalities. Am. J. Respir. Crit. Care Med. 2016, 194, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Vittal, R.; Mickler, E.A.; Fisher, A.J.; Zhang, C.; Rothhaar, K.; Gu, H.; Brown, K.M.; Emtiazdjoo, A.; Lott, J.M.; Frye, S.B.; et al. Type V collagen induced tolerance suppresses collagen deposition, TGF-beta and associated transcripts in pulmonary fibrosis. PLoS ONE 2013, 8, e76451. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla, J.L.; Love, R.B.; Jankowska-Gan, E.; Xu, Q.; Haynes, L.D.; Braun, R.K.; Hayney, M.S.; del Rio, A.M.; Meyer, K.; Greenspan, D.S.; et al. Th-17, monokines, collagen type V, and primary graft dysfunction in lung transplantation. Am. J. Respir. Crit. Care Med. 2008, 177, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Burlingham, W.J.; Love, R.B.; Jankowska-Gan, E.; Haynes, L.D.; Xu, Q.; Bobadilla, J.L.; Meyer, K.C.; Hayney, M.S.; Braun, R.K.; Greenspan, D.S.; et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J. Clin. Investig. 2007, 117, 3498–3506. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, D.S.; Chew, T.; Flaherty, K.R.; Frye, S.; Gibson, K.F.; Kaminski, N.; Klemsz, M.J.; Lange, W.; Noth, I.; Rothhaar, K. Oral immunotherapy with type V collagen in idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1393–1402. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Name | Function |
|---|---|---|
| TERT | Telomerase reverse transcriptase | Encodes the protein subunit of telomerase (hTERT) |
| TERC | Telomerase RNA component | Encodes the RNA subunit of telomerase (hTR) |
| DKC-1 | Dyskerin pseudouridine synthase 1 | Active role in telomerase stabilisation |
| TINF2 | TRF1 (Telomerase repeat factor 1) interacting nuclear factor 2 | Provides instructions of making part of the shelterin protein complex |
| RTEL1 | Regulator of telomere elongation helicase 1 | Encodes a DNA helicase that helps stabilize telomeres during RNA replication |
| PARN | Poly(A) specific ribonuclease | Involved in TERC RNA maturation |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. https://doi.org/10.3390/jcm7080201
Barratt SL, Creamer A, Hayton C, Chaudhuri N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. Journal of Clinical Medicine. 2018; 7(8):201. https://doi.org/10.3390/jcm7080201
Chicago/Turabian StyleBarratt, Shaney L., Andrew Creamer, Conal Hayton, and Nazia Chaudhuri. 2018. "Idiopathic Pulmonary Fibrosis (IPF): An Overview" Journal of Clinical Medicine 7, no. 8: 201. https://doi.org/10.3390/jcm7080201
APA StyleBarratt, S. L., Creamer, A., Hayton, C., & Chaudhuri, N. (2018). Idiopathic Pulmonary Fibrosis (IPF): An Overview. Journal of Clinical Medicine, 7(8), 201. https://doi.org/10.3390/jcm7080201