Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease



{kind=link}
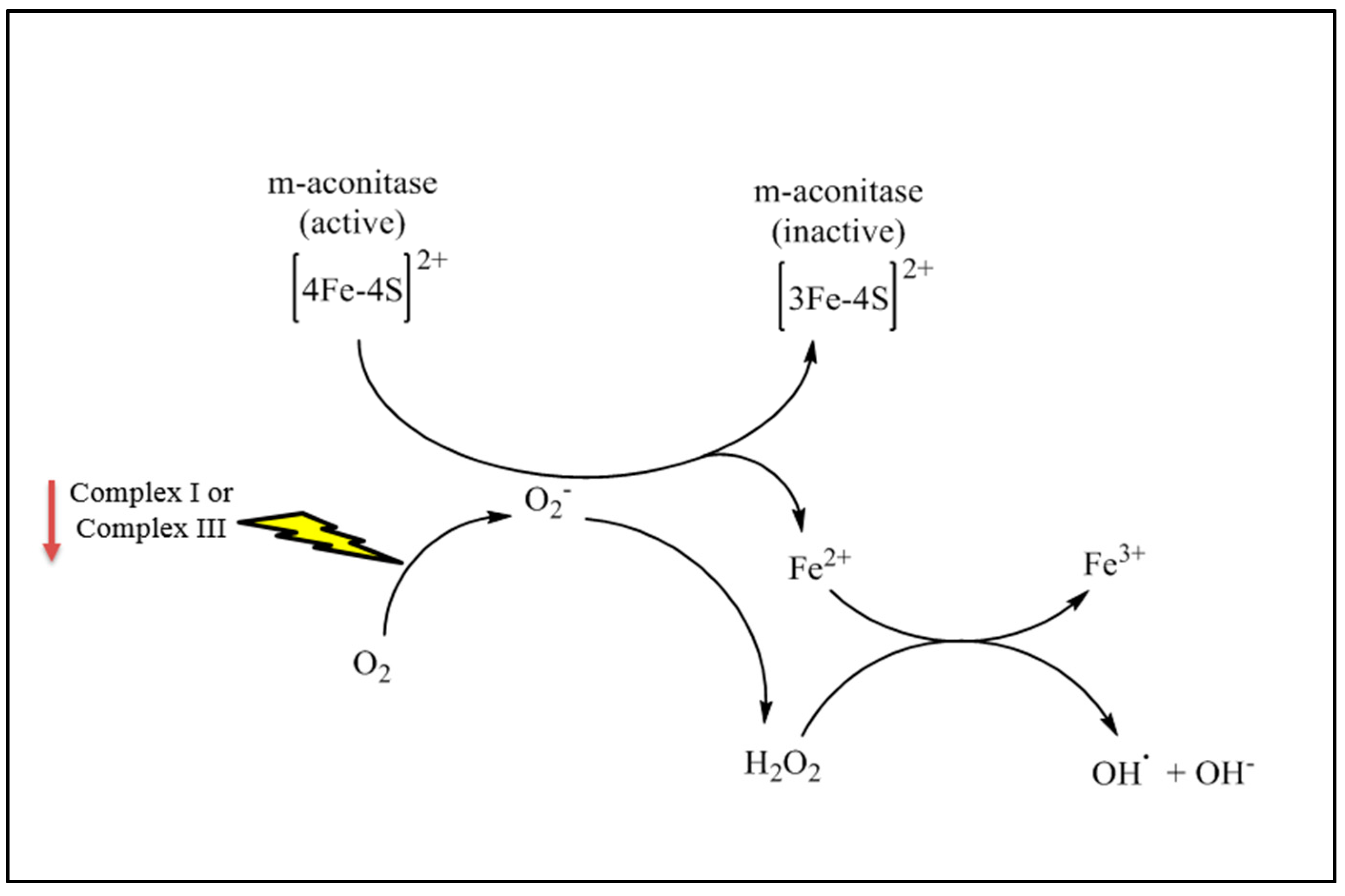{kind=link}
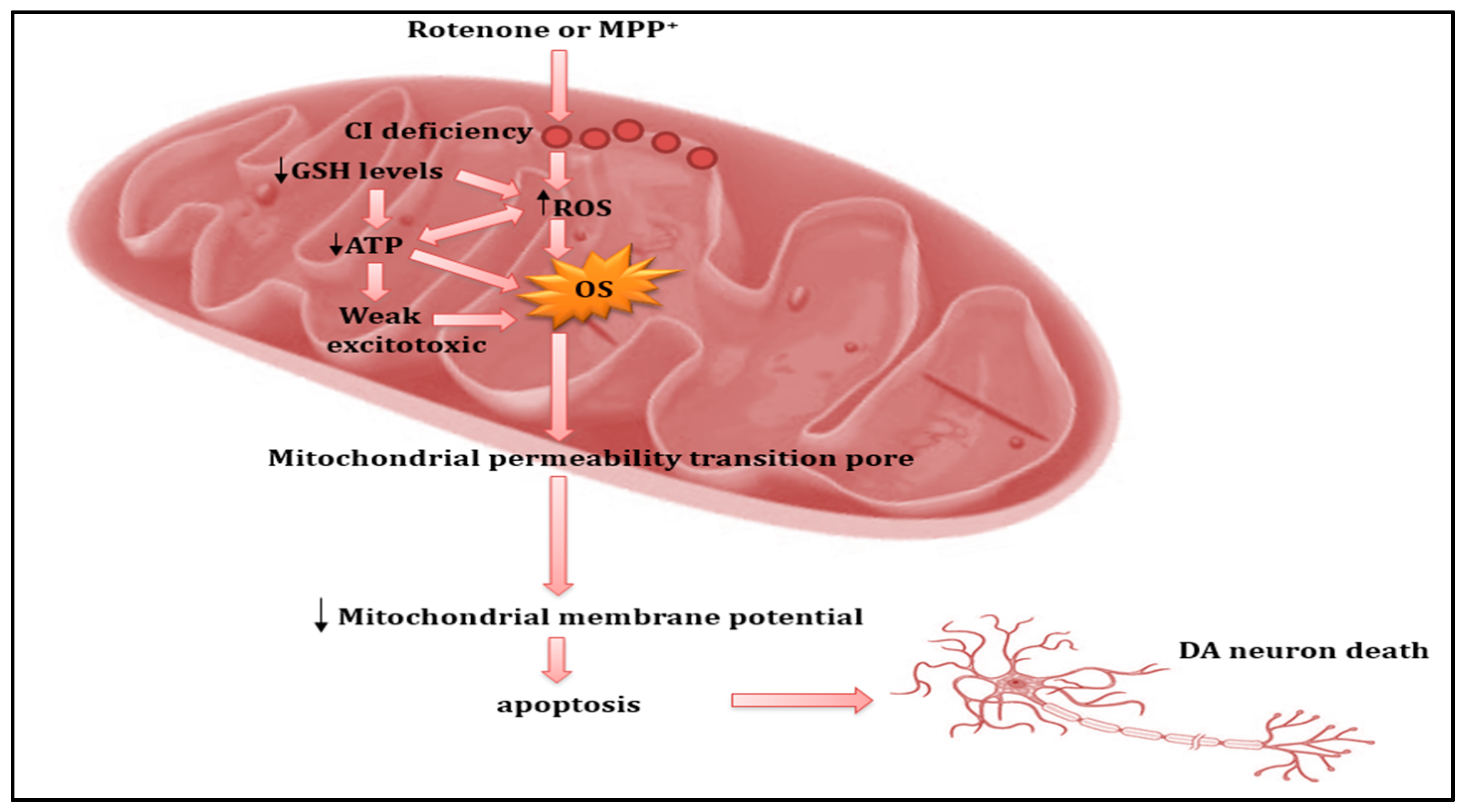{kind=link}
{kind=link}
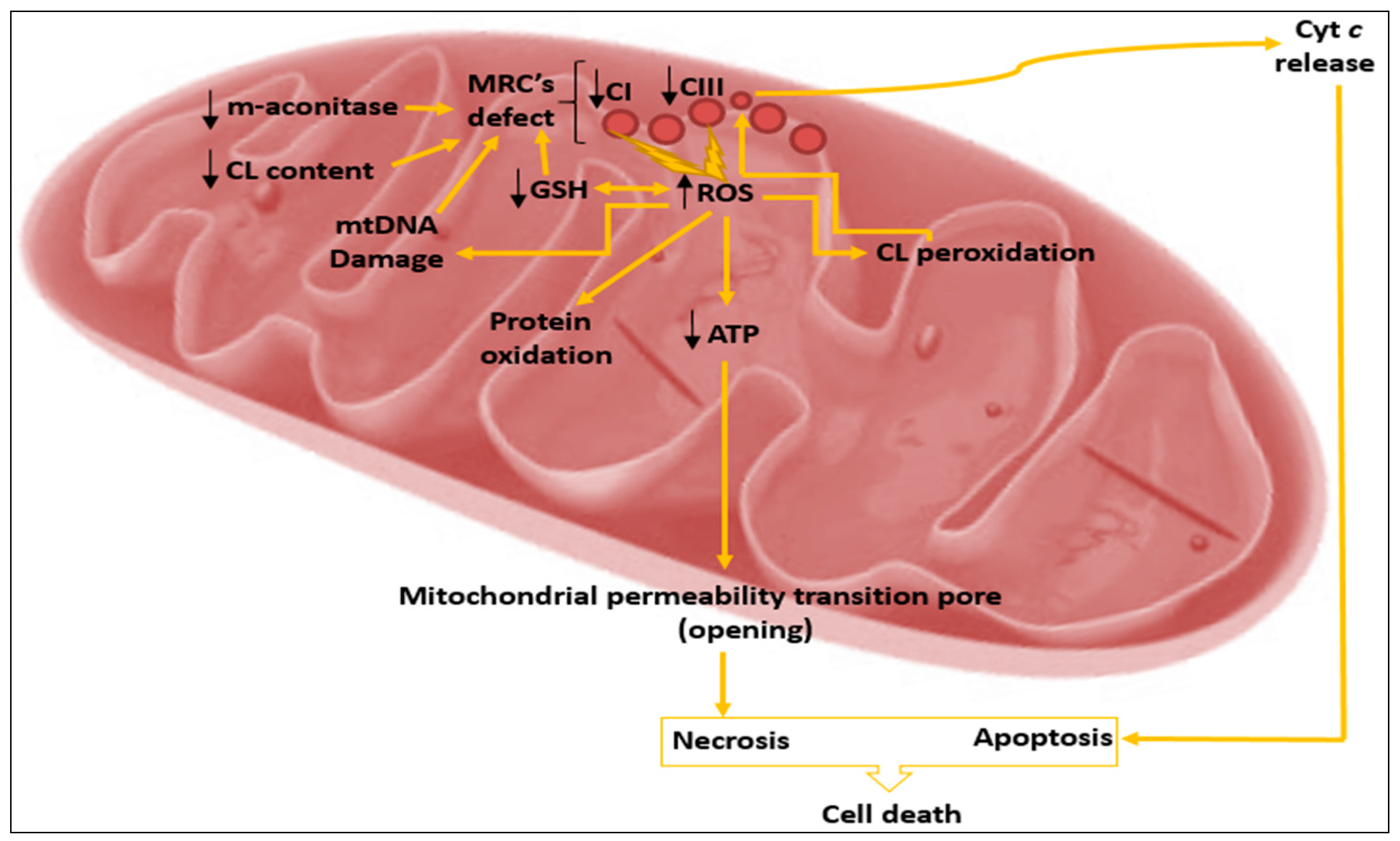{kind=link}
Abstract
1. Introduction
2. Inherited Mitochondrial Disorders
2.1. Inherited Mitochondrial DNA (mtDNA) Disorders
2.2. An Inherited Mitochondrial Lipid Disorder
2.3. An Inherited Mitochondrial Protein Disorder
3. Parkinson’s Disease (PD)
4. The Role of Antioxidants in the Prevention of Oxidative Damage
4.1. Glutathione (GSH)
4.2. Coenzyme Q10
4.3. Other Antioxidants
4.4. Ketogneic Diet (KD)
5. Conclusion Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| PD | Parkinson’s disease |
| MRC | Mitochondrial respiratory chain |
| GPx | Glutathione peroxidase |
| GSH | Reduced glutathione |
| CL | Cardiolipin |
| mtDNA | Mitochondrial DNA |
| LHON | Leber’s hereditary optic neuropathy |
| BTHS | Barth syndrome |
| FRDA | Friedreich ataxia |
| MPTP | 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine |
References
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Dnavies, N.; Cooper, C.; Singer, M. Association between Mitochondrial Dysfunction and Severity and Outcome of Septic Shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef]
- Land, J.; Hughes, J.; Hargreaves, I.; Heales, S. Mitochondrial disease: A historical, biochemical, and London perspective. Neurochem. Res. 2004, 29, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.; Al Shahrani, M.; Wainwright, L.; Heales, S. Drug-Induced Mitochondrial Toxicity. Drug Saf. 2016, 39, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.; Vazquez, E.; Moghaddas, S.; Hoppel, C.; Lesnefsky, E. Production of Reactive Oxygen Species by Mitochondria: Central Role of Complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- Heales, S.; Bolanos, J.; Stewart, V.; Brookes, P.; Land, J.; Clark, J. Nitric Oxide, Mitochondria and Neurological Disease. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 215–228. [Google Scholar] [CrossRef]
- Turrens, J. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Bunker, V. Free radicals, antioxidants and ageing. Med. Lab. Sci. 1992, 49, 299–312. [Google Scholar] [PubMed]
- White, E.; Shannon, J.; Patterson, R. Relationship between Vitamin and Calcium Supplement Use and Colon Cancer. Cancer Epidemiol. Biomark. Prev. 1997, 6, 769–774. [Google Scholar]
- Duberley, K.; Heales, S.; Abramov, A.; Chalasani, A.; Land, J.; Rahman, S.; Hargreaves, I. Effect of Coenzyme Q10 Supplementation on Mitochondrial Electron Transport Chain Activity and Mitochondrial Oxidative Stress in Coenzyme Q10 Deficient Human Neuronal Cells. Int. J. Biochem. Cell Biol. 2014, 50, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Shenvi, S.; Widlansky, M.; Suh, J.; Hagen, T. Lipoic Acid as a Potential Therapy for Chronic Diseases Associated with Oxidative Stress. Curr. Med. Chem. 2004, 11, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Fiedor, J.; Burda, K. Potential Role of Carotenoids as Antioxidants in Human Health and Disease. Nutrients 2014, 6, 466–488. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.; Duchen, M.; Hothersall, J.; Clark, J.; Heales, S. Induction of Mitochondrial Oxidative Stress in Astrocytes by Nitric Oxide Precedes Disruption of Energy Metabolism. J. Neurochem. 2005, 95, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, G.; Cortopassi, G. Oxidative Stress in Inherited Mitochondrial Disease. Free Radic. Biol. Med. 2015, 88, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Stewart, V.; Heales, S. Nitric Oxide-Induced Mitochondrial Dysfunction: Implication for Neurodegeneration. Free Radic. Biol. Med. 2003, 34, 287–303. [Google Scholar] [CrossRef]
- Chinnery, P.; Johnson, M.; Wardell, T.; Singh-Kler, R.; Hayes, C.; Taylor, R.; Bindoff, L.; Turnbull, D. The Epidemiology of Pathogenic Mitochondrial DNA Mutations. Ann. Neurol. 2000, 48, 188–193. [Google Scholar] [CrossRef]
- Lamont, P.; Surtees, R.; Woodward, C.; Leonard, J.; Wood, N.; Harding, A. Clinical and Laboratory Findings in Referrals for Mitochondrial DNA Analysis. Arch. Dis. Child. 1998, 79, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Gillis, L.; Sokol, R. Gastrointestinal Manifestation of Mitochondrial Disease. Gastroenterol. Clin. N. Am. 2003, 32, 789–817. [Google Scholar] [CrossRef]
- Sykora, P.; Wilson, D.; Bohr, V. Repair of Persistent Strand Breaks in the Mitochondrial Genome. Mech. Aging Dev. 2012, 133, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Cline, S. Mitochondrial DNA Damage and Its Consequences for Mitochondrial Gene Expression. Biochim. Biophys. Acta Bioenerg. 2012, 1819, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenburgh, C.; Hiona, A. The Role Mitochondrial DNA Mutations in Aging and Sarcopenia. Exp. Gerontol. 2008, 43, 24–33. [Google Scholar]
- Huoponen, K.; Vilkki, J.; Aula, P.; Nikoskelainen, E.; Savontaus, M. A New mtDNA Mutation Associated with Leber Hereditary Optic Neuroretinopathy. Am. J. Hum. Genet. 1991, 48, 1147–1153. [Google Scholar] [PubMed]
- Sadun, A.; Morgia, C.; Carelli, V. Leber Hereditary Optic Neuropathy. Curr. Treat. Options Neurol. 2011, 13, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Battisti, C.; Formichi, P.; Cardaioli, E.; Bianchi, S.; Mangiavacchi, P.; Tripodi, S.; Tosi, P.; Federico, A. Cell Response to Oxidative Stress Induced Apoptosis in Patients with Leber Hereditary Optic Neuropathy. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Sharpley, M.; Fan, W.; Waymire, K.; Sadun, A.; Carelli, F.; Ross-Cisneros, F.; Baciu, P.; Sung, E.; McManus, M.; et al. Mouse mtDNA Mutant Model of Leber Hereditary Optic Neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef] [PubMed]
- Howell, N. Leber Hereditary Optic Neuropathy: Respiratory Chain Dysfunction and Degeneration of the Optic Nerve. Vis. Res. 1998, 38, 1495–1504. [Google Scholar] [CrossRef]
- Kirches, E. LHON: Mitochondrial Mutation and More. Curr. Genom. 2011, 12, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, Y.; Wang, J.; Tong, Y. Oxidative Stress in Chinese Patients with Leber Hereditary Optic Neuropathy. J. Int. Med. Res. 2008, 36, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, R.; Arumi, E.; Llige, D.; Marti, R.; Solano, A.; Montoya, J.; Arenas, J.; Andreu, A. Free Radicals-mediated Damage in Transmitochondrial Cells Harboring the T14487C Mutation in the ND6 Gene of mtDNA. FEBS Lett. 2005, 579, 6909–6913. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Pitkanen, S.; Kassovska, S.; Robinson, B.; Lehotay. Excessive Formation of Hydroxyl Radicals and Aldehydic Lipid Peroxidation Products in Cultured Skin Fibroblasts From Patients with Complex I Deficiency. J. Clin. Investig. 1997, 99, 2877–2882. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Cavelier, L.; Collins-Schramm, H.; Seldin, M.; McGrogan, M.; Savontaus, M.; Cortopassi, G. Differentiation-specific Effects of LHON Mutation Introduced into Neuronal NT2 Cells. Hum. Mol. Genet. 2002, 11, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.; Kao, S.; Wang, A.; Wei, Y. Increased 8 hydroxy 2′ deoxyguanosine in Leukocyte DNA in Leber Hereditary Optic Neuropathy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1688–1691. [Google Scholar] [CrossRef]
- Sadun, A.; Carelli, V.; Salomao, S.; Berezovsky, A.; Quiros, P.; Sadun, F.; DeNegri, A.; Andrade, R.; Moraes, M.; Passos, A.; et al. Extensive Investigation of a Large Brazilian Pedigree of 11778/haplogroup J Leber Hereditary Optic Neuropathy. Am. J. Ophthalmol. 2004, 136, 231–238. [Google Scholar] [CrossRef]
- Barth, P.; Scholte, H.; Berden, J.; Moorsel, J.; Luyt-Houwen, I.; Veer-Korthof, E.; Harten, J.; Sobotka-Plojhar, M. An X-linked Mitochondrial Disease Affecting Cardiac Muscle, Skeletal Muscle and Neutrophil Leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- Jefferies, J. Barth Syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.; Valianpour, F.; Bowen, V.; Lam, J.; Duran, M.; Vaz, F.; Wanders, R. X-linked Cardioskeletal Myopathy and Neutropenia (Barth Syndrome): An Update. Am. J. Med. Genet. 2004, 126, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Pope, S.; Land, J.; Heales, J. Oxidative Stress and Mitochondrial Dysfunction in Neurodegeneration; Cardiolipin a Critical Target? Biochim. Biophys. Acta Bioenerg. 2008, 1777, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Greenberg, R. The role of cardiolipin in The Structural Organization of Mitochondrial Membranes. Biochim. Biophys. Acta Bioenerg. 2009, 1788, 2080–2083. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Benedictis, V.; Ruggiero, F.; Petrosillo, G. Functional Role of Cardiolipin in Mitochondrial Bioenergetics. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, K.; Gohil, V.; Stuart, R.; Hunte, C.; Brandt, U.; Greenberg, M.; Schagger, H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [PubMed]
- Vladimir, G.; Sten, O.; Boris, Z. Multiple Pathways of Cytochrome c Release from Mitochondria in Apoptosis. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 639–647. [Google Scholar]
- Barth, P.; Wanders, R.; Vreken, P.; Janssen, E.; Lam, J.; Baas, F. X-linked Cardioskeletal Myopathy and Neutropenia (Barth Syndrome) (MIM 302060). J. Inherit. Metab. Dis. 1999, 22, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.; Duncan, A.; Wu, L.; Agrawal, A.; Land, J.; Heales, S. Inhibition of Mitochondrial Complex IV Leads to Secondary Loss Complex II–III Activity: Implication for the Pathogenesis and Treatment of Mitochondrial Encephalomyopathies. Mitochondrion 2007, 7, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.; Forni, M.; Santos, V.; Pinto, N.; Kowaltowski, A. Cardiolipin is a Key Determinant for mtDNA Stability and Segregation during Mitochondrial Stress. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The Maintenance of Mitochondrial DNA Integrity—Critical Analysis and Update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Saric, A.; Andreau, K.; Armand, A.; Moller, I.; Petit, P. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front. Genet. 2016, 6, 359. [Google Scholar] [CrossRef] [PubMed]
- Rotig, A.; Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and Mitochondrial Iron-sulphur Protein Defeciencey in Friedreich Ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A. Friedreich Ataxia: Pathology, Pathogenesis, and Molecular Genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Parkinson, M.; Hargreaves, I.; Angelova, P.; Sandi, C.; Pook, M.; Giunti, P.; Abramov, A. Mitochondrial Energy Imbalance and Lipid Peroxidation Cause Cell Death in Friedreich Ataxia. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of Mitochomdrial Iron Accumulation by Yfh1p, a putative Homolog of Frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.; Muhlenhoff, U.; Lill, R. An Interaction between Frataxin and Isu1/Nfs1 That is Crucial for Fe/S Cluster Synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J. Friedreich Ataxia is a Mitochondrial Disorder. Proc. Natl. Acad. Sci. USA 1999, 96, 10948–10949. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Eckl, P. Oxidative Stress and the Homoeodynamics of Iron Metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich Ataxia Mutation Confers Cellular Sensitivity to Oxidant Stress Which is Rescued by Chelators of Iron and Calcium and Inhibitors of Apoptosis. Hum. Mol. Genet. 1999, 8, 6. [Google Scholar] [CrossRef]
- Yuxi, S.; Schoenfeld, R.; Hayashi, G.; Napoli, E.; Akiyama, T.; Carstens, M.; Carstens, E.; Pook, M.; Cortopa, G. Frataxin Deficiency Leads to Defect in Expression of Antioxidants and Nrf2 Expression in Dorsal Root Ganglia of the Friedreich Ataxia YG8R Mouse Model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar]
- Runko, A.; Griswold, A.; Kyung-Tai, M. Overexpression of Frataxin in the Mitochondria Increases Resistance to Oxidative Stress and Extends Lifespan in Drosophila. FEBS Lett. 2008, 582, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Mandel, J. Deciphering the Cause of Friedreich Ataxia. Curr. Opin. Neurobiol. 1997, 7, 689–694. [Google Scholar] [CrossRef]
- Beinert, H.; Kennedy, M. Aconitase, a Two-faced Protein: Enzyme and Iron Regulatory Factor. FASEB J. 1993, 15, 1442–1449. [Google Scholar]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedyl, M. Mitochondrial Aconitase is a Source of Hydroxyl Radical: An Electron Spin Resonance Investigation. J. Biol. Chem. 2000, 275, 14064–14069. [Google Scholar] [CrossRef] [PubMed]
- Houten, B.; Woshner, V.; Santos, J. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair (Amst.) 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.; Fridovich, I. Superoxide Sensitivity of the Escherichia Coli Aconitase. J. Biol. Chem. 1991, 266, 19328–19333. [Google Scholar] [PubMed]
- Haile, D.; Rouault, T.; Harford, J.; Kennedy, M.; Blondin, G.; Klausner, R. Cellualr Regulation of the Iron-responsive Element Binding Protein: Disassembly of the Cubane Iron-sulfur Cluster Results in High Affinity RNA Binding. Proc. Natl. Acad. Sci. USA 1992, 89, 11735–11739. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Ho, Y.; Patel, M. Mitochondrial Superoxide Production in Kainate-induced Hippocampal Damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef]
- Kim, H.; LaVaute, T.; Iwai, K.; Klausner, R.; Rouault, T. Identification of a Conserved and Functional Iron-responsive Element in the 5′-Untranslated Region of Mammalian Mitochondrial Aconitase. J. Biol. Chem. 1996, 271, 24226–24230. [Google Scholar] [CrossRef] [PubMed]
- Stankiewics, J.; Brass, S. Role of Iron in Neurotoxicity: A cause for Concern in the Elderly? Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Fariss, M.; Chan, C.; Patel, M.; Houten, B.; Orrenius, S. Role of Mitochondria in Toxic Oxidative Stress. Mol. Interv. 2005, 5, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Aging and Parkinson’s disease: Why is Advancing Age the Biggest Risk Factor? Aging Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Eden, S.; Tanner, M.; Bernstein, A.; Fross, R.; Leimpeter, A.; Bloch, D.; Nelson, L. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.; Ballard, P.; Tetrud, J.; Irwin, I. Chronic Parkinsonism in Humans due to a Product of Meperidine-analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Abou-Sleiman, P.; Muqit, M.; Wood, N. Expanding Insights of Mitochondrial Dysfunction in Parkinson’s Disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and New Animal Models of Parkinson’s Disease. J. Biomed. Biotechnol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.; Parks, J.; Swerdlow, R. Complex I Deficiency in Parkinson’s Disease Frontal Cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Nappi, G.; Greenamyre, J. Quantitative Study of Mitochondrial Complex I in Platelets of Parkinsonian Patients. Mov. Disord. 1998, 13, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Blin, O.; Desnuelle, C.; Rascol, O.; Borg, M.; Peyro, H.; Azulay, J.; Bille, F.; Figarella, D.; Coulom, F.; Pellissier, J. Mitochondrial Respiratory Failure in Skeletal Muscle from Patients with Parkinson’s Disease and Multiple System Atrophy. J. Neurol. Sci. 1994, 125, 95–101. [Google Scholar] [CrossRef]
- Haas, R.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C. Low platelet Mitochondrial complex I and Complex II/III Activity in Early Untreated Parkinson’s Disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Acı́n-Pérez, R.; Bayona-Bafaluy, M.; Fernández-Silva, P.; Moreno-Loshuertos, R.; Pérez-Martos, A.; Bruno, C.; Moraes, C.; Enríquez, J. Respiratory Complex III Is Required to Maintain Complex I in Mammalian Mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Keane, P.; Kurzawa, M.; Blain, P.; Morris, C. Mitochondrial Dysfunction in Parkinson’s Disease. Parkinsons Dis. 2011, 2011, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dominic, H.; Scott, A.; Ashley, B.; Peng, L. A Delicate Balance: Iron Metabolism and Disease of the Brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar]
- Moos, T.; Nielsen, T.; Skjorringe, T.; Morgan, E. Iron Trafficking Inside the Brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Winter, W.; Bazydlo, L.; Harris, N. The Molecular Biology of Human Iron Metabolism. Lab. Med. 2014, 45, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.; Benkovic, S. Iron Regulation in the Brain: Histochemical, Biochemical, and Molecular Considerations. Ann. Neurol. 1992, 32, 51–61. [Google Scholar] [CrossRef]
- Brzoska, K.; Meczynska, S.; Kruszewski, M. Iron-sulfur Cluster Proteins: Electron Transfer and Beyond. Acta Biochim. Pol. 2006, 53, 685–691. [Google Scholar] [PubMed]
- Hirsch, E.; Faucheux, B. Iron Metabolism and Parkison’s Disease. Mov. Disord. 1998, 13, 39–45. [Google Scholar] [PubMed]
- Dexter, D.; Wells, F.; Lees, A.; Javoy-Agid, F.; Agid, Y.; Jenner, P.; Marsden, C. Increased Nigral Iron Content and Alterations in Other Metal Ions Occurring in Brain in Parkinson’s Disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.; Wells, F.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C. Incresed Nigral Iron Content in Postmortem Parkinsonian Brain. Lancet 1987, 330, 1219–1220. [Google Scholar] [CrossRef]
- Hirsch, E.; Brandel, J.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and Aluminum Increase in the Substantia Nigra of Patients with Parkinson’s Disease: An X-Ray Microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Mazzio, E.; Soliman, K. Effects of Enhancing Mitochondrial Oxidative Phoshporylation with Reducing Equivalents and Ubiquinone on 1-methyl-4-phenylpyridinium Toxicity and Complex I-IV Damage in Neuroblastoma Cells. Biochem. Pharmacol. 2004, 67, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Shang, T.; Kotamraiu, S.; Kalivendi, S.; Hillard, C.; Kalyanaraman, B. 1-Methyl-4-phenylpyridinium-induced Apoptosis in Cerebellar Granule Neurons is Mediated by Transferrin Receptor Iron-dependent Depletion of Tetrahydrobiopterin and Neuronal Nitric Oxide Synthase-derived Superoxide. J. Biol. Chem. 2004, 279, 19099–19112. [Google Scholar] [CrossRef] [PubMed]
- Li-Ping, L.; Patel, M. Iron-sulfur Enzyme Mediated Mitochondrial Superoxide Toxicity in Experimental Parkinson’s Disease. J. Neurochem. 2004, 90, 1076–1084. [Google Scholar]
- Ebadi, M.; Srinivasan, K.; Baxi, M. Oxidative Stress and Antioxidant Therapy in Parkinson’s Disease. Prog. Neurobiol. 1996, 48, 1–19. [Google Scholar] [CrossRef]
- Obata, T. Dopamine Efflux by MPTP and Hydroxyl Radical Generation. J. Neural Trans. 2002, 109, 1159–1180. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in Metal-induced Oxidative Stress and Human Disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Nunez, T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron Toxicity in Neurodegeneration. Biometals 2012, 25, 761–776. [Google Scholar]
- Owen, J.; Butterfield, D. Measurment of Oxidized/Reduced Glutathione Ratio. Methods Mol. Biol. 2010, 648, 269–277. [Google Scholar] [PubMed]
- Sipos, K.; Lange, H.; Fekete, Z.; Ullmann, P.; Lill, R.; Kispal, G. Maturation of Cytosolic Iron-sulfur Proteins Requires Glutathione. J. Biol. Chem. 2002, 277, 26944–26949. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M. The Role Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [PubMed]
- Heales, S.; Bolanos, J. Impirment of Brain Mitochondrial Function by Reactive Nitrogen Species: The Role of Glutathione in Dictating Susceptibility. Neurochem. Int. 2002, 40, 469–474. [Google Scholar] [CrossRef]
- Dimonte, D.; Chan, P.; Sandy, M. Glutathione in Parkinson’s Disease: A link Between Oxidative Stress and Mitochondrial Damage? Ann. Neurol. 1992, 32, 111–115. [Google Scholar] [CrossRef]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Heales, S.; Davies, S.; Bates, T.; Clark, J. Depletion of Brain Glutathione is Accompanied by Impaired Mitochondrial Function and Decreased N-acetyl Aspartate Concentration. Neurochem. Res. 1995, 20, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Merad-Saidoune, M.; Biotier, E.; Nicole, A.; Marsac, C.; Martinou, J.; Sola, B.; Sinet, P.; Ceballos-Picot, I. Overproduction of Cu/Zn-superoxide Dismutase or Bcl-2 Prevents the Brain Mitochondrial Respiratory Dysfunction Induced by Glutathione Depletion. Exp. Neurol. 1999, 158, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Jha, N.; Jurma, O.; Lalli, G.; Liu, Y.; Pettus, E.; Greenamyrel, J.; Liu, R.; Forman, H.; Anderson, J. Glutathione Depletion in PC12 Results in Selective Inhibition of Mitochondrial Complex I Activity: Implication for Parkinson’s Disease. J. Biol. Chem. 2000, 275, 26096–26101. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.; Sheena, Y.; Land, J.; Heales, S. Glutathione Deficiencey in Patients with Mitochondrial Disease: Implication for Pathogenesis and Treatment. J. Inherit. Dis. 2005, 28, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Dexter, D.; Sian, J.; Schapira, A.; Marsden, C. Oxidative stress as a Cause of Nigral Cell Death in Parkinson’s Disease and Incidental Lewy Body Disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann. Neurol. 1992, 32, 82–87. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Manzino, L.; Sonsalla, P.K.; Bernard, L.P. Characterization of Intracellular Elevation of Glutathione (GSH) with Glutathione Monoethyl Ester and GSH in Brain and Neuronal Cultures: Relevance to Parkinson’s Disease. Exp. Neurol. 2007, 203, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.; Poon, H.; Dogrukol-Ak, D.; Drake, J.; Banks, W.; Eyerman, E.; Butterfield, D.; Morley, J. The Antioxidants α-lipoic Acid and N-Acetylcysteine Reverse Memory Impairment and Brain Oxidative Stress in Aged SAMP8 Mice. J. Neurochem. 2003, 84, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Rey, P.; Soto, R.; Guerra, M.; Labandeira, L. Systemic Administration of N Acetylcysteine Protects Dopaminergic Neurons Against 6 Hydroxydopamine Induced Degeneration. J. Neurosci. Res. 2004, 76, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P. Coenzyme Q10 as a Therapy for Mitochondrial Disease. Int. J. Biochem. Cell Biol. 2014, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.; Heales, S.; Mills, K.; Eaton, S.; Land, J.; Hargreaves, I. Determination of Coenzyme Q10 Status in Blood Mononuclear Cells, Skeletal Muscle, and Plasma by HPLC with Di-Propoxy-Coenzyme Q10. Clin. Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Ogasahara, S.; Engel, A.; Frens, D.; Mack, D. Muscle Coenzymee Q Deficiency in Familial Mitocondrial Encephalomyopathy. Proc. Natl. Acad. Sci. USA 1989, 86, 2379–2382. [Google Scholar] [CrossRef] [PubMed]
- Maldergem, L.; Trijbels, F.; DiMauro, S.; Sindelar, P.; Musumeci, O.; Janssen, A.; Delberghe, X.; Martin, J.; Gillerot, Y. Coenzyme Q-Responsive Leigh’s Encephalopathy in Two Sisters. Ann. Neurol. 2002, 52, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, A.; Hadjigeorgiou, G.; Divari, R.; Papagalanis, N.; Comi, G.; Bresolin, N. The Influence of Coenzyme Q10 on Total Serum Calcium concentration in Two Patients with Kearns—Sayre syndrome and hypoparathyroidism. Neuromuscul. Disord. 1996, 6, 49–53. [Google Scholar] [CrossRef]
- Quinzii, C.; DiMauro, S.; Hirano, M. Human Coenzyme Q10 Deficiency. Neurochem. Res. 2007, 32, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; Smeitink, J. Metabolic Manipulators: A Well Founded Strategy to Combat Mitochondrial Dysfunction. J. Inherit. Metab. Dis. 2011, 34, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Salyetti, S.; Vista, M.; Lombardi, V.; Siciliano, G.; Giraldi, C. Long-Term Treatment with Idebenone and Riboflavin in a Patient with MELAS. Neurol. Sci. 2000, 21, 981–982. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Filosto, M.; Simoncini, C.; Siciliano, G. Drugs and Mitochondrial Diseases: 40 Queries and Answers. Expert Opin. Pharmacother. 2012, 13, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Jaber, S.; Polster, B. Idebenone and Neuroprotection: Antioxidant, Pro-Oxidant, or Electron Carrier? J. Bioenergy Biomembr. 2015, 47, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.; Haas, R.; Passov, D.; Beal, M. Coenzyme Q10 Levels Correlate with the Activities of Complex I and II/III in Mitochondria From Parkinsonian and Nonparkinsonian subjects. Ann. Neurol. 1997, 42, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.; Lane, A.; Sleiman, P. The Coenzyme Q10 Status of the Brain Regions of Parkinson’s Disease Patients. Neurosci. Lett. 2008, 447, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Sohmiya, M.; Tanaka, M.; Tak, N.W.; Yanagisawa, M.; Tanino, Y.; Suzuki, Y.; Okamoto, K.; Yamamoto, Y. Redox Status of Plasma Coenzyme Q10 Indicates Elevated Systemic Oxidative Stress in Parkinson’s Disease. J. Neurol. Sci. 2004, 223, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Gotz, M.; Gerstner, A.; Harth, R.; Dirr, A.; Janetzky, B.; Kuhn, W.; Riederer, P.; Gerlach, M. Altered Redox State of Platelet Coenzyme Q10 in Parkinson’s Disease. J. Neural Transm. (Vienna) 2000, 107, 41–48. [Google Scholar]
- Gille, G.; Hung, S.; Reichmann, H.; Rausch, W. Oxidative Stress to Dopaminergic Neurons as Models of Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2004, 1018, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kooncumchoo, P.; Sharma, S.; Porter, J.; Govitrapong, P.; Ebadi, M. Coenzyme Q (10) Provides Neuroprotection in Iron-Induced Apoptosis in Dopaminergic Neurons. J. Mol. Neurosci. 2006, 28, 125–141. [Google Scholar] [CrossRef]
- Winkler-Stuck, K.; Wiedemann, F.; Wallesch, C.; Kunz, S. Effect of Coenzyme Q10 on the Mitochondrial Function of Skin Fibroblasts from Parkinson Patients. J. Neurol. Sci. 2004, 220, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Zhu, X.; Wang, X.; Lee, Y.-G.; Nunomura, A.; Peterson, R.B.; Perry, G.; Smith, M.A. Mitochondria: A Therapeutic Target in Neurodegeneration. Biochim. Biophys. Acta 2010, 1802, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.; Oakes, D.; Kieburtz, K.; Beal, M.; Haas, R.; Plumb, S.; Juncos, J.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Parkinson Study Group. Effects of Coenzyme Q10 in Early Parkinson Disease: Evidence of Slowing of the Functional Decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Josef, F. Treatment of Mitochondrial Disorders. Eur. J. Pediatr. Neurol. 2010, 14, 29–44. [Google Scholar]
- Josef, F.; Bindu, P. Therapeutic Strategies for Mitochondrial Disorders. Pediatr. Neurol. 2015, 52, 302–313. [Google Scholar]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kaylanaraman, B.; Kanthasamy, G. Mitochondria-targeted Antioxidants for Treatment of Parkinson’s Disease: Preclinical and Clinical Outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, G.; Baraldi, G.; Sampaio, H.; Bomfim, H.; Queiroz, L.; Passos, A.; Carneiro, M.; Alberici, C.; Gomis, R.; Amaral, G.; et al. Melatonin Prevents Mitochondrial Dysfunction and Insulin Resistance in Rat Skeletal Muscle. J. Pineal Res. 2014, 57, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Adlam, V.; Blaikie, F.; Manas, A.; Porteous, C.; James, A.; Ross, M.; Logan, A.; Cochemé, H.; Trnka, J.; et al. Mitochondria-Targeted Antioxidants in the Treatment of Disease. Ann. N. Y. Acad. Sci. 2008, 1147, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.B.; Filloux, F.M.; Alder, S.C.; Lyon, J.L.; Caplin, D.A. Efficacy of the Ketogenic Diet as a treatment Option for Epilepsy: Meta-Analysis. J. Child. Neurol. 2006, 3, 193–198. [Google Scholar]
- Keene, D.L. A Systematic Review of the Use of the Ketogenic Diet in Childhood Epilepsy. Pediatr. Neurol. 2006, 1, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. A Randomized Trial of Classical and Medium-Chain Triglyceride Ketogenic Diets in the Treatment of Childhood Epilepsy. Epilepsia 2009, 5, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial Biogenesis in The Anticonvulsant Mechanism of the Ketogenic Diet. Ann. Neurol. 2006, 2, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The Ketogenic Diet Component Decanoic Acid Increases Mitochondrial Citrate Synthase and Complex I Activity in Neuronal Cells. J. Neurochem. 2014, 3, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Kanabus, M.; Fassone, E.; Hughes, S.D.; Bilooei, S.F.; Rutherford, T.; O’Donnell, M.; Heales, S.J.R.; Rahman, S. The Pleiotropic Effects of Decanoic Acid Treatment on Mitochondrial Function in Fibroblasts from Patients with Complex I Deficient Leigh Syndrome. J. Inherit. Metab. Dis. 2016, 39, 415–426. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Shahrani, M.; Heales, S.; Hargreaves, I.; Orford, M. Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. J. Clin. Med. 2017, 6, 100. https://doi.org/10.3390/jcm6110100
Al Shahrani M, Heales S, Hargreaves I, Orford M. Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. Journal of Clinical Medicine. 2017; 6(11):100. https://doi.org/10.3390/jcm6110100
Chicago/Turabian StyleAl Shahrani, Mesfer, Simon Heales, Iain Hargreaves, and Michael Orford. 2017. "Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease" Journal of Clinical Medicine 6, no. 11: 100. https://doi.org/10.3390/jcm6110100
APA StyleAl Shahrani, M., Heales, S., Hargreaves, I., & Orford, M. (2017). Oxidative Stress: Mechanistic Insights into Inherited Mitochondrial Disorders and Parkinson’s Disease. Journal of Clinical Medicine, 6(11), 100. https://doi.org/10.3390/jcm6110100