
The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart




Abstract
:
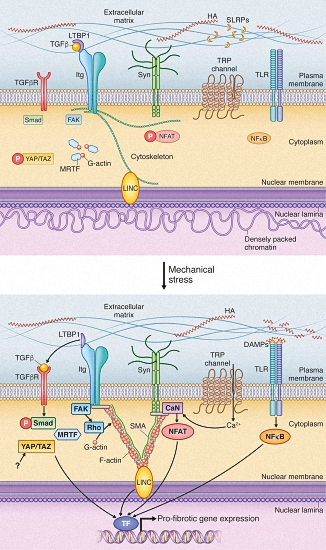{kind=link}
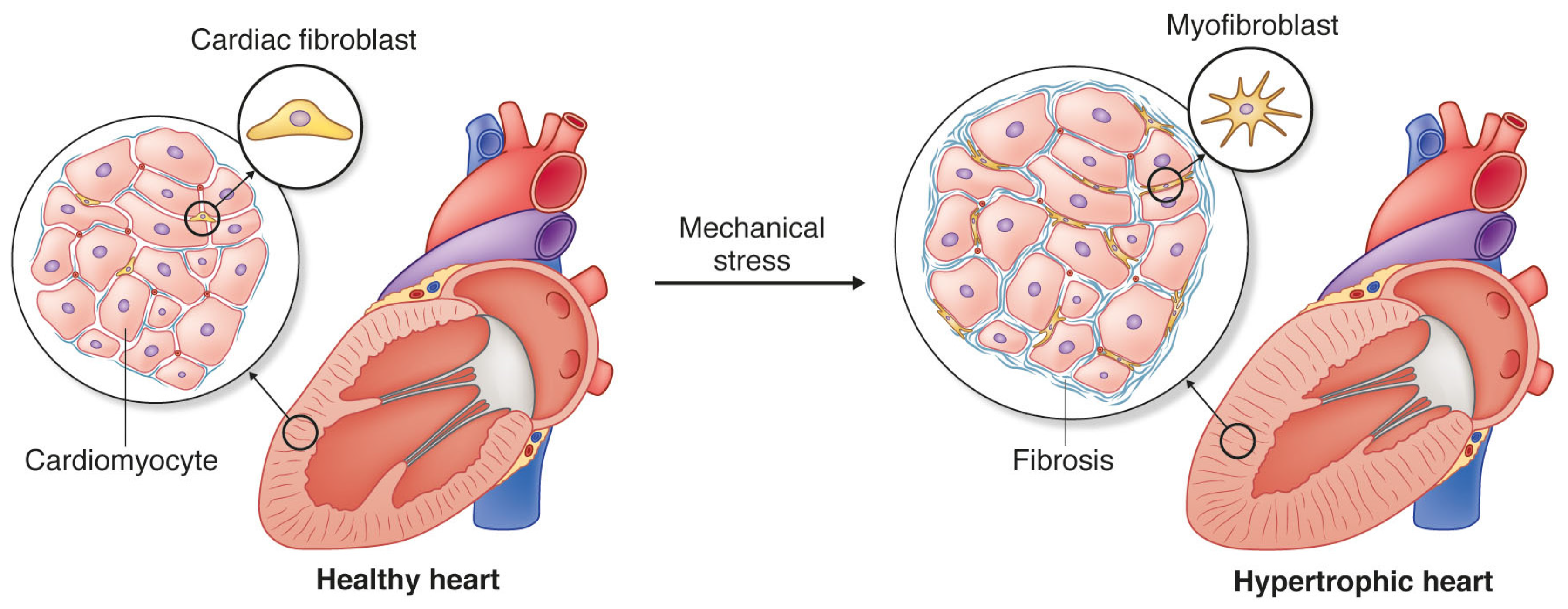{kind=link}
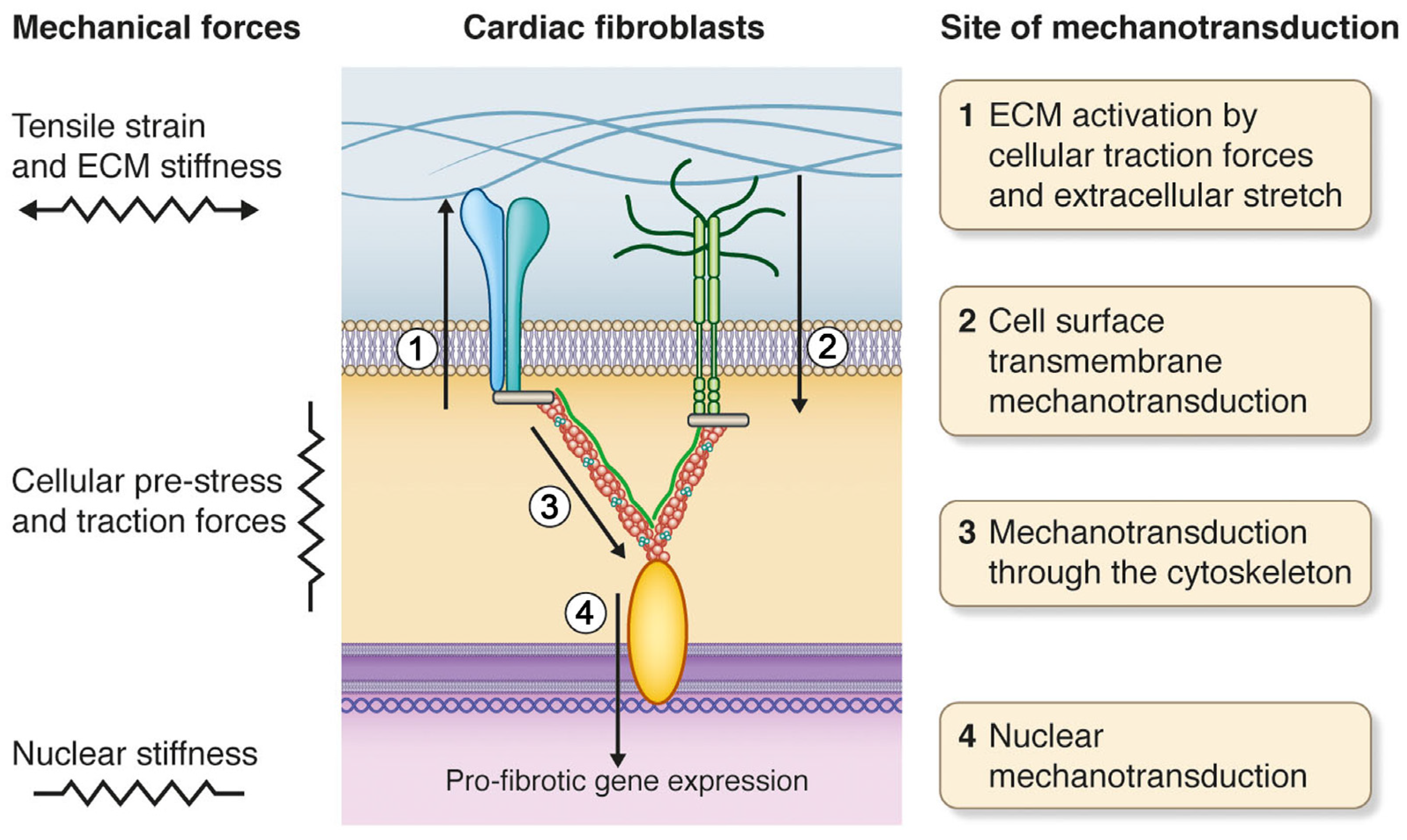{kind=link}
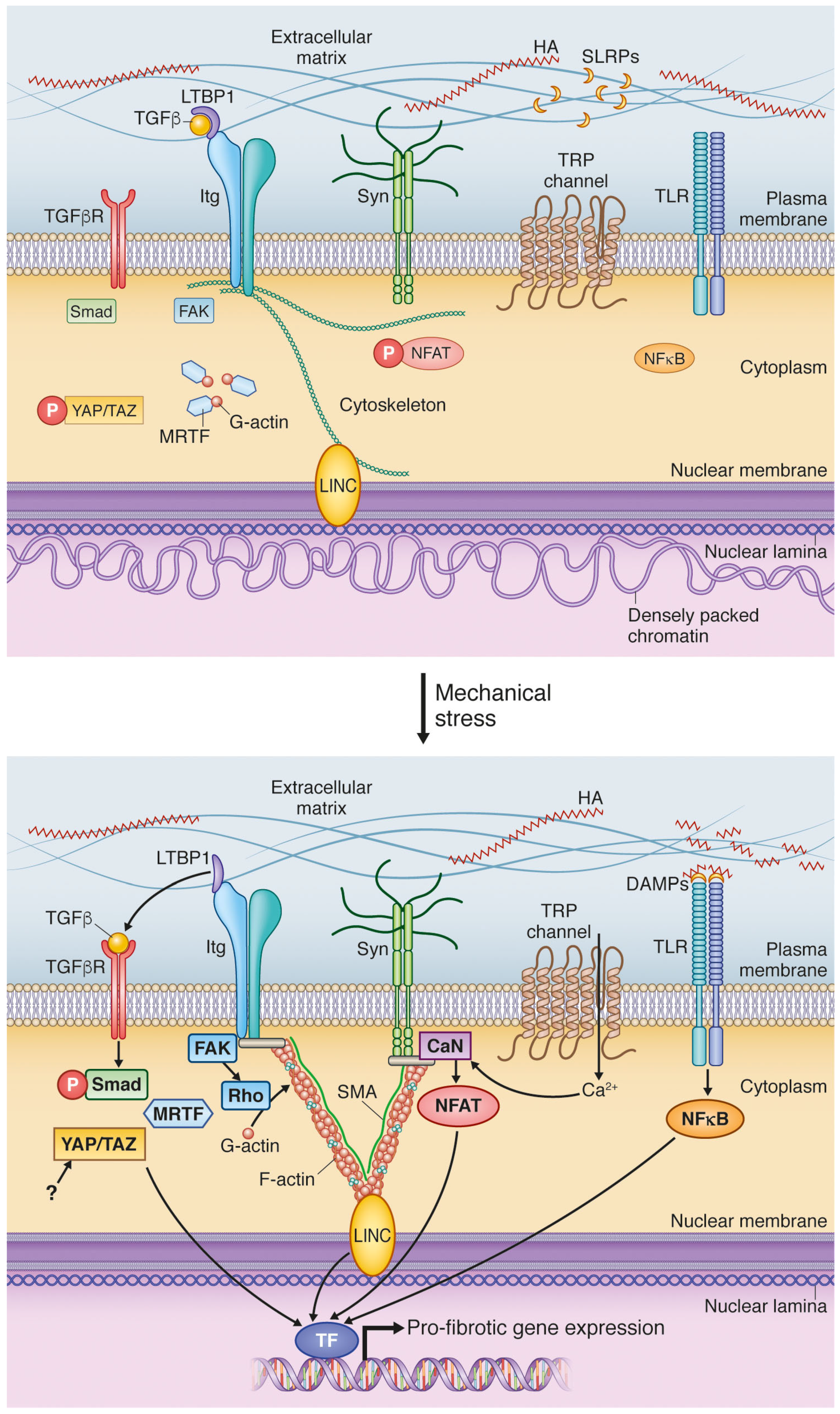{kind=link}
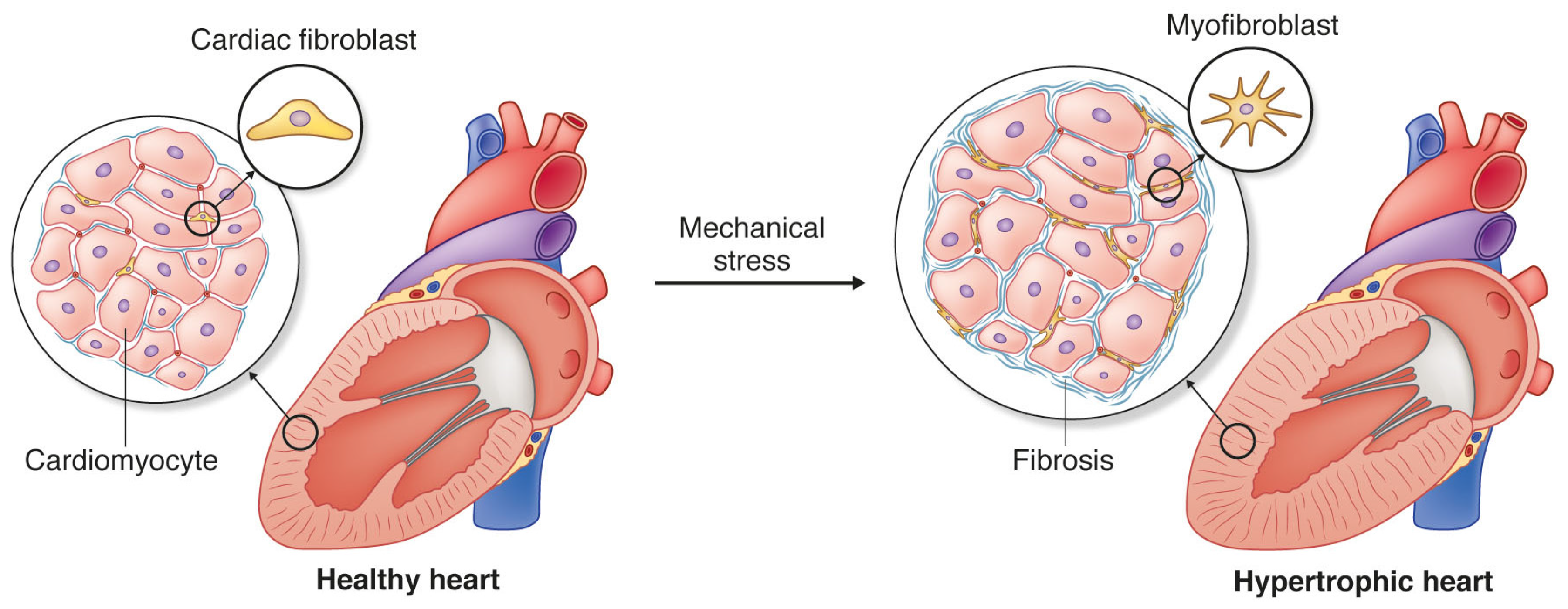
1. Cardiac Fibrosis and Heart Disease
2. Mechanical Forces of the Heart
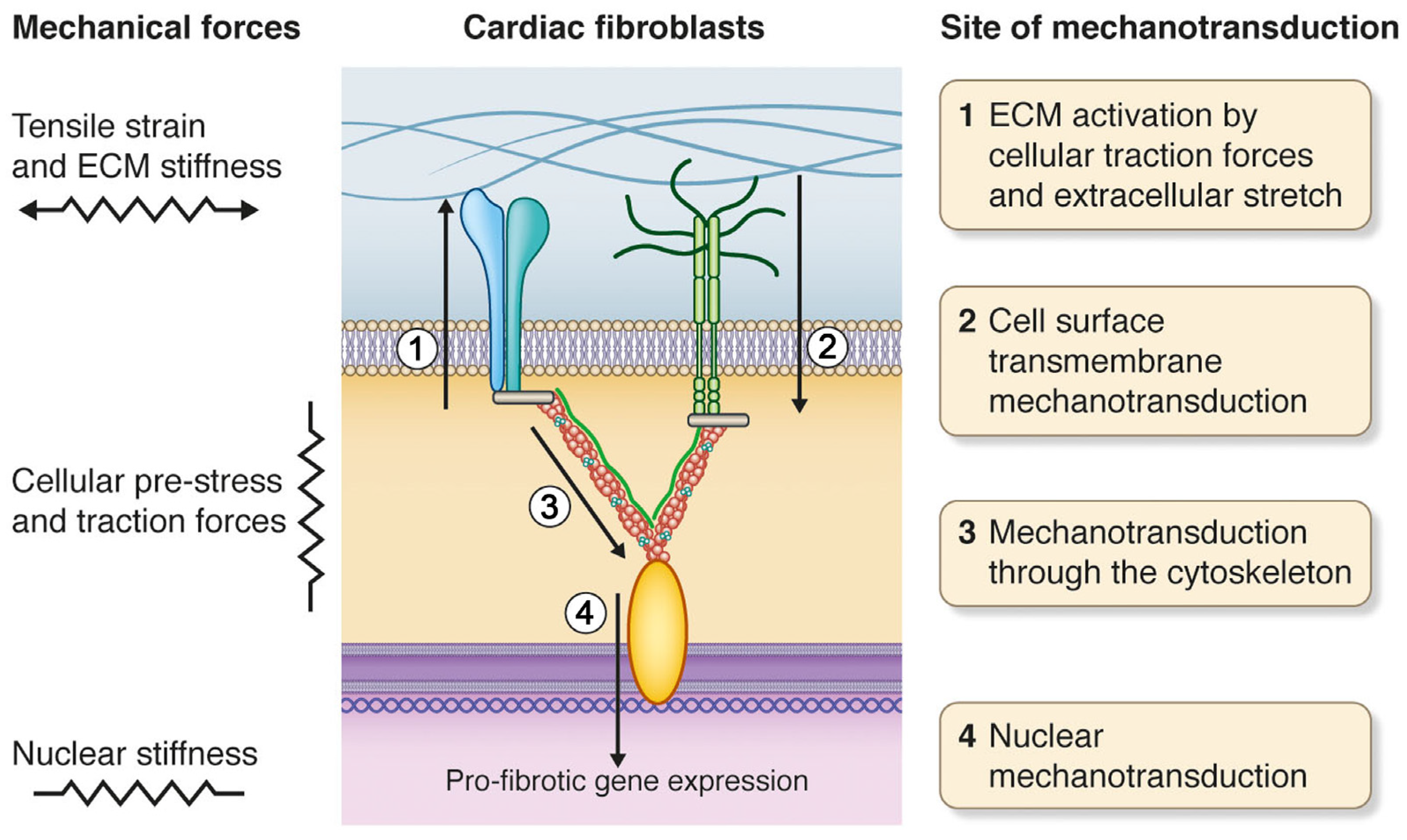
3. Cardiac Fibroblasts and Mechanotransduction
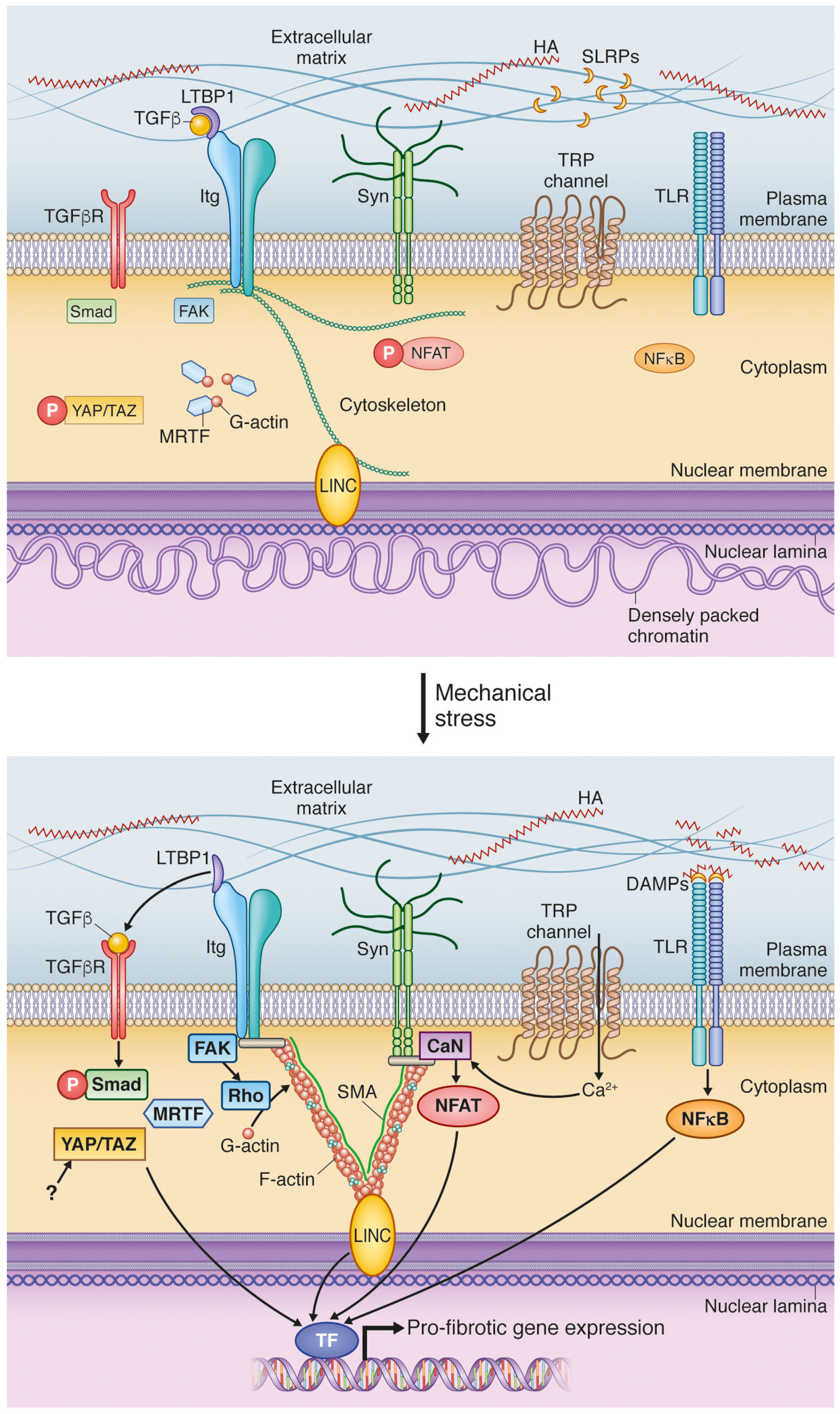
4. Mechanical Activation of Extracellular Matrix: A Vigilant Pro-Fibrotic Reservoir
4.1. Transforming Growth Factor β Activation; A Potent Signal Activated in ECM during Increased Stress
4.2. DAMPs Are Stress-Induced Initiators of Inflammation and Fibrosis
5. Cell Surface Mechanotransduction: A Strained Relationship between the ECM and Cell Interior
5.1. Integrins Are Main Constituents of Focal Adhesions Integrating Extracellular and Intracellular Signals
5.2. Syndecan-4 Is Part of the Mechanosensory Apparatus of Fibroblasts
5.3. Stretch-Activated Ion Channels (SACs); Functional Roles in Fibrosis Still to Be Explored
6. Mechanotransduction by the Cytoskeleton: Actin’ on Nuclear Translocation
6.1. Myocardin-Related Transcription Factors Are Liberated from Actin by Mechanical Stimulation
6.2. Hippo Signaling Pathway Is Regulated by Cytoskeletal Dynamics
7. Nuclear Mechanosensing: Long-Distance Communication
7.1. Linker of Nucleoskeleton and Cytoskeleton; A Complex for Nuclear Detection of Force
7.2. Nuclear Shape and Stiffness Is Associated with Cardiac Fibrosis
8. Manipulating the Soft and Hard-Heartedness of Cardiac Fibroblasts
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Gyongyosi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; Gonzalez, A.; Thum, T.; Diez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.F.; Zawaideh, S.; Hikita, S.; Kumar, V.A.; Cantor, H.; Ashkar, S. Phosphorylation-dependent interaction of osteopontin with its receptors regulates macrophage migration and activation. J. Leukoc. Biol. 2002, 72, 752–761. [Google Scholar] [PubMed]
- Weber, K.T.; Pick, R.; Jalil, J.E.; Janicki, J.S.; Carroll, E.P. Patterns of myocardial fibrosis. J. Mol. Cell. Cardiol. 1989, 21, 121–131. [Google Scholar] [CrossRef]
- Weber, K.T.; Sun, Y.; Bhattacharya, S.K.; Ahokas, R.A.; Gerling, I.C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 2013, 10, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Fail. Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed]
- Schellings, M.W.; Pinto, Y.M.; Heymans, S. Matricellular proteins in the heart: Possible role during stress and remodeling. Cardiovasc. Res. 2004, 64, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Rienks, M.; Papageorgiou, A.P.; Frangogiannis, N.G.; Heymans, S. Myocardial extracellular matrix: An ever-changing and diverse entity. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Tschope, C.; Lam, C.S. Diastolic heart failure: What we still don’t know. Looking for new concepts, diagnostic approaches, and the role of comorbidities. Herz 2012, 37, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Van den Borne, S.W.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Lajiness, J.D.; Conway, S.J. Origin, development, and differentiation of cardiac fibroblasts. J. Mol. Cell. Cardiol. 2014, 70, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Zietsch, C.; Becher, P.M.; Schulze, K.; Schultheiss, H.P.; Tschope, C.; Westermann, D. Differential expression of matrix metalloproteases in human fibroblasts with different origins. Biochem. Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ivey, M.J.; Tallquist, M.D. Defining the cardiac fibroblast. Circ. J. 2016, 80, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Gonzalez, A.; Pizard, A.; Papageorgiou, A.P.; Lopez-Andres, N.; Jaisser, F.; Thum, T.; Zannad, F.; Diez, J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur. J. Heart Fail. 2015, 17, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Phelan, D.; Collier, P.; Horgan, S.; Glezeva, N.; Cooke, G.; Xu, M.; Ledwidge, M.; McDonald, K.; Baugh, J.A. Extracellular matrix sub-types and mechanical stretch impact human cardiac fibroblast responses to transforming growth factor β. Connect. Tissue Res. 2014, 55, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S. TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Maisch, B.; Zhou, G.; Weber, K.T. Hormonal regulation of cardiac fibroblast function. Eur. Heart J. 1995, 16, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.E.; Gay, N.J.; Heymans, S.; Sacre, S.; Schaefer, L.; Midwood, K.S. Advances in toll-like receptor biology: Modes of activation by diverse stimuli. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 359–379. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Villongco, C.T.; Chuang, J.; Frank, L.R.; Nigam, V.; Belezzuoli, E.; Stark, P.; Krummen, D.E.; Narayan, S.; Omens, J.H.; et al. Patient-specific models of cardiac biomechanics. J. Comput. Phys. 2013, 244, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Amundsen, B.H.; Helle-Valle, T.; Edvardsen, T.; Torp, H.; Crosby, J.; Lyseggen, E.; Stoylen, A.; Ihlen, H.; Lima, J.A.; Smiseth, O.A.; et al. Noninvasive myocardial strain measurement by speckle tracking echocardiography: Validation against sonomicrometry and tagged magnetic resonance imaging. J. Am. Coll. Cardiol. 2006, 47, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Axel, L.; Montillo, A.; Kim, D. Tagged magnetic resonance imaging of the heart: A survey. Med. Image Anal. 2005, 9, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, S.; Shafieyan, Y.; Hinz, B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 2016, 93, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Goffin, J.M.; Pittet, P.; Csucs, G.; Lussi, J.W.; Meister, J.J.; Hinz, B. Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers. J. Cell Biol. 2006, 172, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Hameed, F.M.; Yang, B.; Lee, K.; Pan, C.Q.; Park, S.; Sheetz, M. Cyclic stretching of soft substrates induces spreading and growth. Nat. Commun. 2015, 6, 6333. [Google Scholar] [CrossRef] [PubMed]
- Pelham, R.J., Jr.; Wang, Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef] [PubMed]
- Zajaczkowski, M.B.; Cukierman, E.; Galbraith, C.G.; Yamada, K.M. Cell-matrix adhesions on poly(vinyl alcohol) hydrogels. Tissue Eng. 2003, 9, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Carag-Krieger, C.; Johnson, C.P.; Raab, M.; Tang, H.Y.; Speicher, D.W.; Sanger, J.W.; Sanger, J.M.; Discher, D.E. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: Scar-like rigidity inhibits beating. J. Cell Sci. 2008, 121, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.F.; Engler, A.J.; Woo, Y.J.; Pirolli, T.J.; Bish, L.T.; Jayasankar, V.; Morine, K.J.; Gardner, T.J.; Discher, D.E.; Sweeney, H.L. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2196–H2203. [Google Scholar] [CrossRef] [PubMed]
- Solon, J.; Levental, I.; Sengupta, K.; Georges, P.C.; Janmey, P.A. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 2007, 93, 4453–4461. [Google Scholar] [CrossRef] [PubMed]
- Tee, S.Y.; Fu, J.; Chen, C.S.; Janmey, P.A. Cell shape and substrate rigidity both regulate cell stiffness. Biophys. J. 2011, 100, L25–L27. [Google Scholar] [CrossRef] [PubMed]
- Brown, X.Q.; Ookawa, K.; Wong, J.Y. Evaluation of polydimethylsiloxane scaffolds with physiologically-relevant elastic moduli: Interplay of substrate mechanics and surface chemistry effects on vascular smooth muscle cell response. Biomaterials 2005, 26, 3123–3129. [Google Scholar] [CrossRef] [PubMed]
- Caliari, S.R.; Perepelyuk, M.; Cosgrove, B.D.; Tsai, S.J.; Lee, G.Y.; Mauck, R.L.; Wells, R.G.; Burdick, J.A. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ondeck, M.G.; Engler, A.J. Mechanical characterization of a dynamic and tunable methacrylated hyaluronic acid hydrogel. J. Biomech. Eng. 2016, 138, 0210031–0210036. [Google Scholar] [CrossRef] [PubMed]
- Caliari, S.R.; Perepelyuk, M.; Soulas, E.M.; Lee, G.Y.; Wells, R.G.; Burdick, J.A. Gradually softening hydrogels for modeling hepatic stellate cell behavior during fibrosis regression. Integr. Biol. 2016, 8, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Throm Quinlan, A.M.; Sierad, L.N.; Capulli, A.K.; Firstenberg, L.E.; Billiar, K.L. Combining dynamic stretch and tunable stiffness to probe cell mechanobiology in vitro. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical regulation of cardiac fibroblast pro-fibrotic phenotypes. Mol. Biol. Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.; Querejeta, R.; Gonzalez, A.; Larman, M.; Diez, J. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage c heart failure: Potential role of lysyl oxidase. Hypertension 2012, 60, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Kasner, M.; Westermann, D.; Lopez, B.; Gaub, R.; Escher, F.; Kuhl, U.; Schultheiss, H.P.; Tschope, C. Diastolic tissue doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J. Am. Coll. Cardiol. 2011, 57, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.L.; McCulloch, A.D.; Omens, J.H. Glycated collagen cross-linking alters cardiac mechanics in volume-overload hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1277–H1284. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tolic-Norrelykke, I.M.; Chen, J.; Mijailovich, S.M.; Butler, J.P.; Fredberg, J.J.; Stamenovic, D. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am. J. Physiol. Cell Physiol. 2002, 282, C606–C616. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, T.; Ingber, D.E. Mechanical control of tissue and organ development. Development 2010, 137, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- LeGrice, I.J.; Takayama, Y.; Covell, J.W. Transverse shear along myocardial cleavage planes provides a mechanism for normal systolic wall thickening. Circ. Res. 1995, 77, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Hermans, K.C.; Daskalopoulos, E.P.; Blankesteijn, W.M. The janus face of myofibroblasts in the remodeling heart. J. Mol. Cell. Cardiol. 2016, 91, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.; Ubil, E. Cardiac fibroblast in development and wound healing. J. Mol. Cell. Cardiol. 2014, 70, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Pillai, I.C.; Li, S.; Romay, M.; Lam, L.; Lu, Y.; Huang, J.; Dillard, N.; Zemanova, M.; Rubbi, L.; Wang, Y.; et al. Cardiac fibroblasts adopt osteogenic fates and can be targeted to attenuate pathological heart calcification. Cell Stem Cell 2017, 20, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Cattaneo, P.; Puceat, M.; Evans, S.M. Origins of cardiac fibroblasts. J. Mol. Cell. Cardiol. 2016, 91, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, M.J.; Bou-Gharios, G.; Popa, E.; van Ark, J.; Petersen, A.H.; van Dam, G.M.; van Luyn, M.J.; Harmsen, M.C. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J. Pathol. 2008, 214, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Endo, J.; Sano, M.; Fujita, J.; Hayashida, K.; Yuasa, S.; Aoyama, N.; Takehara, Y.; Kato, O.; Makino, S.; Ogawa, S.; et al. Bone marrow derived cells are involved in the pathogenesis of cardiac hypertrophy in response to pressure overload. Circulation 2007, 116, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Flores, L.; Gutierrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Martin-Vasallo, P.; Diaz-Flores, L., Jr. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009, 24, 909–969. [Google Scholar] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Bochaton-Piallat, M.L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The fibronectin domain ed-a is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Takefuji, M.; Ngai, C.Y.; Carvalho, J.; Bayer, J.; Wietelmann, A.; Poetsch, A.; Hoelper, S.; Conway, S.J.; Mollmann, H.; et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 2016, 118, 1906–1917. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.A.; Delhaas, T.; McCulloch, A.D.; Villarreal, F.J. Differential responses of adult cardiac fibroblasts to in vitro biaxial strain patterns. J. Mol. Cell. Cardiol. 1999, 31, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Porter, K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G.; Frangogiannis, N.G.; Hinz, B.; Holmes, J.W.; Kassiri, Z.; Lindsey, M.L. Crossing into the next frontier of cardiac extracellular matrix research. Circ. Res. 2016, 119, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, A.; Orr, A.W.; Tzima, E.; Schwartz, M.A. Integrins in mechanotransduction. J. Biol. Chem. 2004, 279, 12001–12004. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 2012, 92, 635–688. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Hinz, B. Integrins and the activation of latent transforming growth factor β1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. It has to be the αv: Myofibroblast integrins activate latent TGF-β1. Nat. Med. 2013, 19, 1567–1568. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Kelsh, R.; You, R.; Horzempa, C.; Zheng, M.; McKeown-Longo, P.J. Regulation of the innate immune response by fibronectin: Synergism between the iii-1 and eda domains. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor TGF-β signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G. The role of proteases in transforming growth factor-β activation. Int. J. Biochem. Cell Biol. 2008, 40, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, L.; Todorovic, V.; Dabovic, B.; Horiguchi, M.; Courousse, T.; Sakai, L.Y.; Rifkin, D.B. Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: Role of fibrillins and fibronectin. J. Cell. Physiol. 2012, 227, 3828–3836. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.L. Integrin-mediated transforming growth factor-β activation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol. 2009, 175, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, M.M.; Travis, M.A.; Kudo, M.; Sheppard, D. Epithelial cells utilize cortical actin/myosin to activate latent TGF-β through integrin αvβ6-dependent physical force. Exp. Cell Res. 2012, 318, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Chow, M.L.; Koehler, A.; Boo, S.; Buscemi, L.; Quinn, T.M.; Costell, M.; Alman, B.A.; Genot, E.; Hinz, B. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. J. Cell Biol. 2014, 207, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Sakabe, T.; Sunaga, A.; Sakai, K.; Rivera, A.L.; Keene, D.R.; Sasaki, T.; Stavnezer, E.; Iannotti, J.; Schweitzer, R.; et al. Conversion of mechanical force into TGF-β-mediated biochemical signals. Curr. Biol. 2011, 21, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.; Gonzalez, A.; Lindner, D.; Westermann, D.; Ravassa, S.; Beaumont, J.; Gallego, I.; Zudaire, A.; Brugnolaro, C.; Querejeta, R.; et al. Osteopontin-mediated myocardial fibrosis in heart failure: A role for lysyl oxidase? Cardiovasc. Res. 2013, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Huelsz-Prince, G.; Belkin, A.M.; VanBavel, E.; Bakker, E.N. Activation of extracellular transglutaminase 2 by mechanical force in the arterial wall. J. Vasc. Res. 2013, 50, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, A.; Germack, R.; Lortat-Jacob, H.; Muramatsu, T.; Billett, E.; Johnson, T.; Verderio, E.A. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J. Biol. Chem. 2009, 284, 18411–18423. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, A.; Huang, L.; Burhan, I.; Schroeder, N.; Funck, M.; Johnson, T.S.; Verderio, E.A. Syndecan-4 knockout leads to reduced extracellular transglutaminase-2 and protects against tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1013–1027. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Collighan, R.J.; Pytel, K.; Rathbone, D.L.; Li, X.; Griffin, M. Characterization of heparin-binding site of tissue transglutaminase: Its importance in cell surface targeting, matrix deposition, and cell signaling. J. Biol. Chem. 2012, 287, 13063–13083. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Louch, W.E.; Hasic, A.; Boye, S.; Unger, A.; Brorson, S.H.; Sjaastad, I.; Tønnessen, T.; et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 2015, 106, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Zouein, F.A.; Tian, Y.; Padmanabhan Iyer, R.; de Castro Bras, L.E. Osteopontin is proteolytically processed by matrix metalloproteinase 9. Can. J. Physiol. Pharmacol. 2015, 93, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Slimani, H.; Charnaux, N.; Mbemba, E.; Saffar, L.; Vassy, R.; Vita, C.; Gattegno, L. Interaction of rantes with syndecan-1 and syndecan-4 expressed by human primary macrophages. Biochim. Biophys. Acta 2003, 1617, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Bashkin, P.; Doctrow, S.; Klagsbrun, M.; Svahn, C.M.; Folkman, J.; Vlodavsky, I. Basic fibroblast growth factor binds to subendothelial extracellular matrix and is released by heparitinase and heparin-like molecules. Biochemistry 1989, 28, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Bliksoen, M.; Mariero, L.H.; Ohm, I.K.; Haugen, F.; Yndestad, A.; Solheim, S.; Seljeflot, I.; Ranheim, T.; Andersen, G.O.; Aukrust, P.; et al. Increased circulating mitochondrial DNA after myocardial infarction. Int. J. Cardiol. 2012, 158, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial damps cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Vabulas, R.M.; Ahmad-Nejad, P.; da Costa, C.; Miethke, T.; Kirschning, C.J.; Hacker, H.; Wagner, H. Endocytosed hsp60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 2001, 276, 31332–31339. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Cohen, I.R. Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J. Immunol. 2005, 175, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by hmgb1 and rage. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Van Nieuwenhoven, F.A.; Hemmings, K.E.; Porter, K.E.; Turner, N.A. Combined effects of interleukin-1α and transforming growth factor-β1 on modulation of human cardiac fibroblast function. Matrix Biol. 2013, 32, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A. Effects of interleukin-1 on cardiac fibroblast function: Relevance to post-myocardial infarction remodelling. Vascul. Pharmacol. 2014, 60, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, A.; Spary, E.J.; Manfield, I.W.; Ruhmann, M.; Zuliani-Alvarez, L.; Gamboa-Esteves, F.O.; Porter, K.E.; Drinkhill, M.J.; Midwood, K.S.; Turner, N.A. Tenascin c upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. World J. Cardiol. 2016, 8, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Frey, H.; Schroeder, N.; Manon-Jensen, T.; Iozzo, R.V.; Schaefer, L. Biological interplay between proteoglycans and their innate immune receptors in inflammation. FEBS J. 2013, 280, 2165–2179. [Google Scholar] [CrossRef] [PubMed]
- Engebretsen, K.V.; Lunde, I.G.; Strand, M.E.; Waehre, A.; Sjaastad, I.; Marstein, H.S.; Skrbic, B.; Dahl, C.P.; Askevold, E.T.; Christensen, G.; et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J. 2013, 280, 2382–2398. [Google Scholar] [CrossRef] [PubMed]
- Engebretsen, K.V.; Waehre, A.; Bjørnstad, J.L.; Skrbic, B.; Sjaastad, I.; Behmen, D.; Marstein, H.S.; Yndestad, A.; Aukrust, P.; Christensen, G.; et al. Decorin, lumican, and their gag chain-synthesizing enzymes are regulated in myocardial remodeling and reverse remodeling in the mouse. J. Appl. Physiol. 2013, 114, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.E.; Aronsen, J.M.; Braathen, B.; Sjaastad, I.; Kvaløy, H.; Tønnessen, T.; Christensen, G.; Lunde, I.G. Shedding of syndecan-4 promotes immune cell recruitment and mitigates cardiac dysfunction after lipopolysaccharide challenge in mice. J. Mol. Cell. Cardiol. 2015, 88, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Trowbridge, J.M.; Rudisill, J.A.; Termeer, C.C.; Simon, J.C.; Gallo, R.L. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J. Biol. Chem. 2004, 279, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 2006, 177, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Lunde, I.G.; Herum, K.M.; Carlson, C.C.; Christensen, G. Syndecans in heart fibrosis. Cell Tissue Res. 2016, 365, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Waehre, A.; Vistnes, M.; Sjaastad, I.; Nygård, S.; Husberg, C.; Lunde, I.G.; Aukrust, P.; Yndestad, A.; Vinge, L.E.; Behmen, D.; et al. Chemokines regulate small leucine-rich proteoglycans in the extracellular matrix of the pressure-overloaded right ventricle. J. Appl. Physiol. 2012, 112, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.E.; Herum, K.M.; Rana, Z.A.; Skrbic, B.; Askevold, E.T.; Dahl, C.P.; Vistnes, M.; Hasic, A.; Kvaløy, H.; Sjaastad, I.; et al. Innate immune signaling induces expression and shedding of the heparan sulfate proteoglycan syndecan-4 in cardiac fibroblasts and myocytes, affecting inflammation in the pressure-overloaded heart. FEBS J. 2013, 280, 2228–2247. [Google Scholar] [CrossRef] [PubMed]
- Melleby, A.O.; Strand, M.E.; Romaine, A.; Herum, K.M.; Skrbic, B.; Dahl, C.P.; Sjaastad, I.; Fiane, A.E.; Filmus, J.; Christensen, G.; et al. The heparan sulfate proteoglycan glypican-6 is upregulated in the failing heart, and regulates cardiomyocyte growth through ERK1/2 signaling. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Paul, N.R.; Humphries, M.J.; Morgan, M.R. Emerging properties of adhesion complexes: What are they and what do they do? Trends Cell Biol. 2015, 25, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Schroer, A.K.; Merryman, W.D. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J. Cell Sci. 2015, 128, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Teoh, C.M.; Tam, J.K.; Tran, T. Integrin and gpcr crosstalk in the regulation of ASM contraction signaling in asthma. J. Allergy 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Wu, C.Y.; Yamaji, S.; Saegusa, J.; Shi, B.; Ma, Z.; Kuwabara, Y.; Lam, K.S.; Isseroff, R.R.; Takada, Y.K.; et al. Direct binding of integrin αvβ3 to FGF1 plays a role in FGF1 signaling. J. Biol. Chem. 2008, 283, 18066–18075. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K. Integrins and cadherins as therapeutic targets in fibrosis. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- MacKenna, D.A.; Dolfi, F.; Vuori, K.; Ruoslahti, E. Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Investig. 1998, 101, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell. Cardiol. 2016, 93, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Karen, J.; Gardner, H.; Gerdin, B.; Rubin, K.; Sundberg, C. Integrin α1β1 is involved in the differentiation into myofibroblasts in adult reactive tissues in vivo. J. Cell. Mol. Med. 2009, 13, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell α3β1 integrin links β-catenin and smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Collighan, R.J.; Gross, S.R.; Danen, E.H.; Orend, G.; Telci, D.; Griffin, M. RGD-independent cell adhesion via a tissue transglutaminase-fibronectin matrix promotes fibronectin fibril deposition and requires syndecan-4/2 α5β1 integrin co-signaling. J. Biol. Chem. 2010, 285, 40212–40229. [Google Scholar] [CrossRef] [PubMed]
- Bouzeghrane, F.; Mercure, C.; Reudelhuber, T.L.; Thibault, G. α8β1 integrin is upregulated in myofibroblasts of fibrotic and scarring myocardium. J. Mol. Cell. Cardiol. 2004, 36, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Sambandamoorthy, S.; Mathew-Steiner, S.; Varney, S.; Zuidema, J.M.; Gilbert, R.J.; Van De Water, L.; LaFlamme, S.E. Matrix compliance and the regulation of cytokinesis. Biol. Open 2015, 4, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Moriwaki, S.; Imokawa, G.; Takema, Y. Crucial role of fibroblast integrins α2 and β1 in maintaining the structural and mechanical properties of the skin. J. Dermatol. Sci. 2007, 45, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Leask, A. Integrin β1 is required for dermal homeostasis. J. Investig. Dermatol. 2013, 133, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, T.; Westermarck, J.; Koivisto, L.; Broberg, A.; Kahari, V.M.; Heino, J. Integrin α2β1 is a positive regulator of collagenase (MMP-1) and collagen α1(I) gene expression. J. Biol. Chem. 1995, 270, 13548–13552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, Z.; Foolen, J.; Schoen, I.; Santoro, A.; Zenobi-Wong, M.; Vogel, V. Disentangling the multifactorial contributions of fibronectin, collagen and cyclic strain on mmp expression and extracellular matrix remodeling by fibroblasts. Matrix Biol. 2014, 40, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Quinones, L.; Kasiganesan, H.; Zhang, Y.; Pleasant, D.L.; Sundararaj, K.P.; Zile, M.R.; Bradshaw, A.D.; Kuppuswamy, D. β3 Integrin in cardiac fibroblast is critical for extracellular matrix accumulation during pressure overload hypertrophy in mouse. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Sun, Y.; Zhu, Z.; Li, F. Is the change of integrin αvβ3 expression in the infarcted myocardium related to the clinical outcome? Clin Nucl Med 2014, 39, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Roca-Cusachs, P.; Gauthier, N.C.; Del Rio, A.; Sheetz, M.P. Clustering of α5β1 integrins determines adhesion strength whereas αvβ3 and talin enable mechanotransduction. Proc. Natl. Acad. Sci. USA 2009, 106, 16245–16250. [Google Scholar] [CrossRef] [PubMed]
- Tiger, C.F.; Fougerousse, F.; Grundstrom, G.; Velling, T.; Gullberg, D. Alpha11β1 integrin is a receptor for interstitial collagens involved in cell migration and collagen reorganization on mesenchymal nonmuscle cells. Dev. Biol. 2001, 237, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Carracedo, S.; Ranta, J.; Heuchel, R.; Soininen, R.; Gullberg, D. The human α11 integrin promoter drives fibroblast-restricted expression in vivo and is regulated by TGF-β1 in a smad- and sp1-dependent manner. Matrix Biol. 2010, 29, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Talior-Volodarsky, I.; Connelly, K.A.; Arora, P.D.; Gullberg, D.; McCulloch, C.A. Alpha11 integrin stimulates myofibroblast differentiation in diabetic cardiomyopathy. Cardiovasc. Res. 2012, 96, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.N.; Zeltz, C.; Sorensen, I.W.; Barczyk, M.; Carracedo, S.; Hallinger, R.; Niehoff, A.; Eckes, B.; Gullberg, D. Reduced granulation tissue and wound strength in the absence of α11β1 integrin. J. Investig. Dermatol. 2015, 135, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, S.; Lu, N.; Popova, S.N.; Jonsson, R.; Eckes, B.; Gullberg, D. The fibroblast integrin α11β1 is induced in a mechanosensitive manner involving activin a and regulates myofibroblast differentiation. J. Biol. Chem. 2010, 285, 10434–10445. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Gullberg, D. The integrin-collagen connection—A glue for tissue repair? J. Cell Sci. 2016, 129, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Talior-Volodarsky, I.; Arora, P.D.; Wang, Y.; Zeltz, C.; Connelly, K.A.; Gullberg, D.; McCulloch, C.A. Glycated collagen induces α11 integrin expression through TGF-β2 and smad3. J. Cell. Physiol. 2015, 230, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J. Hypertens. 2003, 21, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; To, C.; Pasko, E.; Kim, K.S.; Park, C.J.; Hai, J.; Liu, J.; Jonkman, J.; Barczyk, M.; et al. Integrin α11β1 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 2016, 35, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Civitarese, R.A.; Talior-Volodarsky, I.; Desjardins, J.F.; Kabir, G.; Switzer, J.; Mitchell, M.; Kapus, A.; McCulloch, C.A.; Gullberg, D.; Connelly, K.A. The α11 integrin mediates fibroblast-extracellular matrix-cardiomyocyte interactions in health and disease. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H96–H106. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hunter, T. Integrin signalling and tyrosine phosphorylation: Just the faks? Trends Cell Biol. 1998, 8, 151–157. [Google Scholar] [CrossRef]
- Critchley, D.R. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu. Rev. Biophys. 2009, 38, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Manso, A.M.; Li, R.; Monkley, S.J.; Cruz, N.M.; Ong, S.; Lao, D.H.; Koshman, Y.E.; Gu, Y.; Peterson, K.L.; Chen, J.; et al. Talin1 has unique expression versus talin 2 in the heart and modifies the hypertrophic response to pressure overload. J. Biol. Chem. 2013, 288, 4252–4264. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Perez-Gonzalez, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Carraher, C.L.; Schwarzbauer, J.E. Regulation of matrix assembly through rigidity-dependent fibronectin conformational changes. J. Biol. Chem. 2013, 288, 14805–14814. [Google Scholar] [CrossRef] [PubMed]
- Seong, J.; Tajik, A.; Sun, J.; Guan, J.L.; Humphries, M.J.; Craig, S.E.; Shekaran, A.; Garcia, A.J.; Lu, S.; Lin, M.Z.; et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 19372–19377. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Lighthouse, J.K.; Small, E.M. Transcriptional control of cardiac fibroblast plasticity. J. Mol. Cell. Cardiol. 2016, 91, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J.M. Fibroblast-specific genetic manipulation of p38 MAPK in vivo reveals its central regulatory role in fibrosis. Circulation 2017. [Google Scholar] [CrossRef] [PubMed]
- Rossman, K.L.; Der, C.J.; Sondek, J. Gef means go: Turning on Rho GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Swaminathan, V.; Garcia-Mata, R.; O’Brien, E.T.; Superfine, R.; Burridge, K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 2011, 13, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Liao, J.K. Rho kinases and cardiac remodeling. Circ. J. 2016, 80, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, G.; Zhao, H.; Wang, Z.; Li, F.; Zhang, P.; Zhang, J.; Wang, X.; Wang, W. Targeted inhibition of focal adhesion kinase attenuates cardiac fibrosis and preserves heart function in adverse cardiac remodeling. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- McQuade, K.J.; Beauvais, D.M.; Burbach, B.J.; Rapraeger, A.C. Syndecan-1 regulates αvβ5 integrin activity in b82l fibroblasts. J. Cell Sci. 2006, 119, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, J.R.; Behrends, V.; Kirby, H.; Kusche-Gullberg, M.; Muramatsu, T.; Couchman, J.R. Syndecans promote integrin-mediated adhesion of mesenchymal cells in two distinct pathways. Exp. Cell Res. 2007, 313, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Cody, R.J.; Graf, K.; Goetze, S.; Kawano, Y.; Schnee, J.; Law, R.E.; Hsueh, W.A. Angiotensin II enhances integrin and α-actinin expression in adult rat cardiac fibroblasts. Hypertension 2000, 35, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Couchman, J.R. Transmembrane signaling proteoglycans. Annu. Rev. Cell Dev. Biol. 2010, 26, 89–114. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chaikof, E.L. Mechanical stress regulates syndecan-4 expression and redistribution in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.K.; Tumova, S.; Couchman, J.R.; Woods, A. Syndecan-4 associates with α-actinin. J. Biol. Chem. 2003, 278, 7617–7623. [Google Scholar] [CrossRef] [PubMed]
- Finsen, A.V.; Woldbaek, P.R.; Li, J.; Wu, J.; Lyberg, T.; Tonnessen, T.; Christensen, G. Increased syndecan expression following myocardial infarction indicates a role in cardiac remodeling. Physiol. Genom. 2004, 16, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.R.; Humphries, M.J.; Bass, M.D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Bellin, R.M.; Kubicek, J.D.; Frigault, M.J.; Kamien, A.J.; Steward, R.L., Jr.; Barnes, H.M.; Digiacomo, M.B.; Duncan, L.J.; Edgerly, C.K.; Morse, E.M.; et al. Defining the role of Syndecan-4 in mechanotransduction using surface-modification approaches. Proc. Natl. Acad. Sci. USA 2009, 106, 22102–22107. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Florholmen, G.; Behmen, D.; Sjaastad, I.; Carlson, C.R.; Gomez, M.F.; Christensen, G. Syndecan-4 signaling via nfat regulates extracellular matrix production and cardiac myofibroblast differentiation in response to mechanical stress. J. Mol. Cell. Cardiol. 2013, 54, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Cavalheiro, R.P.; Lima, M.A.; Jarrouge-Boucas, T.R.; Viana, G.M.; Lopes, C.C.; Coulson-Thomas, V.J.; Dreyfuss, J.L.; Yates, E.A.; Tersariol, I.L.; Nader, H.B. Coupling of vinculin to F-actin demands syndecan-4 proteoglycan. Matrix Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Finsen, A.V.; Lunde, I.G.; Sjaastad, I.; Østli, E.K.; Lyngra, M.; Jarstadmarken, H.O.; Hasic, A.; Nygård, S.; Wilcox-Adelman, S.A.; Goetinck, P.F.; et al. Syndecan-4 is essential for development of concentric myocardial hypertrophy via stretch-induced activation of the calcineurin-NFAT pathway. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Samarel, A.M. Syndecan-4: A component of the mechanosensory apparatus of cardiac fibroblasts. J. Mol. Cell. Cardiol. 2013, 56, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Søgaard, P.; Multhaupt, H.A.; Pataki, C.; Okina, E.; Xian, X.; Pedersen, M.E.; Stevens, T.; Griesbeck, O.; Park, P.W.; et al. Transmembrane proteoglycans control stretch-activated channels to set cytosolic calcium levels. J. Cell Biol. 2015, 210, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Roshanravan, H.; Dryer, S.E. Syndecan-4 ectodomain evokes mobilization of podocyte TRPC6 channels and their associated pathways: An essential role for integrin signaling. Biochim. Biophys. Acta 2015, 1853, 2610–2620. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Ikesue, M.; Danzaki, K.; Morimoto, J.; Sato, M.; Tanaka, S.; Kojima, T.; Tsutsui, H.; Uede, T. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ. Res. 2011, 108, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Syndecan-1: A critical mediator in cardiac fibrosis. Hypertension 2010, 55, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, D.; Schellings, M.W.; Gotte, M.; Swinnen, M.; Herias, V.; Wild, M.K.; Vestweber, D.; Chorianopoulos, E.; Cortes, V.; Rigotti, A.; et al. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation 2007, 115, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Schellings, M.W.; Vanhoutte, D.; van Almen, G.C.; Swinnen, M.; Leenders, J.J.; Kubben, N.; van Leeuwen, R.E.; Hofstra, L.; Heymans, S.; Pinto, Y.M. Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension 2010, 55, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, T.; Miyamura, N.; Hata, S.; Miura, R.; Hirayama, J.; Nishina, H. The pdz-binding motif of yes-associated protein is required for its co-activation of tead-mediated CTGF transcription and oncogenic cell transforming activity. Biochem. Biophys. Res. Commun. 2014, 443, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; van der Pol, A.; Klip, I.T.; de Boer, R.A.; Jaarsma, T.; van Gilst, W.H.; Voors, A.A.; van Veldhuisen, D.J.; van der Meer, P. Fibrosis marker syndecan-1 and outcome in patients with heart failure with reduced and preserved ejection fraction. Circ. Heart Fail. 2014, 7, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Chatelier, A.; Mercier, A.; Tremblier, B.; Theriault, O.; Moubarak, M.; Benamer, N.; Corbi, P.; Bois, P.; Chahine, M.; Faivre, J.F. A distinct de novo expression of Nav1.5 sodium channels in human atrial fibroblasts differentiated into myofibroblasts. J. Physiol. 2012, 590, 4307–4319. [Google Scholar] [CrossRef] [PubMed]
- Benamer, N.; Vasquez, C.; Mahoney, V.M.; Steinhardt, M.J.; Coetzee, W.A.; Morley, G.E. Fibroblast katp currents modulate myocyte electrophysiology in infarcted hearts. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1231–H1239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Sung, R.J.; Lin, M.W.; Wu, S.N. Contribution of BKCa-channel activity in human cardiac fibroblasts to electrical coupling of cardiomyocytes-fibroblasts. J. Membr. Biol. 2006, 213, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.A.; Hatano, N.; Ohya, S.; Imaizumi, Y.; Giles, W.R. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide c receptor-mediated signalling. J. Physiol. 2007, 580, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. Trpv4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.; Kohl, P.; Peyronnet, R. Molecular candidates for cardiac stretch-activated ion channels. Glob. Cardiol. Sci. Pract. 2014, 2014, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Kamkin, A.; Kirischuk, S.; Kiseleva, I. Single mechano-gated channels activated by mechanical deformation of acutely isolated cardiac fibroblasts from rats. Acta Physiol. 2010, 199, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Norris, R.A.; Kohl, P.; Markwald, R. The living scar—Cardiac fibroblasts and the injured heart. Trends Mol. Med. 2016, 22, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Numaga-Tomita, T.; Kitajima, N.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; Birnbaumer, L.; et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Goswami, R.; Merth, M.; Cohen, J.; Lei, K.Y.; Zhang, D.X.; Rahaman, S.O. TRPV4 ion channel is a novel regulator of dermal myofibroblast differentiation. Am. J. Physiol. Cell Physiol. 2017, 312, C562–C572. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, S.O.; Grove, L.M.; Paruchuri, S.; Southern, B.D.; Abraham, S.; Niese, K.A.; Scheraga, R.G.; Ghosh, S.; Thodeti, C.K.; Zhang, D.X.; et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J. Clin. Investig. 2014, 124, 5225–5238. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Faucherre, A.; Kissa, K.; Nargeot, J.; Mangoni, M.E.; Jopling, C. Piezo1 plays a role in erythrocyte volume homeostasis. Haematologica 2014, 99, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E.; Wang, N.; Stamenovic, D. Tensegrity, cellular biophysics, and the mechanics of living systems. Rep. Prog. Phys. 2014, 77. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/rock: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from rho to the actin cytoskeleton through protein kinases rock and lim-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control srf activity by regulation of its coactivator mal. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Kuwahara, K.; Barrientos, T.; Pipes, G.C.; Li, S.; Olson, E.N. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol. Cell. Biol. 2005, 25, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Mouilleron, S.; Langer, C.A.; Guettler, S.; McDonald, N.Q.; Treisman, R. Structure of a pentavalent G-actin*MRTF-A complex reveals how G-actin controls nucleocytoplasmic shuttling of a transcriptional coactivator. Sci. Signal. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Weise-Cross, L.; Mangum, K.D.; Medlin, M.D.; Mangiante, L.; Taylor, J.M.; Mack, C.P. Nuclear Rhoa signaling regulates MRTF-dependent smc-specific transcription. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H379–H390. [Google Scholar] [CrossRef] [PubMed]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; Dimaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, X.; Hecker, L.; Kurundkar, D.; Kurundkar, A.; Liu, H.; Jin, T.H.; Desai, L.; Bernard, K.; Thannickal, V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Investig. 2013, 123, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of yap/taz in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Codelia, V.A.; Sun, G.; Irvine, K.D. Regulation of yap by mechanical strain through JNK and hippo signaling. Curr. Biol. 2014, 24, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- Muehlich, S.; Rehm, M.; Ebenau, A.; Goppelt-Struebe, M. Synergistic induction of CTGF by cytochalasin D and TGFβ-1 in primary human renal epithelial cells: Role of transcriptional regulators MK11, YAP/TAZ and Smad2/3. Cell. Signal. 2017, 29, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; She, P.; Chi, F.; Feng, Y.; Liu, H.; Jin, D.; Zhao, Y.; Guo, X.; Jiang, D.; Guan, K.L.; et al. Phosphorylation of angiomotin by LATS1/2 kinases inhibits f-actin binding, cell migration, and angiogenesis. J. Biol. Chem. 2013, 288, 34041–34051. [Google Scholar] [CrossRef] [PubMed]
- Mana-Capelli, S.; Paramasivam, M.; Dutta, S.; McCollum, D. Angiomotins link f-actin architecture to hippo pathway signaling. Mol. Biol. Cell 2014, 25, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; de Rond, S.; Werker, P.M.; Boo, S.; Hinz, B.; van Beuge, M.M.; Bank, R.A. YAP1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am. J. Pathol. 2015, 185, 3326–3337. [Google Scholar] [CrossRef] [PubMed]
- Mitani, A.; Nagase, T.; Fukuchi, K.; Aburatani, H.; Makita, R.; Kurihara, H. Transcriptional coactivator with PDZ-binding motif is essential for normal alveolarization in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J. An emerging role for Hippo-YAP signaling in cardiovascular development. J. Biomed. Res. 2014, 28, 251–254. [Google Scholar] [PubMed]
- Lee, M.J.; Ran Byun, M.; Furutani-Seiki, M.; Hong, J.H.; Jung, H.S. Yap and taz regulate skin wound healing. J. Investig. Dermatol. 2014, 134, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ are mechanoregulators of TGF-β-smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. Tead mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Grannas, K.; Arngarden, L.; Lonn, P.; Mazurkiewicz, M.; Blokzijl, A.; Zieba, A.; Soderberg, O. Crosstalk between hippo and TGFB: Subcellular localization of YAP/TAZ/SMAD complexes. J. Mol. Biol. 2015, 427, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Speight, P.; Kofler, M.; Szaszi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFβ-regulated SMAD3. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.W.; Hinz, B.; McCulloch, C.A. Mechanical induction of gene expression in connective tissue cells. Methods Cell Biol. 2010, 98, 178–205. [Google Scholar] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the linc complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Fedorchak, G.R.; Kaminski, A.; Lammerding, J. Cellular mechanosensing: Getting to the nucleus of it all. Prog. Biophys. Mol. Biol. 2014, 115, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Skepper, J.N.; Yang, F.; Davies, J.D.; Hegyi, L.; Roberts, R.G.; Weissberg, P.L.; Ellis, J.A.; Shanahan, C.M. Nesprins: A novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J. Cell Sci. 2001, 114, 4485–4498. [Google Scholar] [PubMed]
- Starr, D.A.; Fischer, J.A. Kash ’n karry: The KASH domain family of cargo-specific cytoskeletal adaptor proteins. Bioessays 2005, 27, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.; Litjens, S.H.; Kuikman, I.; Tshimbalanga, N.; Janssen, H.; van den Bout, I.; Raymond, K.; Sonnenberg, A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 2005, 171, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Hodzic, D.M.; Yeater, D.B.; Bengtsson, L.; Otto, H.; Stahl, P.D. SUN2 is a novel mammalian inner nuclear membrane protein. J. Biol. Chem. 2004, 279, 25805–25812. [Google Scholar] [CrossRef] [PubMed]
- Raff, J.W. The missing (L) UNC? Curr. Biol. 1999, 9, R708–R710. [Google Scholar] [CrossRef]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin a and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell. Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, Y.; Imaizumi, H.; Imada, J.; Katahira, J.; Matsuura, N.; Hieda, M. SUN1 splice variants, SUN1_888, SUN1_785, and predominant SUN1_916, variably function in directional cell migration. Nucleus 2016, 7, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Caputo, S.; Couprie, J.; Duband-Goulet, I.; Konde, E.; Lin, F.; Braud, S.; Gondry, M.; Gilquin, B.; Worman, H.J.; Zinn-Justin, S. The carboxyl-terminal nucleoplasmic region of MAN1 exhibits a DNA binding winged helix domain. J. Biol. Chem. 2006, 281, 18208–18215. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Worman, H.J.; Gundersen, G.G. Accessorizing and anchoring the linc complex for multifunctionality. J. Cell Biol. 2015, 208, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Brosig, M.; Ferralli, J.; Gelman, L.; Chiquet, M.; Chiquet-Ehrismann, R. Interfering with the connection between the nucleus and the cytoskeleton affects nuclear rotation, mechanotransduction and myogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 1717–1728. [Google Scholar] [CrossRef] [PubMed]
- Puckelwartz, M.J.; Kessler, E.; Zhang, Y.; Hodzic, D.; Randles, K.N.; Morris, G.; Earley, J.U.; Hadhazy, M.; Holaska, J.M.; Mewborn, S.K.; et al. Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice. Hum. Mol. Genet. 2009, 18, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynska, A.; Gonzalo, S.; Shanahan, C.; Askjaer, P. The nuclear lamina in health and disease. Nucleus 2016, 7, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, V.; Leimena, C.; McMahon, A.C.; Tan, J.C.; Chandar, S.; Jogia, D.; Kesteven, S.H.; Michalicek, J.; Otway, R.; Verheyen, F.; et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Investig. 2004, 113, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Arsenovic, P.T.; Ramachandran, I.; Bathula, K.; Zhu, R.; Narang, J.D.; Noll, N.A.; Lemmon, C.A.; Gundersen, G.G.; Conway, D.E. Nesprin-2G, a component of the nuclear LINC complex, is subject to myosin-dependent tension. Biophys. J. 2016, 110, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, R.N.; Eckes, B.; Glockner, G.; Groth, M.; Neumann, S.; Gloy, J.; Sellin, L.; Walz, G.; Schneider, M.; Karakesisoglou, I.; et al. The nuclear envelope protein nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus 2012, 3, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Noegel, A.A. Inner nuclear envelope protein sun1 plays a prominent role in mammalian mRNA export. Nucleic Acids Res. 2015, 43, 9874–9888. [Google Scholar] [CrossRef] [PubMed]
- Alsarraj, J.; Faraji, F.; Geiger, T.R.; Mattaini, K.R.; Williams, M.; Wu, J.; Ha, N.H.; Merlino, T.; Walker, R.C.; Bosley, A.D.; et al. BRD4 short isoform interacts with RRP1B, SIPA1 and components of the linc complex at the inner face of the nuclear membrane. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Peng, R.; Ren, Y.; Apparsundaram, S.; Deguzman, J.; Bauer, C.M.; Hoffman, A.F.; Hamilton, S.; Liang, Z.; Zeng, H.; et al. BET bromodomain proteins mediate downstream signaling events following growth factor stimulation in human lung fibroblasts and are involved in bleomycin-induced pulmonary fibrosis. Mol. Pharmacol. 2013, 83, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. Brd4 is a novel therapeutic target for liver fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, J.; Song, K. Bet protein inhibition mitigates acute myocardial infarction damage in rats via the tlr4/traf6/nf-kappab pathway. Exp. Ther. Med. 2015, 10, 2319–2324. [Google Scholar] [PubMed]
- Ding, X.; Xu, R.; Yu, J.; Xu, T.; Zhuang, Y.; Han, M. SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev. Cell 2007, 12, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Stewart-Hutchinson, P.J.; Hale, C.M.; Wirtz, D.; Hodzic, D. Structural requirements for the assembly of linc complexes and their function in cellular mechanical stiffness. Exp. Cell Res. 2008, 314, 1892–1905. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.N.; Ribeiro, A.J.; Lammerding, J. Nuclear shape, mechanics, and mechanotransduction. Circ. Res. 2008, 102, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Caille, N.; Thoumine, O.; Tardy, Y.; Meister, J.J. Contribution of the nucleus to the mechanical properties of endothelial cells. J. Biomech. 2002, 35, 177–187. [Google Scholar] [CrossRef]
- Guilak, F.; Tedrow, J.R.; Burgkart, R. Viscoelastic properties of the cell nucleus. Biochem. Biophys. Res. Commun. 2000, 269, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.H.; Collier, J.H.; Sfeir, C.S.; Healy, K.E. Engineering gene expression and protein synthesis by modulation of nuclear shape. Proc. Natl. Acad. Sci. USA 2002, 99, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Lovett, D.B.; Shekhar, N.; Nickerson, J.A.; Roux, K.J.; Lele, T.P. Modulation of nuclear shape by substrate rigidity. Cell. Mol. Bioeng. 2013, 6, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Hubner, M.R.; Eckersley-Maslin, M.A.; Spector, D.L. Chromatin organization and transcriptional regulation. Curr. Opin. Genet. Dev. 2013, 23, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Tajik, A.; Zhang, Y.; Wei, F.; Sun, J.; Jia, Q.; Zhou, W.; Singh, R.; Khanna, N.; Belmont, A.S.; Wang, N. Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater. 2016, 15, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, T.; Rochman, M.; Taher, L.; Dimitriadis, E.K.; Nagashima, K.; Anderson, S.; Bustin, M. Chromatin decompaction by the nucleosomal binding protein HMGN5 impairs nuclear sturdiness. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Rochman, M.; Postnikov, Y.; Correll, S.; Malicet, C.; Wincovitch, S.; Karpova, T.S.; McNally, J.G.; Wu, X.; Bubunenko, N.A.; Grigoryev, S.; et al. The interaction of NSBP1/HMGN5 with nucleosomes in euchromatin counteracts linker histone-mediated chromatin compaction and modulates transcription. Mol. Cell 2009, 35, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Murphy, A.J.; Dart, A.M. A clinical perspective of anti-fibrotic therapies for cardiovascular disease. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Ponikowski, P.; Unemori, E.; Voors, A.A.; Adams, K.F., Jr.; et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (relax-AHF): A randomised, placebo-controlled trial. Lancet 2013, 381, 29–39. [Google Scholar] [CrossRef]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Gabbiani, G.; Chaponnier, C. The NH2-terminal peptide of α-smooth muscle actin inhibits force generation by the myofibroblast in vitro and in vivo. J. Cell Biol. 2002, 157, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, C.; Spencer-Dene, B.; Lee, R.M.; Hutchings, K.; Lisabeth, E.M.; Treisman, R.; Bailly, M.; Larsen, S.D.; Neubig, R.R.; Khaw, P.T. Local delivery of novel MRTF/SRF inhibitors prevents scar tissue formation in a preclinical model of fibrosis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Ajayi, I.O.; Subbotina, N.; Dodi, A.E.; Rodansky, E.S.; Chibucos, L.N.; Kim, K.K.; Keshamouni, V.G.; White, E.S.; Zhou, Y.; et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am. J. Pathol. 2015, 185, 969–986. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Del Re, D.P.; Nakano, N.; Sciarretta, S.; Zhai, P.; Park, J.; Sayed, D.; Shirakabe, A.; Matsushima, S.; Park, Y.; et al. Mir-206 mediates yap-induced cardiac hypertrophy and survival. Circ. Res. 2015, 117, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Kinoshita, H.; Kuwabara, Y.; Nakagawa, Y.; Usami, S.; Minami, T.; Yamada, Y.; Fujiwara, M.; Nakao, K. Myocardin-related transcription factor a is a common mediator of mechanical stress- and neurohumoral stimulation-induced cardiac hypertrophic signaling leading to activation of brain natriuretic peptide gene expression. Mol. Cell. Biol. 2010, 30, 4134–4148. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sweeney, H.L.; Discher, D.E.; Schwarzbauer, J.E. Extracellular matrix elasticity directs stem cell differentiation. J. Musculoskelet. Neuronal Interact. 2007, 7, 335. [Google Scholar] [PubMed]
- Driesen, R.B.; Nagaraju, C.K.; Abi-Char, J.; Coenen, T.; Lijnen, P.J.; Fagard, R.H.; Sipido, K.R.; Petrov, V.V. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc. Res. 2014, 101, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, B.; Liu, T.; Chen, X. Reversal of myofibroblast differentiation: A review. Eur. J. Pharmacol. 2014, 734, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Talele, N.P.; Boo, S.; Koehler, A.; Knee-Walden, E.; Balestrini, J.L.; Speight, P.; Kapus, A.; Hinz, B. MicroRNA-21 preserves the fibrotic mechanical memory of mesenchymal stem cells. Nat. Mater. 2017, 16, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Enemchukwu, N.O.; Cruz-Acuna, R.; Bongiorno, T.; Johnson, C.T.; Garcia, J.R.; Sulchek, T.; Garcia, A.J. Synthetic matrices reveal contributions of ECM biophysical and biochemical properties to epithelial morphogenesis. J. Cell Biol. 2016, 212, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, J.W.; Gaetani, R.; Garcia, J.J.; Braden, R.L.; Luo, C.G.; Huang, D.; DeMaria, A.N.; Omens, J.H.; Christman, K.L. Evidence for mechanisms underlying the functional benefits of a myocardial matrix hydrogel for post-mi treatment. J. Am. Coll. Cardiol. 2016, 67, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Lee, M.E.; Wang, H.; Takebayashi, S.; Takayama, T.; Kawamura, T.; Arkles, J.S.; Dusaj, N.N.; Dorsey, S.M.; Witschey, W.R.; et al. Injectable shear-thinning hydrogels for minimally invasive delivery to infarcted myocardium to limit left ventricular remodeling. Circ. Cardiovasc. Interv. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Perea-Gil, I.; Prat-Vidal, C.; Bayes-Genis, A. In vivo experience with natural scaffolds for myocardial infarction: The times they are a-changin’. Stem Cell Res. Ther. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.M.; Christman, K.L. Decellularized myocardial matrix hydrogels: In basic research and preclinical studies. Adv. Drug Deliv. Rev. 2016, 96, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Zhu, Y.; Jiang, H.; D’Amore, A.; Sakaguchi, H.; Tchao, J.; Tobita, K.; Wagner, W.R. Timing effect of intramyocardial hydrogel injection for positively impacting left ventricular remodeling after myocardial infarction. Biomaterials 2016, 83, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, S.B.; Rane, A.A.; Liu, C.J.; Rao, N.; Agmon, G.; Suarez, S.; Wang, R.; Munoz, A.; Bajaj, V.; Zhang, S.; et al. Delivery of an engineered hgf fragment in an extracellular matrix-derived hydrogel prevents negative lv remodeling post-myocardial infarction. Biomaterials 2015, 45, 56–63. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herum, K.M.; Lunde, I.G.; McCulloch, A.D.; Christensen, G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J. Clin. Med. 2017, 6, 53. https://doi.org/10.3390/jcm6050053
Herum KM, Lunde IG, McCulloch AD, Christensen G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. Journal of Clinical Medicine. 2017; 6(5):53. https://doi.org/10.3390/jcm6050053
Chicago/Turabian StyleHerum, Kate M., Ida G. Lunde, Andrew D. McCulloch, and Geir Christensen. 2017. "The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart" Journal of Clinical Medicine 6, no. 5: 53. https://doi.org/10.3390/jcm6050053
APA StyleHerum, K. M., Lunde, I. G., McCulloch, A. D., & Christensen, G. (2017). The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. Journal of Clinical Medicine, 6(5), 53. https://doi.org/10.3390/jcm6050053