
Stromal Modulators of TGF-β in Cancer

 ,
,
Abstract
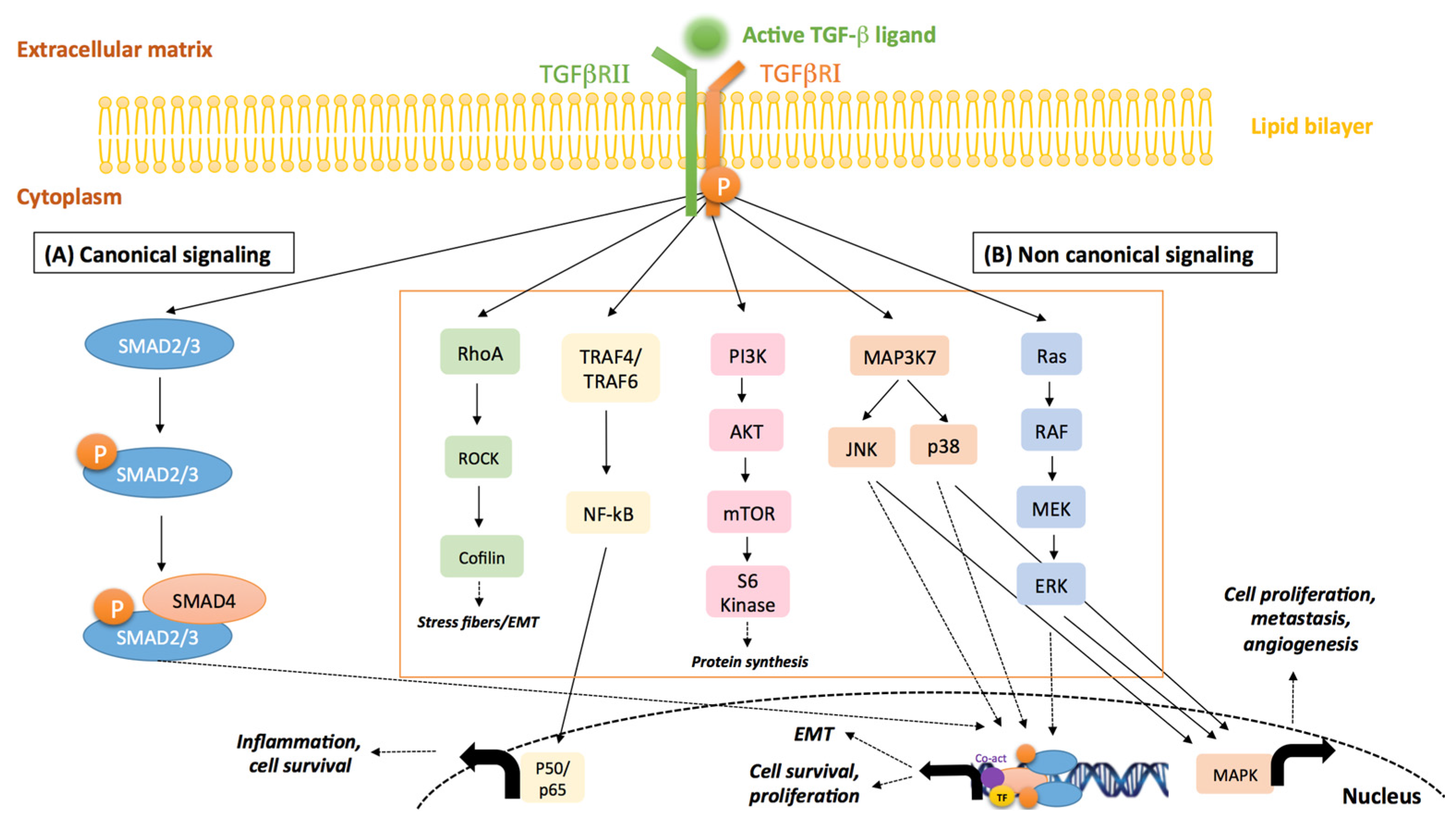
:1. Introduction
2. Cancer Cell/Stroma Crosstalk and TGF-β Activity
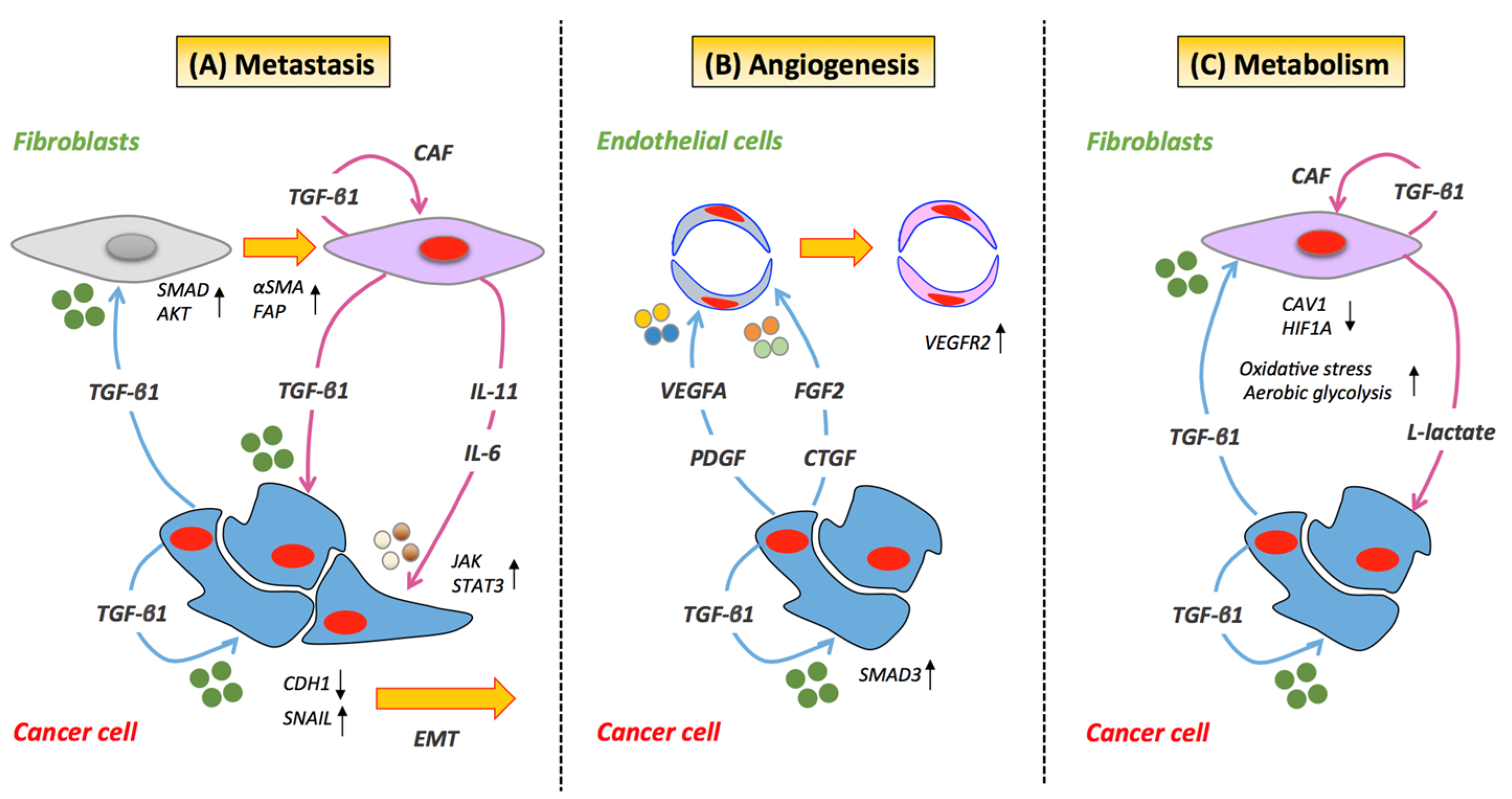
2.1. Metastasis
2.2. Angiogenesis
2.3. Metabolism
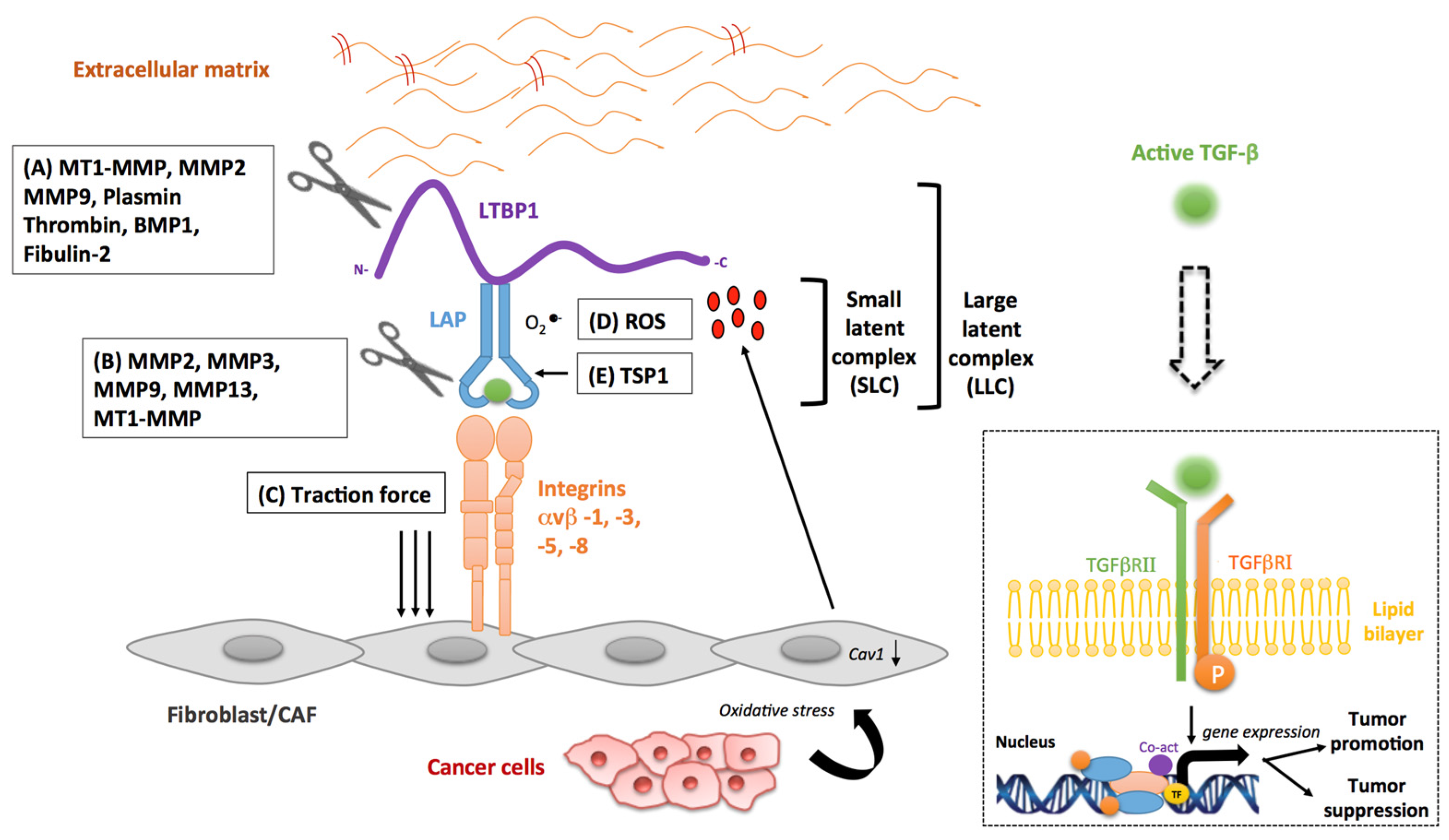
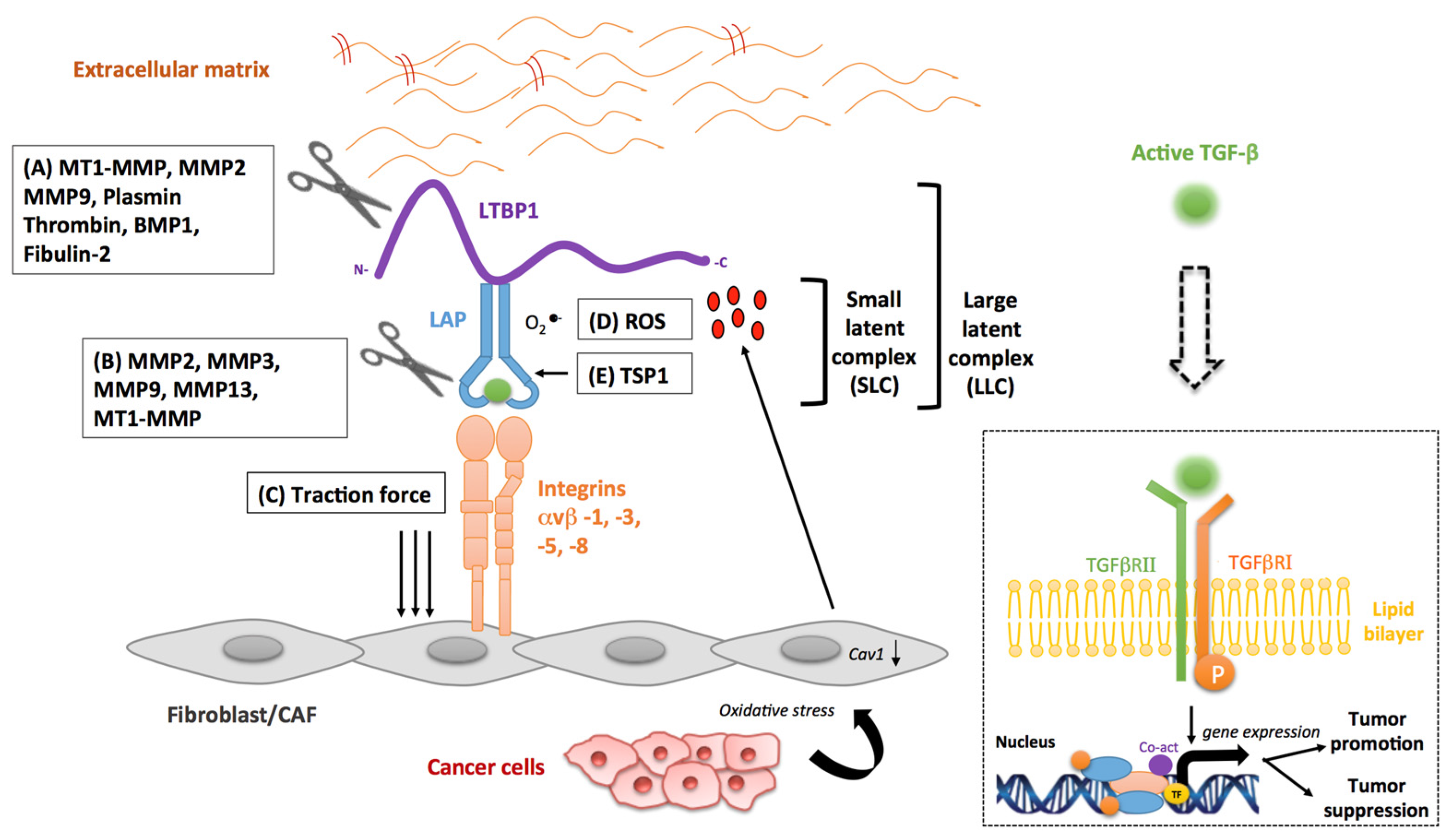
3. Stromal Activators of TGF-β in Cancer
3.1. Matrix Metalloproteinases
3.2. Integrins
3.3. Reactive Oxygen Species
3.4. Other Stromal Activators of TGF-β
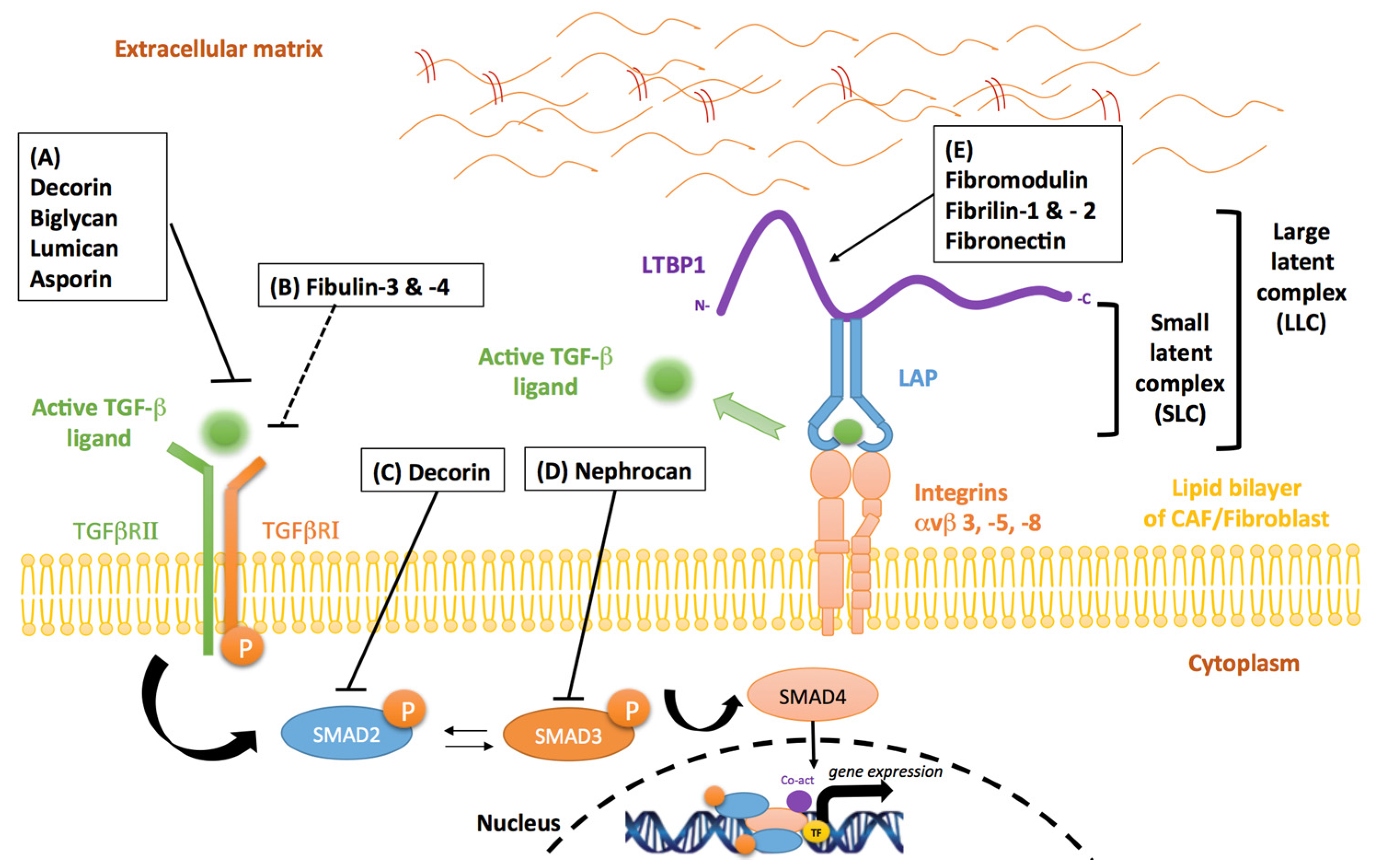
4. Stromal Inhibitors of TGF-β in Cancer
4.1. Proteoglycans
4.2. Fibrillins
4.3. Fibulins
4.4. Fibronectin
5. Clinical Outlook: Targeting Stromal Modulators of TGF-β in Cancer
5.1. MMPs
5.2. Integrins
5.3. Fibronectin
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Imamura, T.; Hikita, A.; Inoue, Y. The roles of TGF-β signaling in carcinogenesis and breast cancer metastasis. Breast Cancer 2012, 19, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFβ: The molecular jekyll and hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Derynck, R. TGF-β signaling in cancer—A double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [CrossRef]
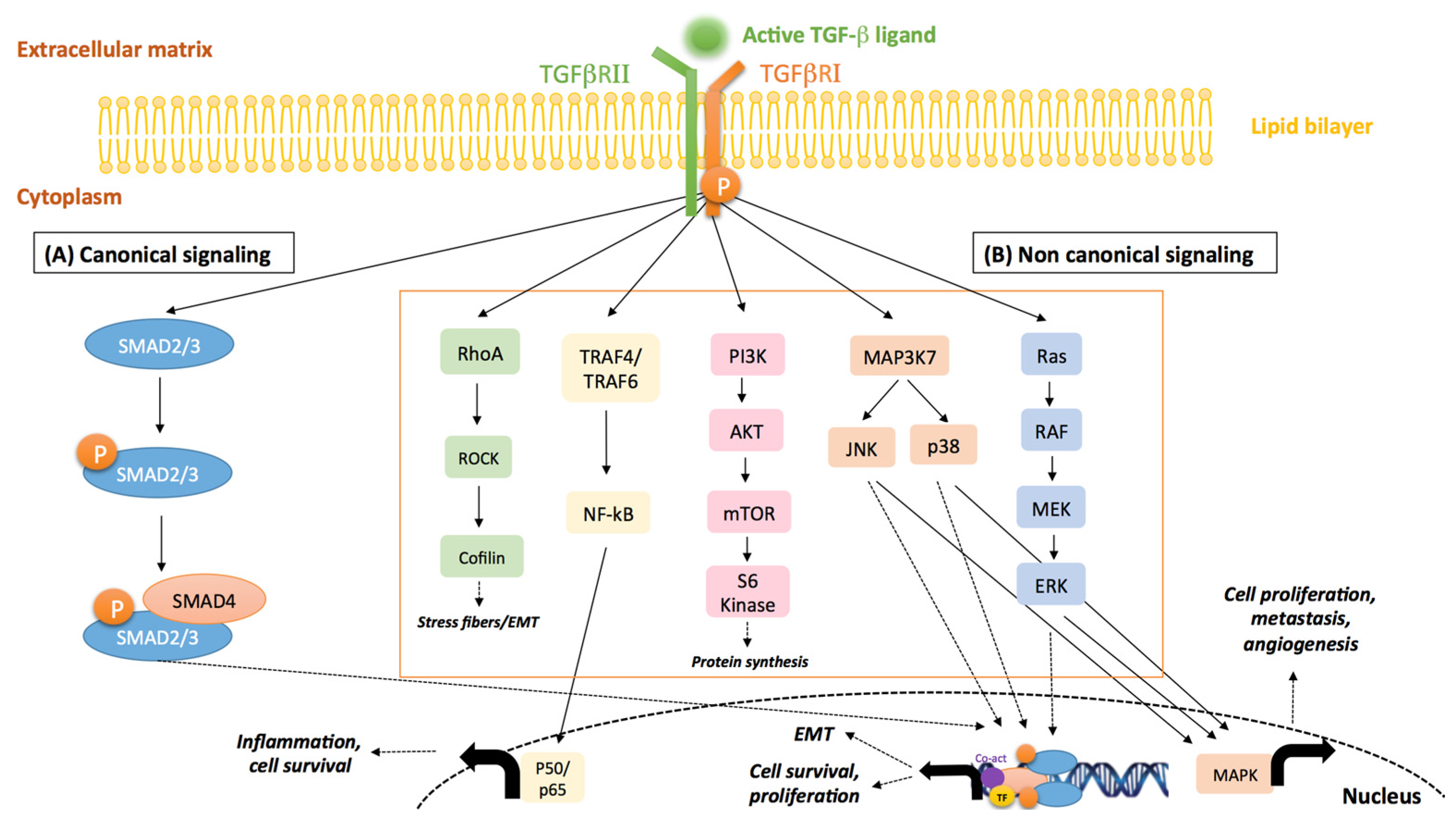
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFβ signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Massague, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massague, J. The TGFβ receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Kretzschmar, M.; Massague, J. Smads: Mediators and regulators of TGF-β signaling. Curr. Opin. Genet. Dev. 1998, 8, 103–111. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. TGF-β and cancer. Cytokine Growth Factor Rev. 2006, 17, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Muller-Pillasch, F.; Menke, A.; Yamaguchi, H.; Elsasser, H.P.; Bachem, M.; Adler, G.; Gress, T.M. TGFβ and the extracellular matrix in pancreatitis. Hepatogastroenterology 1999, 46, 2751–2756. [Google Scholar] [PubMed]
- Ikushima, H.; Miyazono, K. TGFβ signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Vu, M.; Booker, T.; Santner, S.J.; Miller, F.R.; Anver, M.R.; Wakefield, L.M. TGF-β switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J. Clin. Investig. 2003, 112, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-β induces apoptosis through Smad-mediated expression of dap-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Perlman, R.; Schiemann, W.P.; Brooks, M.W.; Lodish, H.F.; Weinberg, R.A. TGF-β-induced apoptosis is mediated by the adapter protein daxx that facilitates jnk activation. Nat. Cell Biol. 2001, 3, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Ghiassi, M.; Jirmanova, L.; Balliet, A.G.; Hoffman, B.; Fornace, A.J., Jr.; Liebermann, D.A.; Bottinger, E.P.; Roberts, A.B. Transforming growth factor-β-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 2003, 278, 43001–43007. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Agajanian, M.; Campeau, A.; Hoover, M.; Hou, A.; Brambilla, D.; Kim, S.L.; Klemke, R.L.; Kelber, J.A. Peak1 acts as a molecular switch to regulate context-dependent TGFβ responses in breast cancer. PLoS ONE 2015, 10, e0135748. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Singh, R.K. Proteases as modulators of tumor-stromal interaction: Primary tumors to bone metastases. Biochim. Biophys. Acta 2008, 1785, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-β signaling in development and disease. FEBS Lett. 2012, 586, 1833. [Google Scholar] [CrossRef] [PubMed]
- Prakash, J. Cancer-associated fibroblasts: Perspectives in cancer therapy. Trends Cancer 2016, 2, 277–279. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Cespedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.; et al. Dependency of colorectal cancer on a TGF-β -driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGF-β activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Ota, M.; Rifkin, D.B. Matrix control of transforming growth factor-β function. J. Biochem. 2012, 152, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Richardsen, E.; Uglehus, R.D.; Johnsen, S.H.; Busund, L.T. Immunohistochemical expression of epithelial and stromal immunomodulatory signalling molecules is a prognostic indicator in breast cancer. BMC Res. Notes 2012, 5, 110. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, H.; Kawata, S.; Tamura, S.; Ito, N.; Shirai, Y.; Kiso, S.; Imai, Y.; Shimomukai, H.; Nomura, Y.; Matsuda, Y.; et al. High levels of transforming growth factor β1 in patients with colorectal cancer: Association with disease progression. Gastroenterology 1996, 110, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.S.; Barrack, E.R. Transforming growth factor-β1 overproduction in prostate cancer: Effects on growth in vivo and in vitro. Mol. Endocrinol. 1992, 6, 15–25. [Google Scholar] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sterling, J.A.; Fan, K.H.; Vessella, R.L.; Shyr, Y.; Hayward, S.W.; Matrisian, L.M.; Bhowmick, N.A. Loss of TGF-β responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol. Cancer Res. 2012, 10, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Transforming growth factor β (TGF-β) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “warburg-like” cancer metabolism and l-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed]
- Elenbaas, B.; Weinberg, R.A. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 2001, 264, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Novitskiy, S.V.; Forrester, E.; Pickup, M.W.; Gorska, A.E.; Chytil, A.; Aakre, M.; Polosukhina, D.; Owens, P.; Yusupova, D.R.; Zhao, Z.; et al. Attenuated transforming growth factor β signaling promotes metastasis in a model of her2 mammary carcinogenesis. Breast Cancer Res. 2014, 16, 425. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Baek, K.E.; Saika, S.; Jeong, M.J.; Yoo, J. Snail is required for transforming growth factor-β-induced epithelial-mesenchymal transition by activating pi3 kinase/akt signal pathway. Biochem. Biophys. Res. Commun. 2007, 353, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Naber, H.P.; Drabsch, Y.; Snaar-Jagalska, B.E.; ten Dijke, P.; van Laar, T. Snail and slug, key regulators of TGF-β-induced emt, are sufficient for the induction of single-cell invasion. Biochem. Biophys. Res. Commun. 2013, 435, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Franci, C.; Gallen, M.; Alameda, F.; Baro, T.; Iglesias, M.; Virtanen, I.; Garcia de Herreros, A. Snail1 protein in the stroma as a new putative prognosis marker for colon tumours. PLoS ONE 2009, 4, e5595. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Liu, X.; Han, Z.; Zhou, L.; Sui, H.; Yan, L.; Jiang, H.; Ren, J.; Cai, J.; Li, Q. Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-β1/Smads signaling pathway mediated snail/e-cadherin expression. BMC Cancer 2015, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, A.; Phadke, D.; Mav, D.; Shah, R.R.; Wade, P.A. The transcription factors snail and slug activate the transforming growth factor-β signaling pathway in breast cancer. PLoS ONE 2011, 6, e26514. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Y.; Zeng, Y.; Lei, Z.; Wang, L.; Yang, H.; Liu, Z.; Zhao, J.; Zhang, H.T. Jak/stat3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int. J. Oncol. 2014, 44, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Kokudo, T.; Suzuki, Y.; Yoshimatsu, Y.; Yamazaki, T.; Watabe, T.; Miyazono, K. Snail is required for TGFβ -induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J. Cell Sci. 2008, 121, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Matise, L.A.; Palmer, T.D.; Ashby, W.J.; Nashabi, A.; Chytil, A.; Aakre, M.; Pickup, M.W.; Gorska, A.E.; Zijlstra, A.; Moses, H.L. Lack of transforming growth factor-β signaling promotes collective cancer cell invasion through tumor-stromal crosstalk. Breast Cancer Res. 2012, 14, R98. [Google Scholar] [CrossRef] [PubMed]
- Orlova, V.V.; Liu, Z.; Goumans, M.J.; ten Dijke, P. Controlling angiogenesis by two unique TGF-β type I receptor signaling pathways. Histol. Histopathol. 2011, 26, 1219–1230. [Google Scholar] [PubMed]
- Bertolino, P.; Deckers, M.; Lebrin, F.; ten Dijke, P. Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest 2005, 128, 585S–590S. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Strand, D.W.; Rowley, D.R. Fibroblast growth factor-2 mediates transforming growth factor-β action in prostate cancer reactive stroma. Oncogene 2008, 27, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Donovan, D.; Harmey, J.H.; Toomey, D.; Osborne, D.H.; Redmond, H.P.; Bouchier-Hayes, D.J. TGF-β-1 regulation of vegf production by breast cancer cells. Ann. Surg. Oncol. 1997, 4, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Lan, H.Y.; Zhu, H.J.; Kang, D.H.; Schreiner, G.F.; Johnson, R.J. Differential regulation of vegf by TGF-β and hypoxia in rat proximal tubular cells. Am. J. Physiol. Renal. Physiol. 2004, 287, F658–F664. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, A.; Okochi, H.; Bradham, D.M.; Grotendorst, G.R. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell 1993, 4, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.N.; Fuchs, E.; Mikula, M.; Huber, H.; Beug, H.; Mikulits, W. Pdgf essentially links TGF-β signaling to nuclear β-catenin accumulation in hepatocellular carcinoma progression. Oncogene 2007, 26, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-β1 (TGF-β1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Chaudhuri, A.; Talmon, G.; Wisecarver, J.L.; Wang, J. TGF-β suppresses VEGFA-mediated angiogenesis in colon cancer metastasis. PLoS ONE 2013, 8, e59918. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Li, J.H.; Garcia, G.; Mu, W.; Piek, E.; Bottinger, E.P.; Chen, Y.; Zhu, H.J.; Kang, D.H.; Schreiner, G.F.; et al. TGF-β induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int. 2004, 66, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type i receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.B.; Canfield, A.E.; Schor, S.L.; Grant, M.E.; Schor, A.M. The response of endothelial cells to TGF β-1 is dependent upon cell shape, proliferative state and the nature of the substratum. J. Cell Sci. 1991, 99, 777–787. [Google Scholar] [PubMed]
- Pavlides, S.; Tsirigos, A.; Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010, 9, 3485–3505. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Howell, A.; Pestell, R.G.; Pavlides, S.; Lisanti, M.P. Caveolin-1 and cancer metabolism in the tumor microenvironment: Markers, models, and mechanisms. Annu. Rev. Pathol. 2012, 7, 423–467. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Power surge: Supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012, 15, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Miyazono, K.; Heldin, C.H.; Keski-Oja, J. Latent transforming growth factor-β1 associates to fibroblast extracellular matrix via latent TGF-β binding protein. J. Cell Biol. 1994, 124, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Lyons, R.M.; Keski-Oja, J.; Moses, H.L. Proteolytic activation of latent transforming growth factor-β from fibroblast-conditioned medium. J. Cell Biol. 1988, 106, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Santibanez, J.F. Transforming growth factor-β and matrix metalloproteinases: Functional interactions in tumor stroma-infiltrating myeloid cells. Sci. World J. 2014, 2014, 521754. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chippada-Venkata, U.D.; Oh, W.K. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers 2014, 6, 1298–1327. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.; Billings, P.C.; Pacifici, M.; Leboy, P.S.; Kirsch, T. Authentic matrix vesicles contain active metalloproteases (MMP). A role for matrix vesicle-associated MMP-13 in activation of transforming growth factor-β. J. Biol. Chem. 2001, 276, 11347–11353. [Google Scholar] [CrossRef] [PubMed]
- Ge, G.; Greenspan, D.S. Bmp1 controls TGFβ1 activation via cleavage of latent TGFβ-binding protein. J. Cell Biol. 2006, 175, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Dean, D.D.; Gomez, R.; Schwartz, Z.; Boyan, B.D. The first stage of transforming growth factor β1 activation is release of the large latent complex from the extracellular matrix of growth plate chondrocytes by matrix vesicle stromelysin-1 (MMP-3). Calcif. Tissue Int. 2002, 70, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin α(v)β8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-β1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes. Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor-β (TGF-β)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-β from bone matrix. J. Biol. Chem. 2002, 277, 21352–21360. [Google Scholar] [CrossRef] [PubMed]
- Tatti, O.; Vehvilainen, P.; Lehti, K.; Keski-Oja, J. Mt1-MMP releases latent TGF-β1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp. Cell Res. 2008, 314, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Dayer, C.; Stamenkovic, I. Recruitment of matrix metalloproteinase-9 (MMP-9) to the fibroblast cell surface by lysyl hydroxylase 3 (LH3) triggers transforming growth factor-β (TGF-β) activation and fibroblast differentiation. J. Biol. Chem. 2015, 290, 13763–13778. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Hinz, B. Integrins and the activation of latent transforming growth factor β1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin αvβ6 binds and activates latent TGF β1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The αvβ1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra279. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased expression of integrin αvβ3 contributes to the establishment of autocrine TGF-β signaling in scleroderma fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Involvement of αvβ5 integrin-mediated activation of latent transforming growth factor β1 in autocrine transforming growth factor β signaling in systemic sclerosis fibroblasts. Arthritis Rheum. 2005, 52, 2897–2905. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, M.; Tramacere, I.; Pomponio, G.; Gabrielli, B.; Avvedimento, E.V.; La Vecchia, C.; Negri, E.; Gabrielli, A. Systemic sclerosis (scleroderma) and cancer risk: Systematic review and meta-analysis of observational studies. Rheumatology 2013, 52, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Trivanovic, D.; Mojsilovic, S.; Santibanez, J.F. Transforming growth factor-β and oxidative stress interplay: Implications in tumorigenesis and cancer progression. Oxid. Med. Cell. Longev. 2015, 2015, 654594. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-β1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [PubMed]
- Ayala, G.; Morello, M.; Frolov, A.; You, S.; Li, R.; Rosati, F.; Bartolucci, G.; Danza, G.; Adam, R.M.; Thompson, T.C.; et al. Loss of caveolin-1 in prostate cancer stroma correlates with reduced relapse-free survival and is functionally relevant to tumour progression. J. Pathol. 2013, 231, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Lee, Y.S.; Han, I.O.; Park, S.H. Expression of caveolin-1 reduces cellular responses to TGF-β 1 through down-regulating the expression of TGF-β type II receptor gene in NIH3T3 fibroblast cells. Biochem. Biophys. Res. Commun. 2007, 359, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Jobling, M.F.; Mott, J.D.; Finnegan, M.T.; Jurukovski, V.; Erickson, A.C.; Walian, P.J.; Taylor, S.E.; Ledbetter, S.; Lawrence, C.M.; Rifkin, D.B.; et al. Isoform-specific activation of latent transforming growth factor β (LTGF-β ) by reactive oxygen species. Radiat. Res. 2006, 166, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E.; Poczatek, M. Activation of latent TGF-β by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 2000, 11, 59–69. [Google Scholar] [CrossRef]
- Schultz-Cherry, S.; Ribeiro, S.; Gentry, L.; Murphy-Ullrich, J.E. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-β in a chemically defined system. J. Biol. Chem. 1994, 269, 26775–26782. [Google Scholar] [PubMed]
- Ribeiro, S.M.; Poczatek, M.; Schultz-Cherry, S.; Villain, M.; Murphy-Ullrich, J.E. The activation sequence of thrombospondin-1 interacts with the latency-associated peptide to regulate activation of latent transforming growth factor-β. J. Biol. Chem. 1999, 274, 13586–13593. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef]
- Yehualaeshet, T.; O’Connor, R.; Green-Johnson, J.; Mai, S.; Silverstein, R.; Murphy-Ullrich, J.E.; Khalil, N. Activation of rat alveolar macrophage-derived latent transforming growth factor β-1 by plasmin requires interaction with thrombospondin-1 and its cell surface receptor, CD36. Am. J. Pathol. 1999, 155, 841–851. [Google Scholar] [CrossRef]
- Miyanaga, K.; Kato, Y.; Nakamura, T.; Matsumura, M.; Amaya, H.; Horiuchi, T.; Chiba, Y.; Tanaka, K. Expression and role of thrombospondin-1 in colorectal cancer. Anticancer Res. 2002, 22, 3941–3948. [Google Scholar] [PubMed]
- Kawataki, T.; Naganuma, H.; Sasaki, A.; Yoshikawa, H.; Tasaka, K.; Nukui, H. Correlation of thrombospondin-1 and transforming growth factor-β expression with malignancy of glioma. Neuropathology 2000, 20, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Nistala, H.; Lee-Arteaga, S.; Smaldone, S.; Siciliano, G.; Carta, L.; Ono, R.N.; Sengle, G.; Arteaga-Solis, E.; Levasseur, R.; Ducy, P.; et al. Fibrillin-1 and -2 differentially modulate endogenous TGF-β and BMP bioavailability during bone formation. J. Cell Biol. 2010, 190, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.N.; Sengle, G.; Charbonneau, N.L.; Carlberg, V.; Bachinger, H.P.; Sasaki, T.; Lee-Arteaga, S.; Zilberberg, L.; Rifkin, D.B.; Ramirez, F.; et al. Latent transforming growth factor β-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J. Biol. Chem. 2009, 284, 16872–16881. [Google Scholar] [CrossRef] [PubMed]
- Sengle, G.; Ono, R.N.; Lyons, K.M.; Bachinger, H.P.; Sakai, L.Y. A new model for growth factor activation: Type II receptors compete with the prodomain for bmp-7. J. Mol. Biol. 2008, 381, 1025–1039. [Google Scholar] [CrossRef] [PubMed]
- Radice, P.D.; Mathieu, P.; Leal, M.C.; Farias, M.I.; Ferrari, C.; Puntel, M.; Salibe, M.; Chernomoretz, A.; Pitossi, F.J. Fibulin-2 is a key mediator of the pro-neurogenic effect of TGF-β1 on adult neural stem cells. Mol. Cell. Neurosci. 2015, 67, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Rifkin, D.B. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: Activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J. Cell. Biol. 1989, 109, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Blakytny, R.; Ludlow, A.; Martin, G.E.; Ireland, G.; Lund, L.R.; Ferguson, M.W.; Brunner, G. Latent TGF-β 1 activation by platelets. J. Cell Physiol. 2004, 199, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Abe, M.; Sato, Y.; Miyazono, K.; Harpel, J.; Heldin, C.H.; Rifkin, D.B. Role of the latent TGF-β binding protein in the activation of latent TGF-β by co-cultures of endothelial and smooth muscle cells. J. Cell. Biol. 1993, 120, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Antonelli-Orlidge, A.; Saunders, K.B.; Smith, S.R.; D’Amore, P.A. An activated form of transforming growth factor β is produced by cocultures of endothelial cells and pericytes. Proc. Natl. Acad. Sci. USA 1989, 86, 4544–4548. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [PubMed]
- Kalamajski, S.; Oldberg, A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010, 29, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Grant, D.S.; Yenisey, C.; Rose, R.W.; Tootell, M.; Santra, M.; Iozzo, R.V. Decorin suppresses tumor cell-mediated angiogenesis. Oncogene 2002, 21, 4765–4777. [Google Scholar] [CrossRef] [PubMed]
- Troup, S.; Njue, C.; Kliewer, E.V.; Parisien, M.; Roskelley, C.; Chakravarti, S.; Roughley, P.J.; Murphy, L.C.; Watson, P.H. Reduced expression of the small leucine-rich proteoglycans, lumican, and decorin is associated with poor outcome in node-negative invasive breast cancer. Clin. Cancer Res. 2003, 9, 207–214. [Google Scholar] [PubMed]
- Baghy, K.; Dezso, K.; Laszlo, V.; Fullar, A.; Peterfia, B.; Paku, S.; Nagy, P.; Schaff, Z.; Iozzo, R.V.; Kovalszky, I. Ablation of the decorin gene enhances experimental hepatic fibrosis and impairs hepatic healing in mice. Lab. Investig. 2011, 91, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, N.; Wicks, S.J.; Mason, R.M.; Chantry, A. Decorin suppresses transforming growth factor-β-induced expression of plasminogen activator inhibitor-1 in human mesangial cells through a mechanism that involves Ca2+-dependent phosphorylation of Smad2 at serine-240. Biochem. J. 2002, 362, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Stander, M.; Naumann, U.; Dumitrescu, L.; Heneka, M.; Loschmann, P.; Gulbins, E.; Dichgans, J.; Weller, M. Decorin gene transfer-mediated suppression of TGF-β synthesis abrogates experimental malignant glioma growth in vivo. Gene. Ther. 1998, 5, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Biglari, A.; Bataille, D.; Naumann, U.; Weller, M.; Zirger, J.; Castro, M.G.; Lowenstein, P.R. Effects of ectopic decorin in modulating intracranial glioma progression in vivo, in a rat syngeneic model. Cancer Gene Ther. 2004, 11, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Merline, R.; Moreth, K.; Beckmann, J.; Nastase, M.V.; Zeng-Brouwers, J.; Tralhao, J.G.; Lemarchand, P.; Pfeilschifter, J.; Schaefer, R.M.; Iozzo, R.V.; et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through pdcd4 and microrna-21. Sci. Signal. 2011, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, A.; Romaris, M.; Rasmussen, L.M.; Heinegard, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor β. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Mochida, Y.; Parisuthiman, D.; Yamauchi, M. Biglycan is a positive modulator of BMP-2 induced osteoblast differentiation. Adv. Exp. Med. Biol. 2006, 585, 101–113. [Google Scholar] [PubMed]
- Kolb, M.; Margetts, P.J.; Sime, P.J.; Gauldie, J. Proteoglycans decorin and biglycan differentially modulate TGF-β -mediated fibrotic responses in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1327–L1334. [Google Scholar] [PubMed]
- Westergren-Thorsson, G.; Antonsson, P.; Malmstrom, A.; Heinegard, D.; Oldberg, A. The synthesis of a family of structurally related proteoglycans in fibroblasts is differently regulated by TFG-β. Matrix 1991, 11, 177–183. [Google Scholar] [CrossRef]
- Schmidt, M.; Bohm, D.; von Torne, C.; Steiner, E.; Puhl, A.; Pilch, H.; Lehr, H.A.; Hengstler, J.G.; Kolbl, H.; Gehrmann, M. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008, 68, 5405–5413. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Piette, F.; Loi, S.; Wang, Y.; Lallemand, F.; Haibe-Kains, B.; Viale, G.; Delorenzi, M.; Zhang, Y.; d’Assignies, M.S.; et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the transbig multicenter independent validation series. Clin. Cancer Res. 2007, 13, 3207–3214. [Google Scholar] [CrossRef] [PubMed]
- LaTulippe, E.; Satagopan, J.; Smith, A.; Scher, H.; Scardino, P.; Reuter, V.; Gerald, W.L. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002, 62, 4499–4506. [Google Scholar] [PubMed]
- Lee, Y.H.; Schiemann, W.P. Fibromodulin suppresses nuclear factor-kappab activity by inducing the delayed degradation of ikba via a jnk-dependent pathway coupled to fibroblast apoptosis. J. Biol. Chem. 2011, 286, 6414–6422. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, D.; Butzow, R.; Hildebrand, A.; Ruoslahti, E. Localization of transforming growth factor β binding site in betaglycan. Comparison with small extracellular matrix proteoglycans. J. Biol. Chem. 1993, 268, 22710–22715. [Google Scholar] [PubMed]
- Zheng, Z.; Nguyen, C.; Zhang, X.; Khorasani, H.; Wang, J.Z.; Zara, J.N.; Chu, F.; Yin, W.; Pang, S.; Le, A.; et al. Delayed wound closure in fibromodulin-deficient mice is associated with increased TGF-β 3 signaling. J. Investig. Dermatol. 2011, 131, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Adini, I.; Ghosh, K.; Adini, A.; Chi, Z.L.; Yoshimura, T.; Benny, O.; Connor, K.M.; Rogers, M.S.; Bazinet, L.; Birsner, A.E.; et al. Melanocyte-secreted fibromodulin promotes an angiogenic microenvironment. J. Clin. Investig. 2014, 124, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Maris, P.; Blomme, A.; Palacios, A.P.; Costanza, B.; Bellahcene, A.; Bianchi, E.; Gofflot, S.; Drion, P.; Trombino, G.E.; Di Valentin, E.; et al. Asporin is a fibroblast-derived TGF-β1 inhibitor and a tumor suppressor associated with good prognosis in breast cancer. PLoS Med. 2015, 12, e1001871. [Google Scholar] [CrossRef] [PubMed]
- Satoyoshi, R.; Kuriyama, S.; Aiba, N.; Yashiro, M.; Tanaka, M. Asporin activates coordinated invasion of scirrhous gastric cancer and cancer-associated fibroblasts. Oncogene 2015, 34, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Kou, I.; Nakajima, M.; Ikegawa, S. Binding characteristics of the osteoarthritis-associated protein asporin. J. Bone Miner. Metab 2010, 28, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Cho, K.; Kawahara, K.; Yamamoto, T.; Fujiwara, Y.; Uchida, E.; Tajiri, T.; Naito, Z. Role of lumican in cancer cells and adjacent stromal tissues in human pancreatic cancer. Oncol. Rep. 2007, 18, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E.; Snell, L.; Dotzlaw, H.; Hole, K.; Hiller-Hitchcock, T.; Roughley, P.J.; Watson, P.H.; Murphy, L.C. Expression of lumican in human breast carcinoma. Cancer Res. 1998, 58, 1348–1352. [Google Scholar] [PubMed]
- Nikitovic, D.; Berdiaki, A.; Zafiropoulos, A.; Katonis, P.; Tsatsakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican expression is positively correlated with the differentiation and negatively with the growth of human osteosarcoma cells. FEBS J. 2008, 275, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Kao, W.W. Ocular surface tissue morphogenesis in normal and disease states revealed by genetically modified mice. Cornea 2006, 25, S7–S19. [Google Scholar] [CrossRef] [PubMed]
- Mochida, Y.; Parisuthiman, D.; Kaku, M.; Hanai, J.; Sukhatme, V.P.; Yamauchi, M. Nephrocan, a novel member of the small leucine-rich repeat protein family, is an inhibitor of transforming growth factor-beta signaling. J. Biol. Chem. 2006, 281, 36044–36051. [Google Scholar] [CrossRef] [PubMed]
- Kielty, C.M.; Wess, T.J.; Haston, L.; Ashworth, J.L.; Sherratt, M.J.; Shuttleworth, C.A. Fibrillin-rich microfibrils: Elastic biopolymers of the extracellular matrix. J. Muscle Res. Cell Motil. 2002, 23, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, V.; Warburton, D. Fibrillin controls TGF-β activation. Nat. Genet. 2003, 33, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Lind, G.E.; Danielsen, S.A.; Ahlquist, T.; Merok, M.A.; Andresen, K.; Skotheim, R.I.; Hektoen, M.; Rognum, T.O.; Meling, G.I.; Hoff, G.; et al. Identification of an epigenetic biomarker panel with high sensitivity and specificity for colorectal cancer and adenomas. Mol. Cancer 2011, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Song, Y.; Zhang, H.; Wu, X.; Xia, P.; Dang, C. Detection of hypermethylated fibrillin-1 in the stool samples of colorectal cancer patients. Med. Oncol. 2013, 30, 695. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Melotte, V.; Vire, E.; Langenkamp, E.; Molema, G.; Fuks, F.; Herman, J.G.; Van Criekinge, W.; Griffioen, A.W.; van Engeland, M. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 2007, 67, 4138–4148. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Lee, S.Y.; Woo, M.K.; Choi, S.I.; Kim, T.R.; Kim, M.J.; Kim, K.C.; Cho, E.W.; Kim, I.G. Fibulin-3 promoter methylation alters the invasive behavior of non-small cell lung cancer cell lines via MMP-7 and MMP-2 regulation. Int. J. Oncol. 2012, 40, 402–408. [Google Scholar] [PubMed]
- Wang, Z.; Yuan, X.; Jiao, N.; Zhu, H.; Zhang, Y.; Tong, J. Cdh13 and flbn3 gene methylation are associated with poor prognosis in colorectal cancer. Pathol. Oncol. Res. 2012, 18, 263–270. [Google Scholar] [PubMed]
- Tian, H.; Liu, J.; Chen, J.; Gatza, M.L.; Blobe, G.C. Fibulin-3 is a novel TGF-β pathway inhibitor in the breast cancer microenvironment. Oncogene 2015, 34, 5635–5647. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.D.; Jiao, N.L.; Wang, Y.X.; Zhang, Y.W.; Han, F. Downregulation of fibulin-3 gene by promoter methylation in colorectal cancer predicts adverse prognosis. Neoplasma 2011, 58, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.W.; Chen, L.B. Aberrant promoter methylation of fbln-3 gene and clinicopathological significance in non-small cell lung carcinoma. Lung Cancer 2010, 69, 239–244. [Google Scholar] [PubMed]
- Nomoto, S.; Kanda, M.; Okamura, Y.; Nishikawa, Y.; Li, Q.; Fujii, T.; Sugimoto, H.; Takeda, S.; Nakao, A. Epidermal growth factor-containing fibulin-like extracellular matrix protein 1, efemp1, a novel tumor-suppressor gene detected in hepatocellular carcinoma using double combination array analysis. Ann. Surg. Oncol. 2010, 17, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Renard, M.; Holm, T.; Veith, R.; Callewaert, B.L.; Ades, L.C.; Baspinar, O.; Pickart, A.; Dasouki, M.; Hoyer, J.; Rauch, A.; et al. Altered TGFβ signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type i caused by fibulin-4 deficiency. Eur J. Hum. Genet. 2010, 18, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Davis, E.C.; Chapman, S.L.; Budatha, M.; Marmorstein, L.Y.; Word, R.A.; Yanagisawa, H. Fibulin-4 deficiency results in ascending aortic aneurysms: A potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ. Res. 2010, 106, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.J.; Sage, H.; Briscoe, G.; Erickson, H.P. The compact conformation of fibronectin is determined by intramolecular ionic interactions. J. Biol. Chem. 1999, 274, 15473–15479. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Engler, A.J.; Slone, R.D.; Galante, L.L.; Schwarzbauer, J.E. Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 2008, 68, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Sivakumar, P.; Jones, C.J.; Chen, Q.; Peters, D.M.; Mosher, D.F.; Humphries, M.J.; Kielty, C.M. Fibronectin regulates latent transforming growth factor-β (TGFβ) by controlling matrix assembly of latent TGFβ-binding protein-1. J. Biol. Chem. 2005, 280, 18871–18880. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [PubMed]
- Collo, G.; Pepper, M.S. Endothelial cell integrin α5β1 expression is modulated by cytokines and during migration in vitro. J. Cell Sci. 1999, 112, 569–578. [Google Scholar] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Saunier, E.F.; Akhurst, R.J. TGFβ inhibition for cancer therapy. Curr. Cancer Drug Targets 2006, 6, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J. The TGFβ pathway as a therapeutic target in cancer. Clin. Transl. Oncol. 2008, 10, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Tralhao, J.G.; Schaefer, L.; Micegova, M.; Evaristo, C.; Schonherr, E.; Kayal, S.; Veiga-Fernandes, H.; Danel, C.; Iozzo, R.V.; Kresse, H.; et al. In vivo selective and distant killing of cancer cells using adenovirus-mediated decorin gene transfer. FASEB J. 2003, 17, 464–466. [Google Scholar] [PubMed]
- Rasmussen, H.S.; McCann, P.P. Matrix metalloproteinase inhibition as a novel anticancer strategy: A review with special focus on batimastat and marimastat. Pharmacol. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Rosemurgy, A.; Brown, P.D.; Bowry, C.; Buckels, J.A. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: A randomized trial. J. Clin. Oncol. 2001, 19, 3447–3455. [Google Scholar] [PubMed]
- Levitt, N.C.; Eskens, F.A.; O’Byrne, K.J.; Propper, D.J.; Denis, L.J.; Owen, S.J.; Choi, L.; Foekens, J.A.; Wilner, S.; Wood, J.M.; et al. Phase i and pharmacological study of the oral matrix metalloproteinase inhibitor, MMI270 (CGS27023A), in patients with advanced solid cancer. Clin. Cancer Res. 2001, 7, 1912–1922. [Google Scholar] [PubMed]
- Bissett, D.; O’Byrne, K.J.; von Pawel, J.; Gatzemeier, U.; Price, A.; Nicolson, M.; Mercier, R.; Mazabel, E.; Penning, C.; Zhang, M.H.; et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Hirte, H.; Vergote, I.B.; Jeffrey, J.R.; Grimshaw, R.N.; Coppieters, S.; Schwartz, B.; Tu, D.; Sadura, A.; Brundage, M.; Seymour, L. A phase III randomized trial of bay 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: A national cancer institute of canada clinical trials group study. Gynecol. Oncol. 2006, 102, 300–308. [Google Scholar] [PubMed]
- Leighl, N.B.; Paz-Ares, L.; Douillard, J.Y.; Peschel, C.; Arnold, A.; Depierre, A.; Santoro, A.; Betticher, D.C.; Gatzemeier, U.; Jassem, J.; et al. Randomized phase iii study of matrix metalloproteinase inhibitor bms-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute Of Canada-Clinical Trials Group Study BR.18. J. Clin. Oncol. 2005, 23, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.; Takimoto, C.; Hidalgo, M.; Rizzo, J.; Kuhn, J.G.; Hammond, L.A.; Schwartz, G.; Tolcher, A.; Patnaik, A.; Eckhardt, S.G.; et al. A phase i and pharmacokinetic study of Col-3 (Metastat), an oral tetracycline derivative with potent matrix metalloproteinase and antitumor properties. Clin. Cancer Res. 2004, 10, 6512–6521. [Google Scholar] [CrossRef] [PubMed]
- Duivenvoorden, W.C.; Popovic, S.V.; Lhotak, S.; Seidlitz, E.; Hirte, H.W.; Tozer, R.G.; Singh, G. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Cancer Res. 2002, 62, 1588–1591. [Google Scholar] [PubMed]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin αvβ3, ± dacarbazine in patients with stage IV metastatic melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, A.; Rawal, S.K.; Szkarlat, K.; Bogdanova, N.; Dirix, L.; Stenzl, A.; Welslau, M.; Wang, G.; Dawkins, F.; de Boer, C.J.; et al. A randomized, double-blind, multicenter, phase 2 study of a human monoclonal antibody to human alphanu integrins (intetumumab) in combination with docetaxel and prednisone for the first-line treatment of patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2013, 24, 329–336. [Google Scholar] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated mgmt promoter (centric eortc 26071–22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet. Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, pharmacokinetic, and pharmacodynamic phase i dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiong, W.; Baca-Regen, L.; Nagase, H.; Baxter, B.T. Mechanism of inhibition of matrix metalloproteinase-2 expression by doxycycline in human aortic smooth muscle cells. J. Vasc. Surg. 2003, 38, 1376–1383. [Google Scholar] [CrossRef]
- Son, K.; Fujioka, S.; Iida, T.; Furukawa, K.; Fujita, T.; Yamada, H.; Chiao, P.J.; Yanaga, K. Doxycycline induces apoptosis in panc-1 pancreatic cancer cells. Anticancer Res. 2009, 29, 3995–4003. [Google Scholar] [PubMed]
- Lamb, R.; Ozsvari, B.; Lisanti, C.L.; Tanowitz, H.B.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget 2015, 6, 4569–4584. [Google Scholar] [CrossRef] [PubMed]
- Delbaldo, C.; Raymond, E.; Vera, K.; Hammershaimb, L.; Kaucic, K.; Lozahic, S.; Marty, M.; Faivre, S. Phase i and pharmacokinetic study of etaracizumab (abegrin), a humanized monoclonal antibody against αvβ3 integrin receptor, in patients with advanced solid tumors. Investig. New Drugs 2008, 26, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Salvo, E.; Garasa, S.; Dotor, J.; Morales, X.; Pelaez, R.; Altevogt, P.; Rouzaut, A. Combined targeting of TGF-β1 and integrin β3 impairs lymph node metastasis in a mouse model of non-small-cell lung cancer. Mol. Cancer 2014, 13, 112. [Google Scholar] [CrossRef] [PubMed]
- O’Day, S.; Pavlick, A.; Loquai, C.; Lawson, D.; Gutzmer, R.; Richards, J.; Schadendorf, D.; Thompson, J.A.; Gonzalez, R.; Trefzer, U.; et al. A randomised, phase ii study of intetumumab, an anti-alphav-integrin mab, alone and with dacarbazine in stage iv melanoma. Br. J. Cancer 2011, 105, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; O’Neill, A.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. 2008, 26, 5610–5617. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.H.; Lafrenie, R.M.; Miyamoto, S.; Yamada, K.M. Integrin signaling: Cytoskeletal complexes, MAP kinase activation, and regulation of gene expression. Cell Adhes. Commun. 1998, 6, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, M.; Zardi, L.; Neri, D. Fibronectin as target for tumor therapy. Int. J. Cancer 2006, 118, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Santimaria, M.; Moscatelli, G.; Viale, G.L.; Giovannoni, L.; Neri, G.; Viti, F.; Leprini, A.; Borsi, L.; Castellani, P.; Zardi, L.; et al. Immunoscintigraphic detection of the ED-B domain of fibronectin, a marker of angiogenesis, in patients with cancer. Clin. Cancer Res. 2003, 9, 571–579. [Google Scholar] [PubMed]
- Fabbrini, M.; Trachsel, E.; Soldani, P.; Bindi, S.; Alessi, P.; Bracci, L.; Kosmehl, H.; Zardi, L.; Neri, D.; Neri, P. Selective occlusion of tumor blood vessels by targeted delivery of an antibody-photosensitizer conjugate. Int. J. Cancer 2006, 118, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Carnemolla, B.; Borsi, L.; Balza, E.; Castellani, P.; Meazza, R.; Berndt, A.; Ferrini, S.; Kosmehl, H.; Neri, D.; Zardi, L. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood 2002, 99, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Borsi, L.; Balza, E.; Carnemolla, B.; Sassi, F.; Castellani, P.; Berndt, A.; Kosmehl, H.; Biro, A.; Siri, A.; Orecchia, P.; et al. Selective targeted delivery of TNFα to tumor blood vessels. Blood 2003, 102, 4384–4392. [Google Scholar] [CrossRef] [PubMed]
- Danielli, R.; Patuzzo, R.; Di Giacomo, A.M.; Gallino, G.; Maurichi, A.; Di Florio, A.; Cutaia, O.; Lazzeri, A.; Fazio, C.; Miracco, C.; et al. Intralesional administration of L19-IL2/L19-TNF in stage III or stage IVM1a melanoma patients: Results of a phase II study. Cancer Immunol. Immunother. 2015, 64, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kokura, S.; Enoki, T.; Sakamoto, N.; Okayama, T.; Ideno, M.; Mineno, J.; Uno, K.; Yoshida, N.; Kamada, K.; et al. Phase I clinical trial of fibronectin CH296-stimulated T cell therapy in patients with advanced cancer. PLoS ONE 2014, 9, e83786. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Class | Target | Developmental Stage |
|---|---|---|---|
| Matrix Metalloproteinases | |||
| Batimastat [162] | Peptidomimetic | Broad spectrum | Cancelled in phase III |
| Marimastat (BB-2516) [163] | Peptidomimetic | Broad spectrum | Cancelled in phase III |
| CGS-27023A (MMI270) [164] | Small molecule | MMP-2, 8, 9 | Cancelled in phase I |
| Prinomastat (AG3340) [165] | Small molecule | MMP-2, 3, 9, 14 | Cancelled in phase III |
| Tanomastat (BAY-129566) [166] | Small molecule | MMP-2, 3, 9, 13 | Cancelled in phase III |
| BMS-275291 (D2163) [167] | Peptidomimetic | MMP-1, 2, 9 | Cancelled in phase III |
| Metastat (COL-3) [168] | Tetracycline derivative | Broad spectrum | Phase I/II |
| Doxycycline [169] | Tetracycline derivative | N/A | Approved/ongoing |
| Integrins | |||
| Vitaxin/etaracizumab [170] | Monoclonal antibody | β7 | Cancelled in phase II |
| Intetumumab (CTNO 95) [171] | Monoclonal antibody | αvβ3, αvβ5 | Phase I/II |
| Cilengitide [172] | Cyclic RGD peptide | αvβ3, αvβ5 | Cancelled in phase III |
| PF-00562271 (VS-6062) [173] | Small molecule | FAK | Phase I |
| GSK2256098 (NCT00996671) | Small molecule | FAK | Phase I |
| VS-4718 (PND-1186) [174] | Small molecule | N/A | Phase II |
| PF-04554878 (VS-6063) (NCT01951690) | Small molecule | FAK, PYK2 | Phase II |
| Fibronectin | |||
| L-19 (NCT02076633) | Monoclonal antibody | ED-B domain of fibronectin | Phase II |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costanza, B.; Umelo, I.A.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal Modulators of TGF-β in Cancer. J. Clin. Med. 2017, 6, 7. https://doi.org/10.3390/jcm6010007
Costanza B, Umelo IA, Bellier J, Castronovo V, Turtoi A. Stromal Modulators of TGF-β in Cancer. Journal of Clinical Medicine. 2017; 6(1):7. https://doi.org/10.3390/jcm6010007
Chicago/Turabian StyleCostanza, Brunella, Ijeoma Adaku Umelo, Justine Bellier, Vincent Castronovo, and Andrei Turtoi. 2017. "Stromal Modulators of TGF-β in Cancer" Journal of Clinical Medicine 6, no. 1: 7. https://doi.org/10.3390/jcm6010007
APA StyleCostanza, B., Umelo, I. A., Bellier, J., Castronovo, V., & Turtoi, A. (2017). Stromal Modulators of TGF-β in Cancer. Journal of Clinical Medicine, 6(1), 7. https://doi.org/10.3390/jcm6010007