
Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases


Abstract
:1. Introduction
2. Mitochondria—The Power House
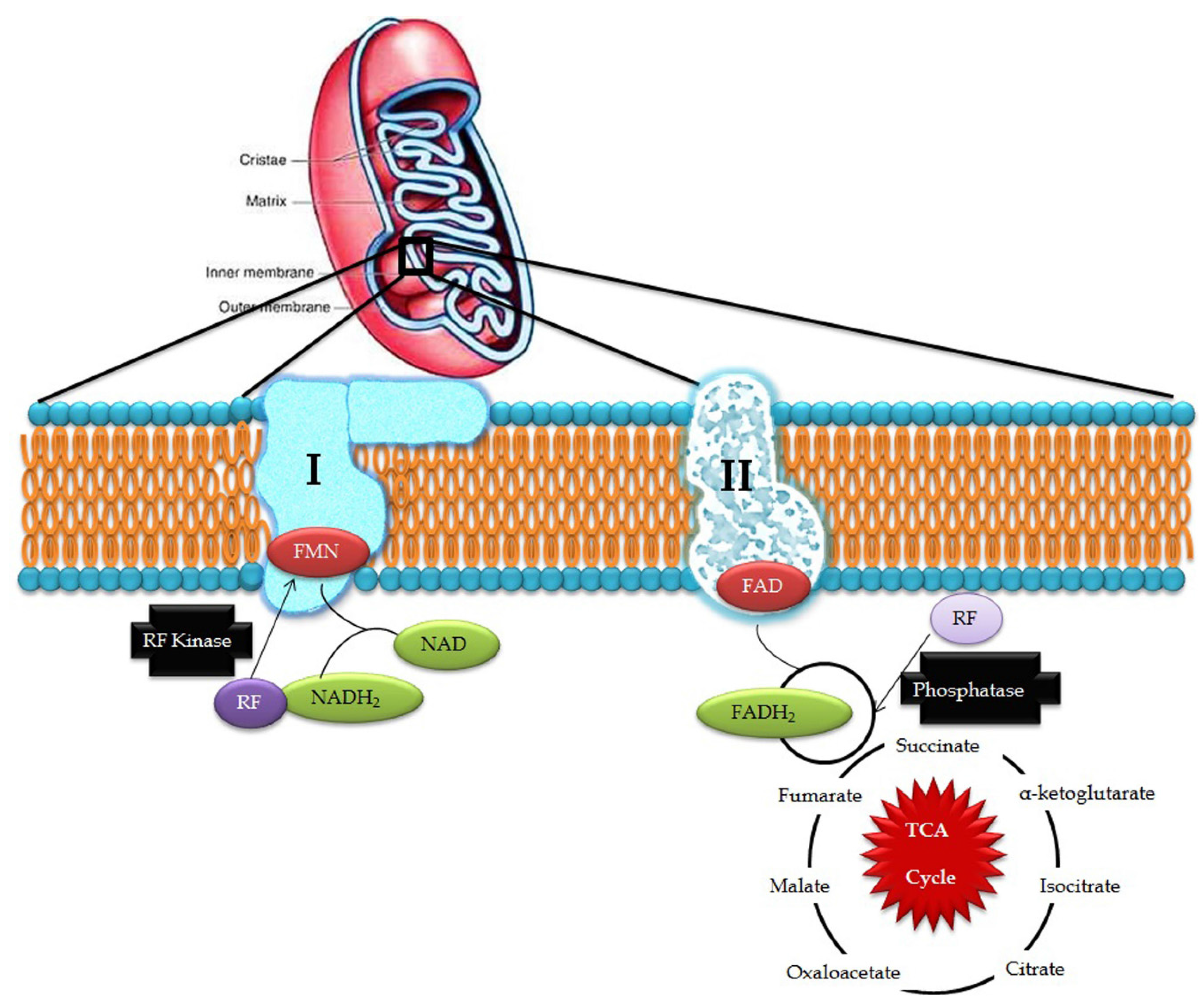
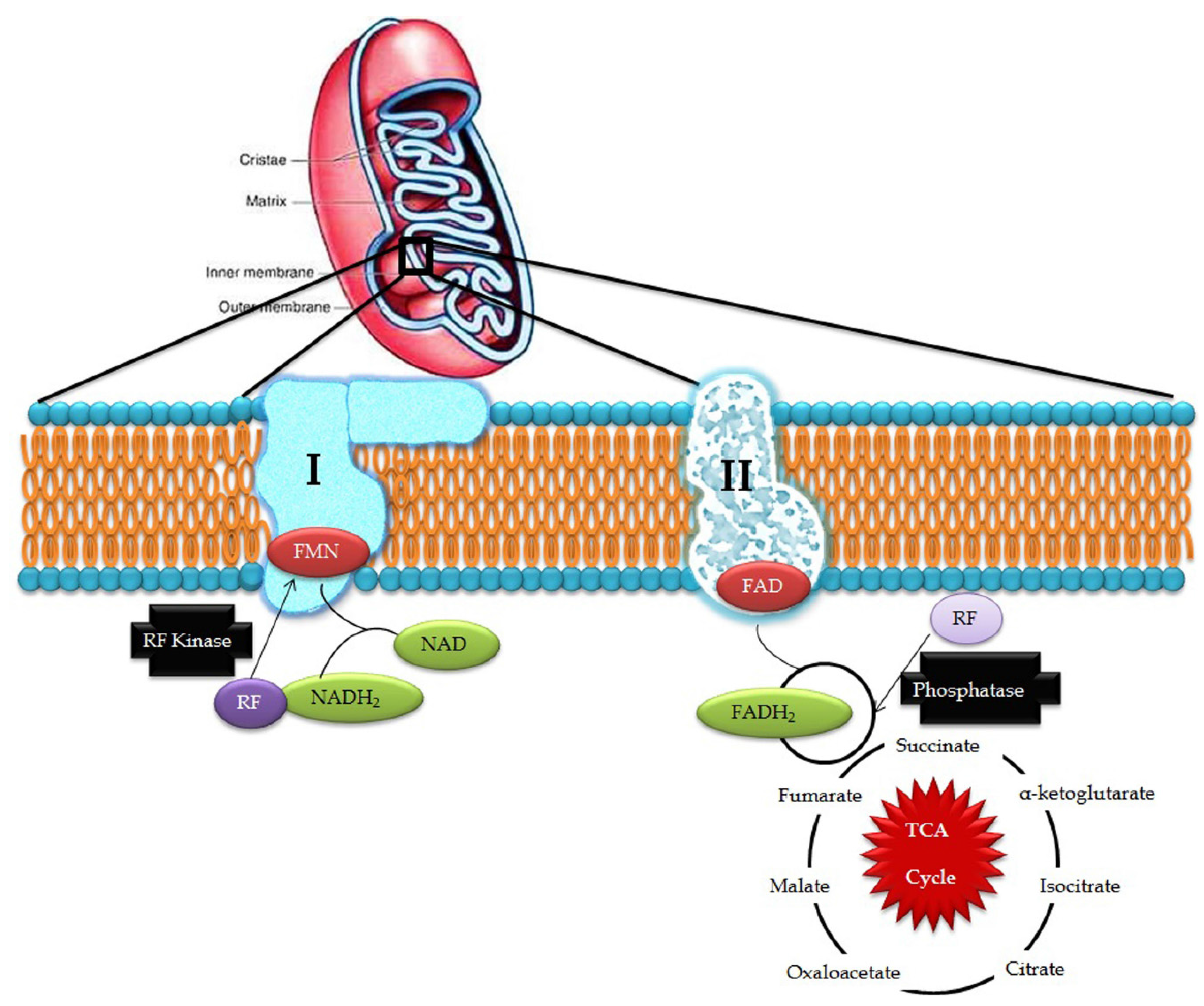
3. Riboflavin in Mitochondrial Pathways
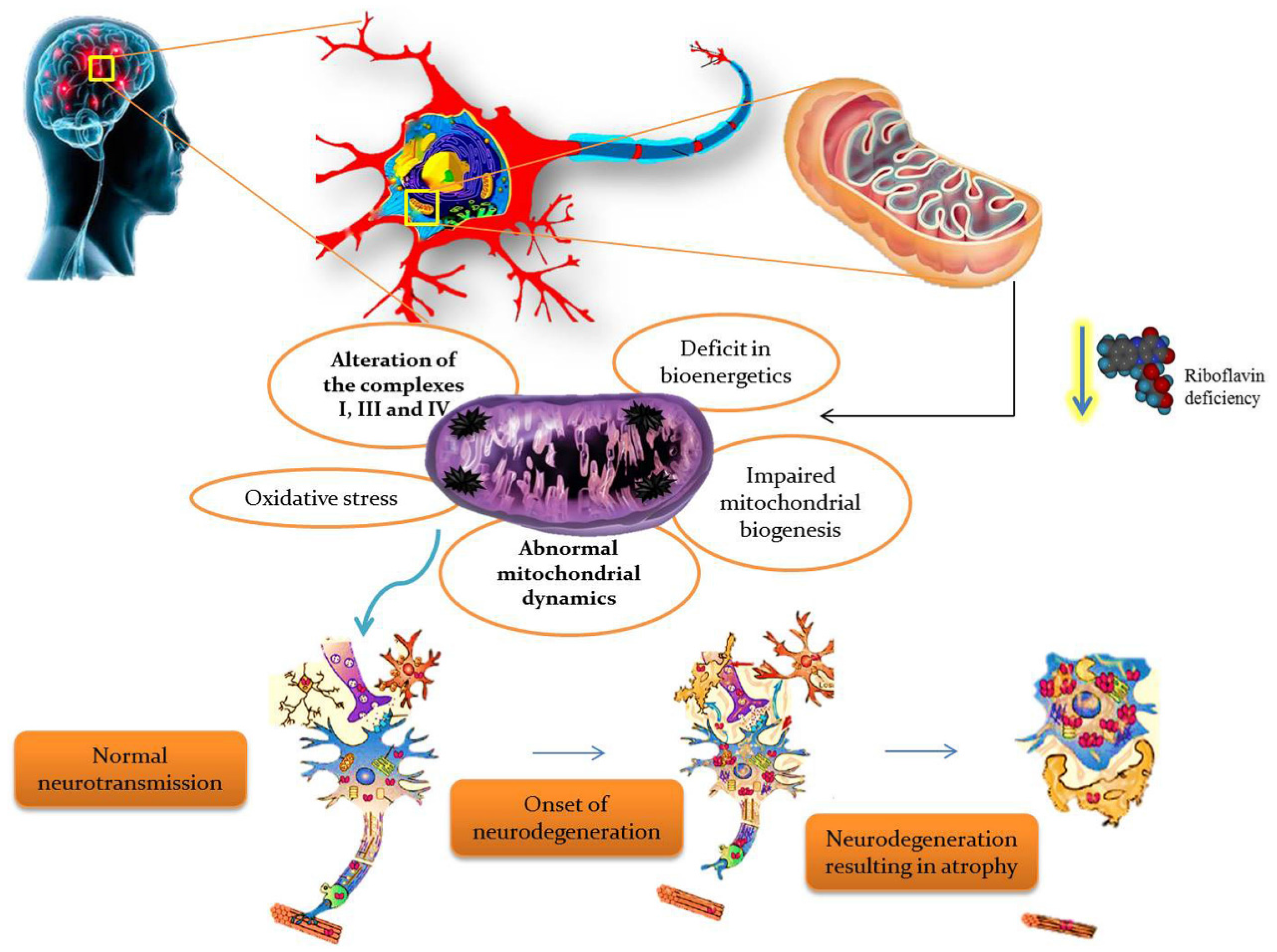
4. Riboflavin Pathogenesis in Mitochondrial Dysfunction
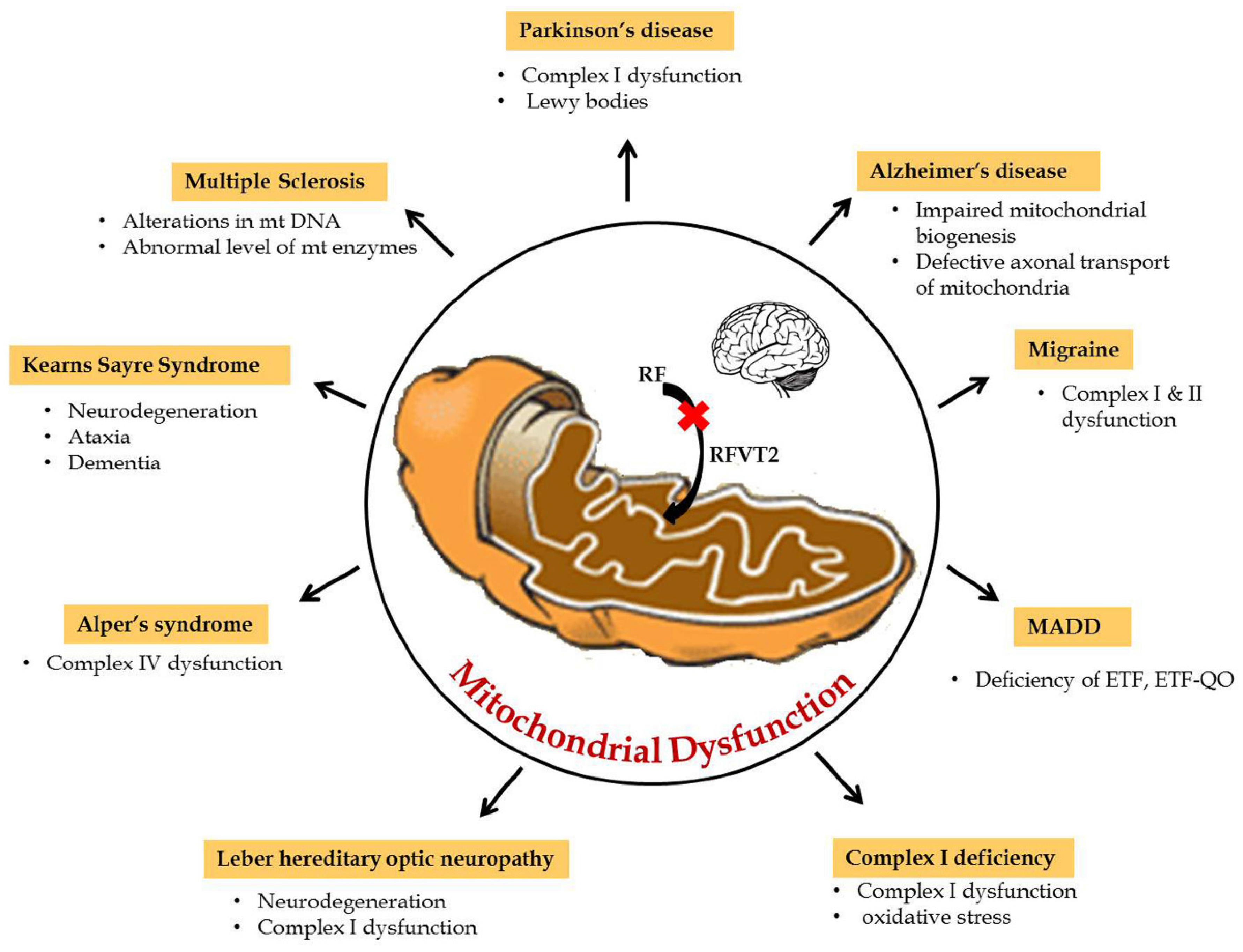
5. Riboflavin Related Mitochondrial Dysfunction in Neurological Disorders
6. Neurological Disorders of Mitochondrial Dysfunction
6.1. Multiple Acyl-CoA Dehydrogenase Deficiency (OMIM 231680)
6.2. Brown-Vialetto-Van Laere Syndrome (OMIM 211530)
6.3. Complex I Deficiency (OMIM 252010)
6.4. Leber Hereditary Optic Neuropathy (LHON; OMIM 535000)
6.5. KearnsSayre Syndrome (OMIM 530000)
6.6. Alper’s Syndrome (OMIM 203700)
6.7. Multiple Sclerosis
6.8. Parkinson’s Disease (OMIM 168600)
6.9. Alzheimer’s Disease (OMIM 104300)
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–410. [Google Scholar] [CrossRef] [PubMed]
- Nass, M.M.K.; Nass, S. Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J. Cell Biol. 1962, 19, 593–612. [Google Scholar] [CrossRef]
- Donald, V.; Voet, J.G.; Pratt, C.W. Fundamentals of Biochemistry, 2nd ed.; John Wiley and Sons, Inc.: Somerset, NJ, USA, 2006; Volume 547, p. 556. [Google Scholar]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [PubMed]
- Powers, H.J. Riboflavin (Vitamin B-2) and health1,2. Am. J. Clin. Nutr. 2003, 77, 1352–1360. [Google Scholar] [PubMed]
- Powers, H.J. Current knowledge concerning optimum nutritional status of riboflavin, niacin and pyridoxine. Proc. Nutr. Soc. 1999, 58, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Massey, V. The chemical and biological versatility of Riboflavin. Biochem. Soc. Trans. 2000, 28, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Bafunno, V.; Giancaspero, T.A.; Brizio, C.; Bufano, D.; Passarella, S.; Boles, E.; Barile, M. Riboflavin uptake and FAD synthesis in saccharomyces cerevisiae mitochondria. Involvement of the FLX1p carrier in FAD export. J. Biol. Chem. 2004, 279, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Barile, M.; Brizio, C.; Valenti, D.; De Virgilio, C.; Passarella, S. The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem. 2000, 267, 4888–4900. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Coppede, F.; Migliore, L.; Siciliano, G.; Murri, L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J. Alzheimers Dis. 2006, 10, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Stewart, V.C.; Heales, S.J.R. Nitric oxide-induced mitochondrial dysfunction: Implications for neurodegeneration. Free Radic. Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Ruiter, J.P.N.; IJLst, L.; Waterham, H.R.; Houten, S.M. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Lamattina, L.; García-Mata, C.; Graziano, M.; Pagnussat, G. Nitric oxide: The versatility of an extensive signal molecule. Annu. Rev. Plant. Biol. 2003, 54, 109–136. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.M.; Abeling, N.G.; IJLst, L.; Knoester, H.; van der Pol, W.L.; Stroomer, A.E.; Wanders, R.J.; Visser, G.; Wijburg, F.A.; Duran, M.; et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: A new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2011, 34, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Roodhooft, A.M.; Van Acker, K.J.; Martin, J.J.; Ceuterick, C.; Scholte, H.R.; Luyt Houwen, I.E.M. Benign mitochondrial myopathy with deficiency of NADH-CoQ reductase and cytochrome c oxidase. Neuropediatrics 1986, 17, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1996, 204, 53–56. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2000, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Cherian, S.; Silver, I.A. Energy metabolism in mammalian brain during development. Prog. Neurobiol. 2004, 73, 397–445. [Google Scholar] [CrossRef] [PubMed]
- Ogunleye, A.J.; Odutuga, A.A. The effect of riboflavin deficiency on cerebrum and cerebellum of developing rat brain. J. Nutr. Sci. Vitaminol. 1989, 35, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Kondo, H.; Itakura, C. Peripheral neuropathy of dietary riboflavin deficiency in racing pigeons. J. Vet. Med. Sci. 1996, 58, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Beard, S.E.; Spector, E.B.; Seltzer, W.K.; Frerman, F.E.; Goodman, S.I. Mutations in electron transfer flavoprotein: Ubiquinone oxidoreductase (ETF:QO) in glutaric acidemia type II (GA2). Clin. Res. 1993, 41, 271. [Google Scholar]
- Frerman, F.E.; Goodman, S.I. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: Glutaric acidemia type II. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2357–2365. [Google Scholar]
- Frerman, F.E.; Goodman, S.I. Deficiency of electron transfer flavoprotein or electron transfer flavoprotein: Ubiquinone oxidoreductase in glutaric academia type II fibroblasts. Proc. Natl. Acad. Sci. USA 1985, 82, 4517–4520. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.; Yonezawa, A.; Masuda, S.; Inui, K.; Sim, K.G.; Carpenter, K.; Olsen, R.K.; Mitchell, J.J.; Rhead, W.J.; Peters, G.; et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum. Mutat. 2010, 32, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Veauville-Merllie, A.; Su, C.H.; Tzagoloff, A.; Rak, M.; Ogier de Baulny, H.; Boutron, A.; Smedts-Walters, H.; Romero, N.B.; Rigal, O.; et al. SLC25A32 mutations and riboflavin-responsive exercise intolerance. N. Engl. J. Med. 2016, 374, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.K.; Konarikova, E.; Giancaspero, T.A.; Mosegaard, S.; Boczonadi, V.; Matakovic, L.; Veauville-Merllie, A.; Terrile, C.; Schwarzmayr, T.; Haack, T.B.; et al. Riboflavin-Responsive and Non-responsive Mutations in FAD Synthase Cause Multiple Acyl-CoA Dehydrogenase and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2016, 98, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Triggs, W.J.; Roe, C.R.; Rhead, W.J.; Hanson, S.K.; Lin, S.N.; Willmore, L.J. Neuropsychiatric manifestations of defect in mitochondrial beta oxidation response to riboflavin. J. Neurol. Neurosurg. Psychiatry 1992, 55, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, N.; Corydon, T.J.; Gregersen, N.; Olsen, R.K. Cellular consequences of oxidative stress in riboflavin responsive multiple acyl-CoA dehydrogenation deficiency patient fibroblasts. Hum. Mol. Genet. 2014, 23, 4285–4301. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, D.; Passadore, P.; Chinaglia, M.; Brazzo, F.; Ravenni, R.; Cudia, P. Clinical features and neurophysiological follow-up in a case of Brown-Vialetto-Van Laere syndrome. Neuromuscul. Disorder. 2005, 15, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, S.A.; Nevin, N.C.; Harding, A.E. Pontobulbar palsy and neurosensory deafness (Brown-Vialetto-Van Laere syndrome) with possible autosomal dominant inheritance. J. Med. Genet. 1990, 27, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, S. Brown-Vialetto-Van Laere syndrome. Orphanet J. Rare Dis. 2008, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Green, P.; Wiseman, M.; Crow, Y.J.; Houlden, H.; Riphagen, S.; Lin, J.P.; Raymon, F.L.; Childs, A.M.; Sheridan, E.; Edwards, S.; et al. Brown-Vialetto-Van Laere Syndrome, a Ponto-Bulbar Palsy with Deafness, Is Caused by Mutations in C20orf54. Am. J. Hum. Genet. 2010, 86, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Udhayabanu, T.; Subramanian, V.S.; Teafatiller, T.; Vykunta Raju, K.N.; Raghavan, V.S.; Varalakshmi, P.; Said, H.M.; Ashokkumar, B. SLC52A2 [p.P141T] and SLC52A3 [p.N21S] causing Brown-Vialetto-Van Laere Syndrome in an Indian patient: First genetically proven case with mutations in two riboflavin transporters. Clin. Chim. Acta 2016, 462, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Gibbs, J.R.; Megarbane, A.; Urtizberea, J.A.; Hernandez, D.G.; Foley, A.R.; Arepalli, S.; Pandraud, A.; Sanchez, J.S.; Clayton, P.; et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012, 135, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Mann, V.M.; Schapira, H. Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: Effect of ageing. J. Neurol. Sci. 1992, 113, 91–98. [Google Scholar] [CrossRef]
- Swalwell, H.; Kirby, D.M.; Blakely, E.L.; Mitchell, A.; Salemi, R.; Sugiana, C.; Compton, A.G.; Tucker, E.J.; Ke, B.X.; Lamont, P.J.; et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur. J. Hum. Genet. 2011, 19, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Lemire, B.D. Riboflavin enhances the assembly of mitochondrial cytochrome c oxidase in C. elegans NADH-ubiquinone oxidoreductase mutants. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Griebel, V.; Krigeloh-Mann, I.; Ruitenbeek, W.; Trijbels, J.M.F.; Paulus, W. A mitochondrial myopathy in an infant with lactic acidosis. Dev. Med. Child. Neurol. 1990, 32, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Ogle, R.F.; Christodoulou, J.; Fagan, E.; Blok, R.B.; Kirby, D.M.; Seller, K.L.; Dahl, H.H.; Thorburn, D.R. Mitochondrial myopathy with tRNA Leu (UUR) mutation and complex I deficiency responsive to riboflavin. J. Pediatr. 1997, 130, 138–145. [Google Scholar] [CrossRef]
- Feng, D.; Witkowski, A.; Smith, S. Down-regulation of mitochondrial acyl carrier protein in mammalian cells compromises protein lipoylation and respiratory complex I and results in cell death. J. Biol. Chem. 2009, 284, 11436–11445. [Google Scholar] [CrossRef] [PubMed]
- Gerards, M.; van den Bosch, B.J.C.; Danhauser, K.; Serre, V.; van Weeghel, M.; Wanders, R.J.A.; Nicolaes, G.A.F.; Sluiter, W.; Schoonderwoerd, K.; Scholte, H.R.; et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: New function for an old gene. Brain 2011, 134, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Man, P.Y.W.; Turnbull, D.M.; Chinnery, P.F. Leber hereditary optic neuropathy. J. Med. Genet. 2002, 39, 162–169. [Google Scholar] [CrossRef]
- Mashima, Y.; Kigasawa, K.; Wakakura, M.; Oguchi, Y. Do idebenone and vitamin therapy shorten the time to achieve visual recovery in Leber hereditary optic neuropathy? J. Neuroophthalmol. 2000, 20, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Moraes, C.T.; DiMauro, S.; Nakase, H.; Bonilla, E.; Schon, E.A.; Rowland, L.P. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology 1998, 51, 1525. [Google Scholar] [CrossRef] [PubMed]
- Mita, S.; Schmidt, B.; Schon, E.A.; DiMauro, S.; Bonilla, E. Detection of “deleted” mitochondrial genomes in cytochrome-c oxidase-deficient muscle fibers of a patient with Kearns-Sayre syndrome. Proc. Natl. Acad. Sci. USA 1989, 86, 9509–9513. [Google Scholar] [CrossRef] [PubMed]
- Rivner, M.H.; Shamsnia, M.; Swift, T.R.; Trefz, J.; Roesel, R.A.; Carter, A.L.; Yanamura, W.; Hommes, F.A. Kearns-Sayre syndrome and complex II deficiency. Neurology 1989, 39, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, E.; Osari, S.I.; Yamanouchi, H.; Matsuda, H.; Goto, Y.I.; Nonaka, I. Long-term therapy with cytochrome c, flavin mononucleotide and thiamine diphosphate for a patient with Kearns-Sayre syndrome. Brain Dev. 1996, 18, 68–70. [Google Scholar] [CrossRef]
- Bugiani, M.; Lamantea, E.; Invemizzi, F.; Moroni, I.; Bizzi, A.; Zeviani, M.; Uziel, G. Effects of riboflavin in chicken with complex II deficiency. Brain Dev. 2006, 28, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Nyhan, W.L.; Barshop, B.A.; Poulton, J.; Markusic, D.; Karpinski, N.C.; Haas, R.H. Mitochondrial DNA polymerase gamma deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann. Neurol. 1999, 45, 54–58. [Google Scholar] [CrossRef]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Wanrooij, S.; Garrido, N.; Comi, G.P.; Morandi, L.; Santoro, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localised in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Deschauer, M.; Busse, K.; Zierz, S.; Chinnery, P.F. Sensory ataxic neuropathy due to a novel C10Orf2 mutation with probable germline mosaicism. Neurology 2005, 64, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, M.; Mastrolia, V.; Rezaei Haddad, A.; Mosley, A.; Mullali, G.; Schiza, D.; Sajic, M.; Hargreaves, I.; Heales, S.; Duchen, M.R.; et al. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci. Rep. 2016, 6, 33249. [Google Scholar] [CrossRef] [PubMed]
- Naghashpour, M.; Majdinasab, N.; Shakerinejad, G.; Kouchak, M.; Haghighizadeh, M.H.; Jarvandi, F.; Hajinajaf, S. Riboflavin supplementation to patients with multiple sclerosis does not improve disability status nor is riboflavin supplementation correlated to homocysteine. Int. J. Vitam. Nutr. Res. 2013, 83, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Naghashpour, M.; Amani, R.; Sarkaki, A.; Ghadiri, A.; Samarbafzadeh, A.; Jafarirad, S.; Saki Malehi, A. Brain-derived neurotrophic and immunologic factors: Beneficial effects of riboflavin on motor disability in murine model of multiple sclerosis. Iran. J. Basic Med. Sci. 2016, 19, 439–448. [Google Scholar] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, C.G.; Junqueira, V.B.C. High doses of riboflavin and the elimination of dietary red meat promote the recovery of some motor functions in Parkinson’s disease patients. Braz. J. Med. Biol. Res. 2003, 36, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Martinez, B.A.; Berkowitz, L.A.; Caldwell, G.A.; Caldwell, K.A. Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson’s model. Cell Death Dis. 2014, 5, e984. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol Aging. 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Amyloid β-peptide1–42 contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2004, 14, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant Huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Guenther, B.D.; Sheppard, C.A.; Tran, P.; Rozen, R.; Matthews, R.G.; Ludwig, M.L. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat. Struct. Biol. 1999, 6, 359–365. [Google Scholar] [PubMed]
- Kamat, P.K.; Vacek, J.C.; Kalani, A.; Tyagi, N. Homocysteine induced cerebrovascular dysfunction: A link to alzheimer’s disease etiology. Open Neurol. J. 2015, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Enzyme | Neurological Disease | Metabolic Function | Location |
|---|---|---|---|
| Succinate dehydrogenase | Complex II deficiency | Krebs cycle | Mitochondrial inner membrane |
| Acyl Co-A dehydrogenase | Acyl Co-A dehydrogenase deficiency | Beta oxidation | Mitochondrial matrix |
| Electron transferring flavo protein—Ubiquinone oxidoreductase | Glutamic academia II C | Electron transport chain | Mitochondrial inner membrane |
| Electron transferring flavo protein | Glutamic academia II A and II B | Electron transport chain | Mitochondrial matrix |
| NADH - Ubiquinone oxidoreductase | Complex I deficiency | Electron transport chain | Mitochondrial inner membrane |
| Dihydrolipoyl dehydrogenase, Succinate dehydrogenase and NADH-Ubiquinone oxidoreductase | Leigh Syndrome | Energy metabolism | Mitochondrial matrix |
| Riboflavin transporter | BVVLS | Riboflavin uptake | Plasma/ Mitochondrial membrane |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udhayabanu, T.; Manole, A.; Rajeshwari, M.; Varalakshmi, P.; Houlden, H.; Ashokkumar, B. Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Clin. Med. 2017, 6, 52. https://doi.org/10.3390/jcm6050052
Udhayabanu T, Manole A, Rajeshwari M, Varalakshmi P, Houlden H, Ashokkumar B. Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. Journal of Clinical Medicine. 2017; 6(5):52. https://doi.org/10.3390/jcm6050052
Chicago/Turabian StyleUdhayabanu, Tamilarasan, Andreea Manole, Mohan Rajeshwari, Perumal Varalakshmi, Henry Houlden, and Balasubramaniem Ashokkumar. 2017. "Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases" Journal of Clinical Medicine 6, no. 5: 52. https://doi.org/10.3390/jcm6050052
APA StyleUdhayabanu, T., Manole, A., Rajeshwari, M., Varalakshmi, P., Houlden, H., & Ashokkumar, B. (2017). Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. Journal of Clinical Medicine, 6(5), 52. https://doi.org/10.3390/jcm6050052