
Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases

Abstract
:1. Human Fibrotic Disorders
2. Myofibroblasts: The Effector Cells Responsible for Tissue Fibrosis
3. Pathways Involved in Tissue Fibrosis Participate in the Molecular Mechanisms of EndoMT
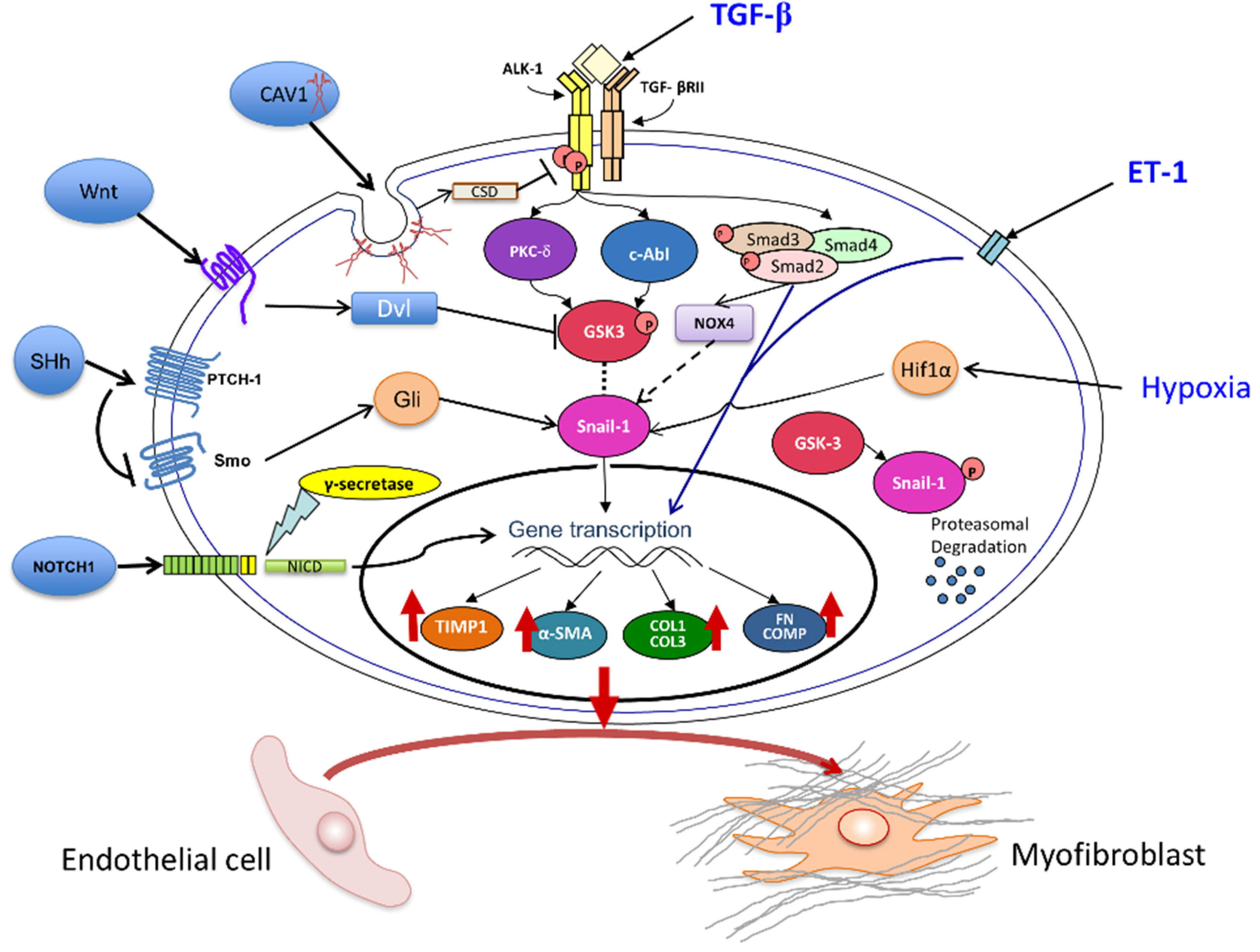
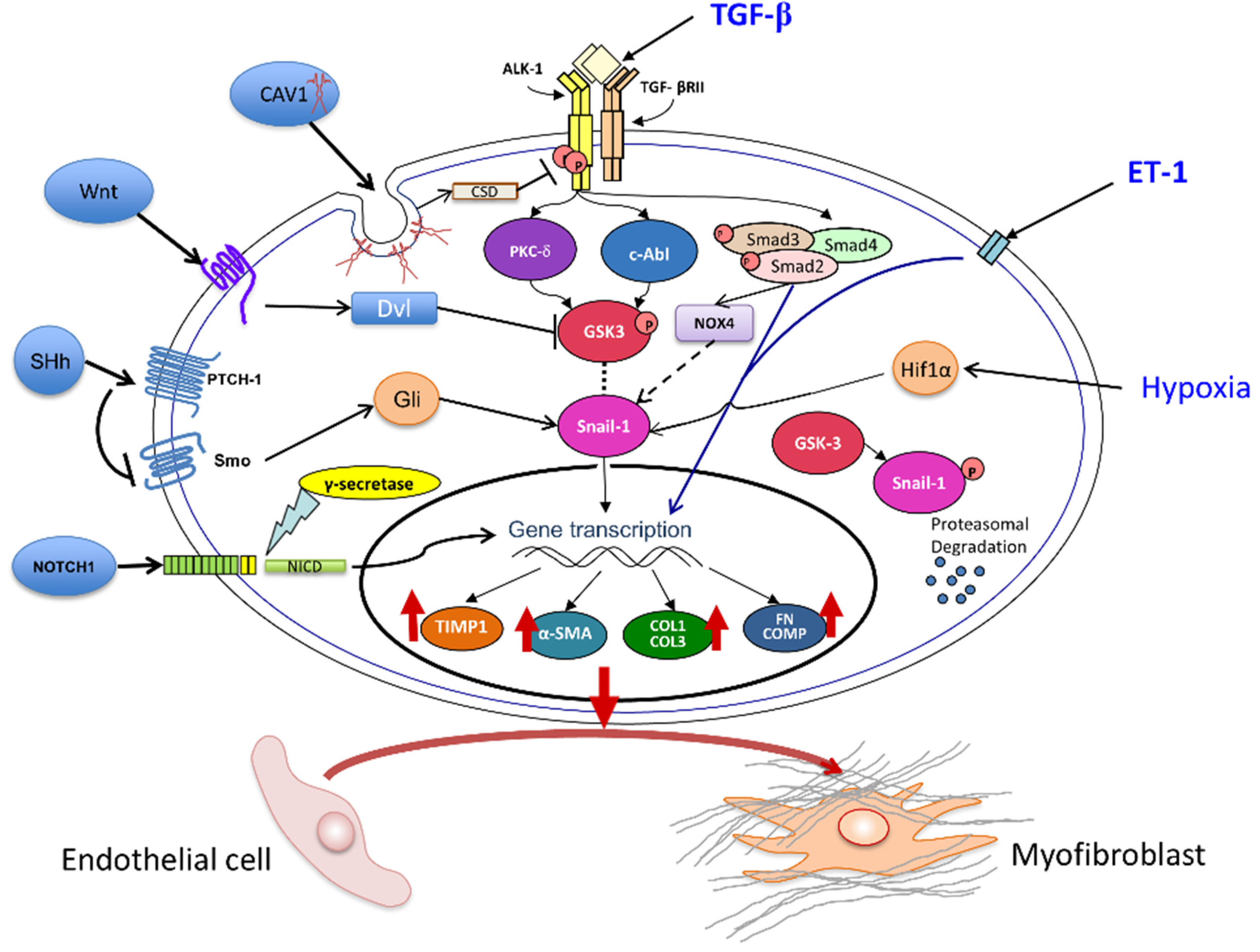
3.1. The Transforming Growth Factor-β (TGF-β) Pathway
3.2. Role of Notch and Hedgehog Signaling Pathways
3.3. Wnt Pathway Effects on EndoMT
3.4. Caveolin-1 (CAV1) Modulation of EndoMT
3.5. The Role of Endothelin-1 (ET-1) in EndoMT
3.6. Involvement of Hypoxia in EndoMT
3.7. Role of NOX4 and Oxidative Stress in EndoMT
3.8. Role of MicroRNA in EndoMT
4. EndoMT in Animal Models of Tissue Fibrosis
4.1. Cardiac Fibrosis
4.2. Renal Fibrosis
4.3. Pulmonary Fibrosis and Pulmonary Arterial Hypertension
5. In Vitro Studies of EndoMT in Human Endothelial Cells
6. EndoMT in Human Fibrotic Diseases
6.1 Idiopathic Pulmonary Fibrosis and Pulmonary Arterial Hypertension
6.2. Systemic Sclerosis
6.3. Radiation Induced Pulmonary Fibrosis
6.4. Cardiac and Renal Fibrosis
6.5. Portal Vein Fibrosis and Other Fibrotic Disorders
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [PubMed]
- Rosenbloom, J.; Castro, S.V.; Jimenez, S.A. Fibrotic Diseases: Cellular and Molecular Mechanisms and Novel Therapies. Ann. Int. Med. 2010, 152, 159–166. [Google Scholar] [PubMed]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Abraham, D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J. Clin. Invest. 2007, 117, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis—A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Cowper, S.E.; Su, L.D.; Bhawan, J.; Robin, H.S.; LeBoit, P.E. Nephrogenic fibrosing dermopathy. Am. J. Dermatopathol. 2001, 23, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.A.; Artlett, C.M.; Sandorfi, N.; Latinis, K.; Piera-Velazquez, S.; Jimenez, S.A. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin. Arthritis Rheum. 2006, 35, 238–249. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Creamer, D.; Du Vivier, A.W.; Pagliuca, A.; Ho, A.Y.; Devereux, S.; Salisbury, J.R.; Mufti, G.J. Sclerodermatous graft-versus-host disease: Clinical spectrum and therapeutic challenges. Br. J. Dermatol. 2007, 156, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-related disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef]
- Ding, N.H.; Li, J.J.; Sun, L.Q. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr. Drug Targets 2013, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.H.; Malhotra, V.; Perry, M.C. Radiation-induced and chemotherapy-induced pulmonary injury. Curr. Opin. Oncol. 2001, 13, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Getting to the heart of the matter: New insights into cardiac fibrosis. Circ. Res. 2015, 116, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Sverzellati, N.; Rossi, G.; Cavazza, A.; Tzouvelekis, A.; Crestani, B.; Vancheri, C. Idiopathic pulmonary fibrosis: An update. Ann. Med. 2015, 47, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Bonder, A.; Tapper, E.B.; Afdhal, N.H. Contemporary assessment of hepatic fibrosis. Clin. Liver Dis. 2015, 19, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Invest. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Rimal, B.; Greenberg, A.K.; Rom, W.N. Basic pathogenetic mechanisms in silicosis: Current understanding. Curr. Opin. Pulm. Med. 2005, 11, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Manning, C.B.; Vallyathan, V.; Mossman, B.T. Diseases caused by asbestos: Mechanisms of injury and disease development. Int. Immunopharmacol. 2002, 2, 191–200. [Google Scholar] [CrossRef]
- Gökmen, M.R.; Cosyns, J.P.; Arlt, V.M.; Stiborová, M.; Phillips, D.H.; Schmeiser, H.H.; Simmonds, M.S.; Cook, H.T.; Vanherweghem, J.L.; Nortier, J.L.; et al. The epidemiology, diagnosis, and management of aristolochic acid nephropathy: A narrative review. Ann. Intern. Med. 2013, 158, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Colley, D.G.; Bustinduy, A.L.; Secor, W.E.; King, C.H. Human schistosomiasis. Lancet 2014, 383, 2253–2264. [Google Scholar] [CrossRef]
- Cunha-Neto, E.; Chevillard, C. Chagas disease cardiomyopathy: Immunopathology and genetics. Mediat. Inflamm. 2014, 2014, 683230. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, S.A.; Hitraya, E.; Varga, J. Pathogenesis of scleroderma. Collagen. Rheum. Dis. Clin. N. Am. 1996, 22, 647–674. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.; Nielsen, M.J.; Sand, J.M.; Hansen, N.; Bay-Jensen, A.C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G807–G830. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Henke, C.A.; Horowitz, J.C.; Noble, P.W.; Roman, J.; Sime, P.J.; Zhou, Y.; Wells, R.G.; White, E.S.; Tschumperlin, D.J. Matrix biology of idiopathic pulmonary fibrosis: A workshop report of the National Heart, Lung, and Blood Institute. Am. J. Pathol. 2014, 184, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Kirk, T.Z.; Mark, M.E.; Chua, C.C.; Chua, B.H.; Mayes, M.D. Myofibroblasts from scleroderma skin synthesize elevated levels of collagen and tissue inhibitor of metalloproteinase (TIMP-1) with two forms of TIMP-1. J. Biol. Chem. 1995, 270, 3423–3428. [Google Scholar] [PubMed]
- Rosenbloom, J.; Mendoza, F.A.; Jimenez, S.A. Strategies for Antifibrotic Therapies. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Castelino, F.V.; Varga, J. Emerging cellular and molecular targets in fibrosis: Implications for scleroderma pathogenesis and targeted therapy. Curr. Opin. Rheumatol. 2014, 26, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P. Systemic sclerosis: From pathogenesis to targeted therapy. Clin. Exp. Rheumatol. 2015, 33 (Suppl. 92), S3–S7. [Google Scholar] [PubMed]
- Gabbiani, G. The myofibroblast: A key cell for wound healing and fibrocontractive diseases. Prog. Clin. Biol. Res. 1981, 54, 183–194. [Google Scholar] [PubMed]
- Abraham, D.J.; Eckes, B.; Rajkumar, V.; Krieg, T. New developments in fibroblast and myofibroblast biology: Implications for fibrosis and scleroderma. Curr. Rheumatol. Rep. 2007, 9, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Gilbane, A.J.; Denton, C.P.; Holmes, A.M. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Arthritis Res. Ther. 2013, 15, 215. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Phan, S.H. Myofibroblasts. Curr. Opin. Rheumatol. 2013, 25, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Discher, D.E. Matrix elasticity, cytoskeletal tension, and TGF-beta: The insoluble and soluble meet. Sci. Signal 2008, 1. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: Implications for the pathogenesis and treatment of fibrosis. Curr. Rheumatol. 2009, 11, 120–126. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell. Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 2011, 23. [Google Scholar] [CrossRef] [PubMed]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Poslethwaite, A.E.; Shigemitsu, H.; Kanagat, S. Cellular origins of fibroblasts: Possible implications for organ fibrosis in systemic sclerosis. Curr. Opin. Rheumatol. 2004, 16, 733–738. [Google Scholar] [CrossRef]
- Strieter, R.M.; Keeley, E.C.; Hughes, M.A.; Burdick, M.D.; Mehrad, B. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of pulmonary fibrosis. J. Leukoc. Biol. 2009, 86, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.L.; Bucala, R. Fibrocytes in health and disease. Exp. Hematol. 2010, 38, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin on myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Karasek, M.A. Does transformation of microvascular endothelial cells into myofibroblasts play a key role in the etiology and pathology of fibrotic disease? Med. Hypotheses 2007, 68, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Lu, M.M.; Jiang, Y.Q.; Epstein, J.A.; Gruber, P.J. Molecular markers of cardiac endocardial cushion development. Dev. Dyn. 2003, 228, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Neves, C.Y.; Carrillo, L.M.; Zambrano, E.A.; Ramírez, R. Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium 2005, 12, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Taranavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; van Zonneveld, A.J.; ten Dijke, P. Transforming growth factor β-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bertram, J.F. Review: Endothelial-Myofibroblast Transition, a new player in diabetic renal fibrosis. Nephrology 2010, 15, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of Endothelial-Mesenchymal Transition (EndoMT) in the Pathogenesis of Fibrotic Disorders. Am. J. Pathol. 2011, 179, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Wang, N.; Zhang, T.C. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB. Life 2012, 64, 717–723. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2012, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Kalluri, R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Cell. Physiol. 2013, 304, C216–C225. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Kessler, S.P.; West, G.A.; Bhilocha, S.; de la Motte, C.; Sadler, T.M.; Gopalan, B.; Stylianou, E.; Fiocchi, C. Inflammation-induced endothelial-to-mesenchymal transition: A novel mechanism of interstitial fibrosis. Am. J. Pathol. 2011, 179, 2660–2673. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Watabe, T. Roles of TGF-β signals in endothelial-mesenchymal transition during cardiac fibrosis. Int. J. Inflam. 2011, 2011, 724080. [Google Scholar] [CrossRef] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β signaling mediates endothelial-to mesenchymal transition (EndoMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef] [PubMed]
- Kitao, A.; Sato, Y.; Sawada-Kitamura, S.; Harada, K.; Sasaki, M.; Morikawa, H.; Shiomi, S.; Honda, M.; Matsui, O.; Nakanuma, Y. Endothelial to mesenchymal transition via transforming growth factor-beta1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am. J. Pathol. 2009, 175, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Mintet, E.; Rannou, E.; Buard, V.; West, G.; Guipaud, O.; Tarlet, G.; Sabourin, J.C.; Benderitter, M.; Fiocchi, C.; Milliat, F.; et al. Identification of endothelial-to-mesenchymal transition as a potential participant in radiation proctitis. Am. J. Pathol. 2015, 185, 2550–2562. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.; Piera-Velazquez, S.; Farber, J.; Feghali-Bostwick, C.; Jimenez, S.A. Endothelial cells expressing endothelial and mesenchymal cell gene products in Systemic Sclerosis-associated interstitial lung disease lung tissues. Arthritis Rheum. 2016, 68, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Invest. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Welch-Reardon, K.M.; Wu, N.; Hughes, C.C. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H.; et al. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. PNAS 1986, 83, 4167–4171. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Noble, N.A. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [PubMed]
- Varga, J.; Whitfield, M.L. Transforming growth factor-beta in systemic sclerosis (scleroderma). Front. Biosci. 2009, 1, 226–235. [Google Scholar] [CrossRef]
- Jiménez, S.A.; Castro, S.V.; Piera-Velázquez, S. Role of growth factors in the pathogenesis of tissue fibrosis in systemic sclerosis. Curr. Rheumatol. Rev. 2010, 6, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Lafyatis, R. Transforming growth factor β—At the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Pohlers, D.; Brenmoehl, J.; Löffler, I.; Müller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-beta and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-β2 promotes Snail-mediated endothelial mesenchymal transition through convergence of Smad-dependent and Smad-independent signaling. Biochem. J. 2011, 433, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; ten Dijke, P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012, 347, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair 2012, 5 (Suppl. 1), S7. [Google Scholar]
- Li, Z.; Jimenez, S.A. Protein kinase Cδ and c-Abl kinase are required for transforming growth factor β induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 2011, 63, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Xavier, S.; Vasko, R.; Matsumoto, K.; Zullo, J.A.; Chen, R.; Maizel, J.; Chander, P.N.; Goligorsky, M.S. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. 2015, 26, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Srivastava, S.P.; Kanasaki, M.; He, J.; Kitada, M.; Nagai, T.; Takagi, S.; Kanasaki, K.; Koya, D. Interactions of DPP-4 and integrin β1 influences endothelial-to mesenchymal transition. Kidney Int. 2015, 88, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Artavanis-Tsakonas, S. Notch and disease: A growing field. Semin. Cell. Dev. Biol. 2012, 23, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Joyner, A.L.; Loomis, C.A.; Munger, J.S. Sonic hedgehog signaling in the lung. From development to disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Distler, A.; Lang, V.; Del Vecchio, T.; Huang, J.; Zhang, Y.; Beyer, C.; Lin, N.Y.; Palumbo-Zerr, K.; Distler, O.; Schett, G.; et al. Combined inhibition of morphogen pathways demonstrates additive antifibrotic effects and improved tolerability. Ann. Rheum. Dis. 2014, 73, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell 2011, 21, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential regulation of transforming growth factor beta signaling pathways by Notch in human endothelial cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhou, D.; Hao, S.; Zhou, L.; He, W.; Nie, J.; Hou, F.F.; Liu, Y. Sonic Hedgehog Signaling Mediates Epithelial-Mesenchymal Communication and Promotes Renal Fibrosis. J. Am. Soc. Nephrol. 2012, 23, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Jung, Y.; Omenetti, A.; Abdelmalek, M.; Guy, C.D.; Yang, L.; Wang, J.; Witek, R.P.; Fearing, C.M.; Pereira, T.A.; et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 2009, 137, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Fabian, S.L.; Penchev, R.R.; Jacques, B.S.; Rao, A.N.; Sipilä, P.; West, K.A.; McMahon, A.P.; Humphreys, B.D. Hedgehog-Gli Pathway Activation during Kidney Fibrosis. Am. J. Pathol. 2012, 180, 2935–2951. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, A.L.; Milla, C.M.; Lira, J.C.; Ramírez, R.; Checa, M.; Barrera, L.; García-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012, 64, 2724–2733. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signaling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Distler, J.H. Canonical Wnt signaling in systemic sclerosis. Lab. Invest. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Fang, F.; Lam, A.P.; Sargent, J.L.; Hamburg, E.; Hinchcliff, M.E.; Gottardi, C.J.; Atit, R.; Whitfield, M.L.; Varga, J. Wnt/β-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012, 64, 2734–2745. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Schramm, A.; Akhmetshina, A.; Dees, C.; Kireva, T.; Gelse, K.; Sonnylal, S.; de Crombrugghe, B.; Taketo, M.M.; Distler, O.; et al. β-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Flozak, A.S.; Russell, S.; Wei, J.; Jain, M.; Mutlu, G.M.; Budinger, G.R.; Feghali-Bostwick, C.A.; Varga, J.; Gottardi, C.J. Nuclear β-catenin is increased in systemic sclerosis pulmonary fibrosis and promotes lung fibroblast migration and proliferation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Model. Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, L.; Zang, J.; Tang, X.; Liu, Y.; Zhang, J.; Bai, L.; Yin, Q.; Lu, Y.; Cheng, J.; et al. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism 2015, 64, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.L.; Shao, J.S.; Behrmann, A.; Krchma, K.; Towler, D.A. Dkk1 and MSX2-Wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zhang, X.L.; Bitzer, M.; von Gersdorff, G.; Böttinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGF-beta type I receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Del Galdo, F.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr. Opin. Rheumatol. 2008, 20, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Gvaramia, D.; Blaauboer, M.E.; Hanemaaijer, R.; Everts, V. Role of caveolin-1 in fibrotic diseases. Matrix Biol. 2013, 32, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Del Galdo, F.; Sotgia, F.; de Almeida, C.J.; Jasmin, J.F.; Musick, M.; Lisanti, M.P.; Jiménez, S.A. Decreased expression of caveolin 1 in patients with systemic sclerosis: Crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. 2008, 58, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Tourkina, E.; Richard, M.; Gööz, P.; Bonner, M.; Pannu, J.; Harley, R.; Bernatchez, P.N.; Sessa, W.C.; Silver, R.M.; Hoffman, S. Antifibrotic properties of caveolin-1 scaffolding domain in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L843–L861. [Google Scholar] [CrossRef] [PubMed]
- Jasmin, J.F.; Mercier, I.; Dupuis, J.; Tanowitz, H.B.; Lisanti, M.P. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation 2006, 114, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wermuth, P.J.; Benn, B.S.; Lisanti, M.P.; Jimenez, S.A. Caveolin-1 deficiency induces spontaneous endothelial-to-mesenchymal transition in murine pulmonary endothelial cells in vitro. Am. J. Pathol. 2013, 182, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Shi-Wen, X.; Denton, C.P.; Dashwood, M.R.; Holmes, A.M.; Bou-Gharios, G.; Pearson, J.D.; Black, C.M.; Abraham, D.J. Fibroblast matrix gene expression and connective tissue remodeling: Role of endothelin-1. J. Invest. Dermatol. 2001, 116, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Dou, T.T.; Yang, J.Q.; Chen, X.B.; Cao, H.L.; Min, M.; Cai, S.Q.; Zheng, M.; Man, X.Y. Role of endothelin-1 in the skin fibrosis of systemic sclerosis. Eur. Cytokine Netw. 2015, 26, 10–14. [Google Scholar] [PubMed]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, P.; Li, Z.; Jimenez, S.A. Synergistic stimulation of Transforming Growth Factor-β1 induced endothelial-to-mesenchymal transition and tissue fibrosis by Endothelin-1 (ET-1): A novel profibrotic effect of ET-1. 2016; under review. [Google Scholar]
- Cipriani, P.; Di Benedetto, P.; Ruscitti, P.; Capece, D.; Zazzeroni, F.; Liakouli, V.; Pantano, I.; Berardicurti, O.; Carubbi, F.; Pecetti, G.; et al. The endothelial-mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J. Rheumatol. 2015, 42, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185. [Google Scholar] [PubMed]
- Hasse, V.H. Pathophysiological consequences of HIF activation; HIF as a modulator of fibrosis. Ann. N. Y. Acad. Sci. 2009, 1177, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Schett, G.; Gay, S.; Distler, O.; Distler, J.H. Hypoxia. Hypoxia in the pathogenesis of systemic sclerosis. Arthritis Res. Ther. 2009, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.; Jüngel, A.; Pileckyte, M.; Zwerina, J.; Michel, B.A.; Gay, R.E.; Kowal-Bielecka, O.; Matucci-Cerinic, M.; Schett, G.; Marti, H.H.; et al. Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum. 2007, 56, 4203–4215. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Elsner, T.; Botella, L.M.; Velasco, B.; Corbí, A.; Attisano, L.; Bernabéu, C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001, 276, 38527–38535. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Invest. 2007, 117, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Sanchez, E.; Zeisberg, M.; Zeisberg, E.M. Snail is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J. Biol. Chem. 2015, 290, 16653–16664. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hong, Z.Y.; Nam, J.K.; Lee, H.J.; Jang, J.; Yoo, R.J.; Lee, Y.J.; Lee, C.Y.; Kim, K.H.; Park, S.; et al. A Hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin. Cancer Res. 2015, 21, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox. Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox. Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Bourji, K.; Meyer, A.; Chatelus, E.; Pincemail, J.; Pigatto, E.; Defraigne, J.O.; Singh, F.; Charlier, C.; Geny, B.; Gottenberg, J.E.; et al. High reactive oxygen species in fibrotic and nonfibrotic skin of patients with diffuse cutaneous systemic sclerosis. Free Radic. Biol. Med. 2015, 87, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Kliment, C.R.; Oury, T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010, 49, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Siani, A.; Tirelli, N. Myofibroblast differentiation: Main features, biomedical relevance, and the role of reactive oxygen species. Antioxid. Redox. Signal 2014, 21, 768–785. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, G.S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox. Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Crestani, B.; Besnard, V.; Boczkowski, J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2011, 43, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernández-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Kim, J.; Aoyama, T.; De Minicis, S.; Bataller, R.; Brenner, D.A. Role of NADPH oxidases in liver fibrosis. Antioxid. Redox Signal 2014, 20, 2854–2872. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, T.; Svegliati Baroni, S.; Amico, D.; Albani, L.; Moroncini, G.; Avvedimento, E.V.; Gabrielli, A. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol. 2015, 67, 1611–1622. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Makul, A.; Jimenez, S.A. Increased expression of NADPH Oxidase 4 (NOX4) in Systemic Sclerosis dermal fibroblasts: Regulation of Transforming Growth Factor β. Arthritis Rheumatol. 2015, 67, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Stricker, H.M.; Gou, D.; Liu, L. miRNA: Past and present. Front. Biosci. 2007, 12, 2316–2329. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Tsitsiou, E.; Herrick, S.E.; Lindsay, M.A. MicroRNAs and the regulation of fibrosis. FEBS J. 2010, 277, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Rutnam, Z.J.; Wight, T.N.; Yang, B. miRNAs regulate expression and function of extracellular matrix molecules. Matrix Biol. 2013, 32, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.; Jenkins, R.H.; Fraser, D.J. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J Pathol. 2013, 229, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Babalola, O.; Mamalis, A.; Lev-Tov, H.; Jagdeo, J. The role of microRNAs in skin fibrosis. Arch. Dermatol. Res. 2013, 305, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Luo, H.; Zuo, X. MicroRNAs: Their involvement in fibrosis pathogenesis and use as diagnostic biomarkers in scleroderma. Exp. Mol. Med. 2013, 45, e41. [Google Scholar] [CrossRef] [PubMed]
- Pandit, K.V.; Milosevic, J. MicroRNA regulatory networks in idiopathic pulmonary fibrosis. Biochem. Cell Biol. 2015, 93, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Thum, T. Noncoding RNAs and myocardial fibrosis. Nat. Rev. Cardiol. 2014, 11, 655–663. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, C.; Zhang, S.P.; Sun, X.; Long, X.R.; Li, J. The potential of microRNAs in liver fibrosis. Cell Signal 2012, 24, 2268–2272. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Koya, D.; Kanasaki, K. MicroRNAs in kidney fibrosis and diabetic nephropathy: Roles on EMT and EndMT. Biomed. Res. Int. 2013, 2013, 125469. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Nagpal, V.; Covington, J.W.; Michaels, M.A.; Vaughan, D.E. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): Differential expression of microRNAs during EndMT. Cell Signal 2012, 24, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b is critical for fibroblast0to-myofibroblast transition and cardiac fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [PubMed]
- Zhang, J.; Zhang, Z.; Zhang, D.Y.; Zhu, J.; Zhang, T.; Wang, C. microRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS ONE 2013, 8, e83294. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Volkmann, I.; Jazbutyte, V.; Dangwal, S.; Park, D.H.; Thum, T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, P.; Bledsoe, G.; Yang, Z.R.; Chao, L.; Chao, J. Kallistatin inhibits TGF-β-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp. Cell Res. 2015, 337, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: Mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Katsura, A.; Suzuki, H.I.; Ueno, T.; Mihira, H.; Yamazaki, T.; Yasuda, T.; Watabe, T.; Mano, H.; Yamada, Y.; Miyazono, K. MicroRNA-31 is a positive modulator of endothelial-mesenchymal transition and associated secretory phenotype induced by TGF-β. Genes Cells 2016, 21, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Barnes, C.; Charisse, K.; Yi, T.; Zhang, X.; Ali, R.; Medina, P.P.; Yu, J.; Slack, F.J.; et al. FGF regulates TGF-β signaling and endothelial-to mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012, 2, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Kanasaki, M.; Srivastava, S.P.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Shi, S.; Kanasaki, K.; Koya, D. N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. Biomed. Res. Int. 2014, 2014, 696475. [Google Scholar] [CrossRef] [PubMed]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S.P.; Koya, D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 2014, 63, 2120–2131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, X.; Cai, J.J.; Chen, L.Z.; Gong, Y.S.; Wang, L.X.; Gao, Z.; Zhang, H.Q.; Huang, W.J.; Zhou, H. Relaxin inhibits cardiac fibrosis and endothelial-mesenchymal transition via the Notch pathway. Drug Des. Devel. Ther. 2015, 9, 4599–4611. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, Y.; Koya, D.; Kanasaki, K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, I.; Lenna, S.; Stawski, L.; Trojanowska, M. Interferon-γ promotes vascular remodeling in human microvascular endothelial cells by upregulating endothelin (ET)-1 and transforming growth factor (TGF) β2. J. Cell Physiol. 2013, 228, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G.; et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc. Res. 2015, 105, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Charytan, D.M.; Padera, R.; Helfand, A.M.; Zeisberg, M.; Xu, X.; Liu, X.; Himmelfarb, J.; Cinelli, A.; Kalluri, R.; Zeisberg, E.M. Increased concentration of circulating angiogenesis and nitric oxide inhibitors induces endothelial to mesenchymal transition and myocardial fibrosis in patients with chronic kidney disease. Int. J. Cardiol. 2014, 176, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, S.A. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN. Rheumatol. 2013, 2013, 835948. [Google Scholar] [CrossRef] [PubMed]
- Sadler, T.; Scarpa, M.; Rieder, F.; West, G.; Stylianou, E. Cytokine-induced chromatin modifications of the type I collagen alpha 2 gene during intestinal endothelial-to-mesenchymal transition. Inflamm. Bowel Dis. 2013, 19, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, S.A.; Piera-Velazquez, S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Duffhues, G.; Orlova, V.; Ten Dijke, P. In Brief: Endothelial-to-mesenchymal transition. J. Pathol. 2016, 238, 378–380. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| A. Systemic Fibrotic Diseases |
|---|
|
| B. Organ-Specific Fibrotic Diseases |
| Cardiac Fibrosis |
|
| Kidney Fibrosis |
|
| Pulmonary Fibrosis |
|
| Liver and Portal Vein Fibrosis |
|
| C. Other Organ-specific Fibrotic Diseases |
|
| Fibrotic Disease | Evidence of EndoMT in Affected Tissues | Source of Data | |
|---|---|---|---|
| Tissue Source | Method(s) | ||
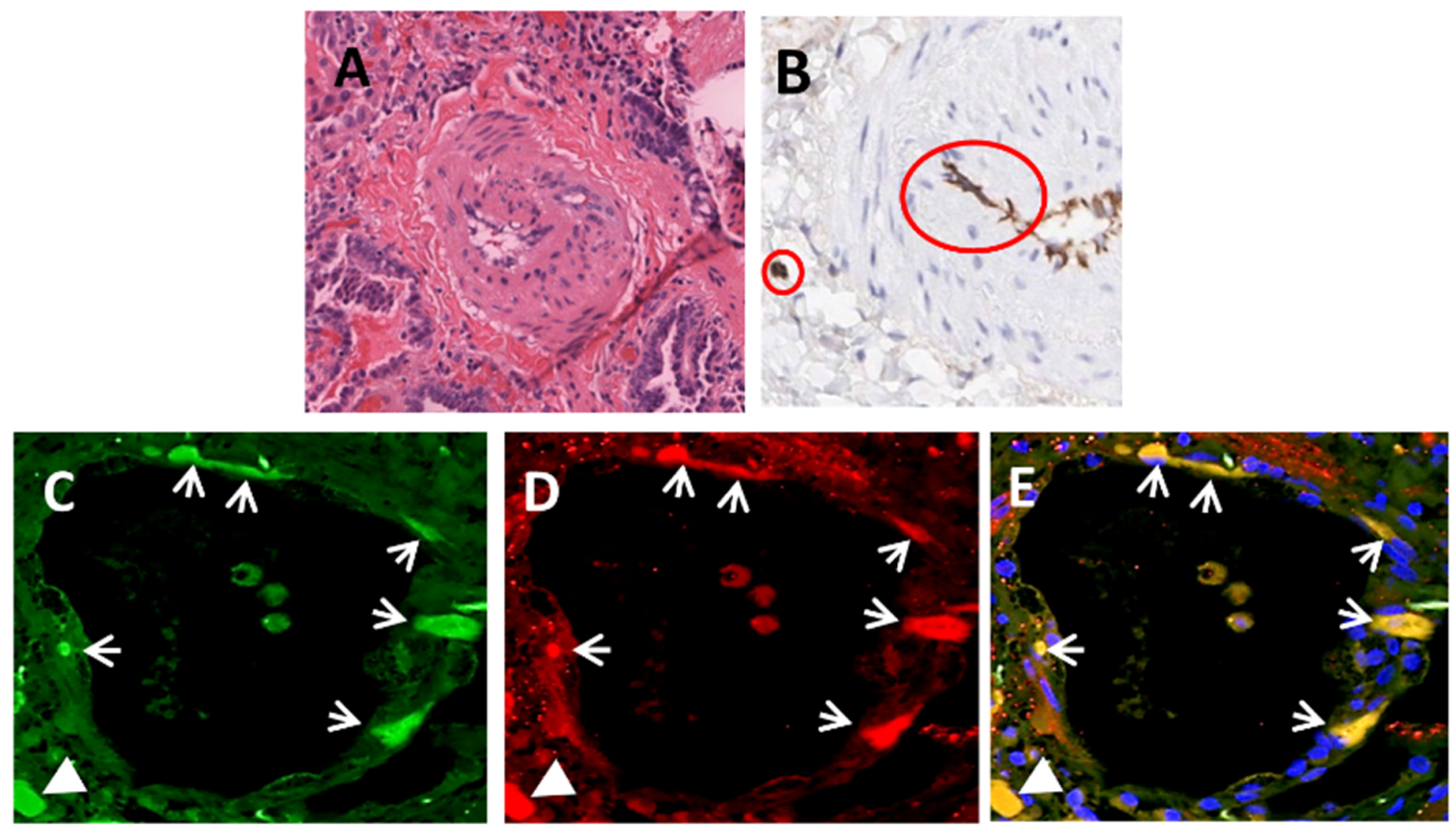
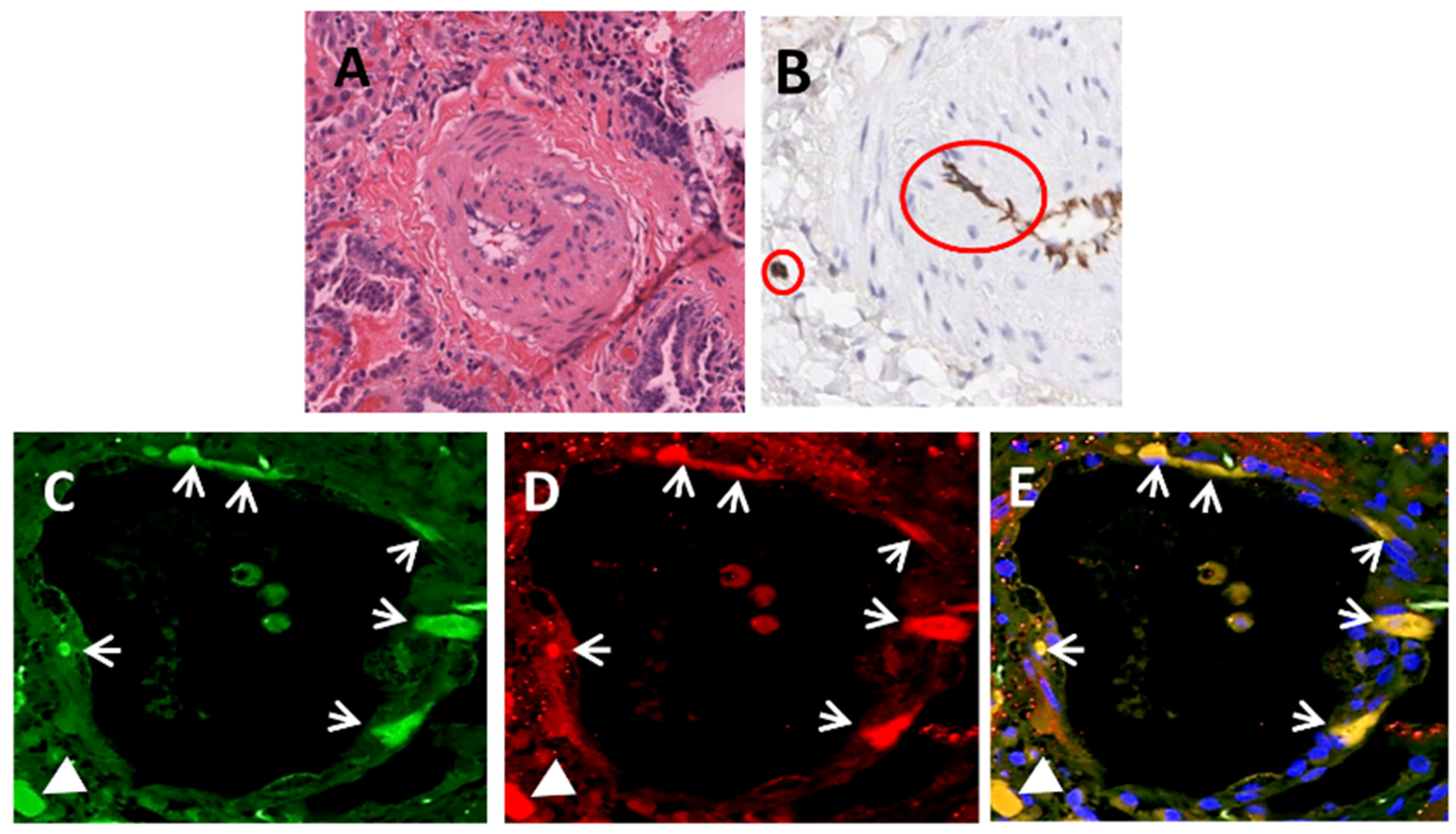
| SSc-associated Pulmonary Fibrosis | Lung transplants | Immunohistochemistry | Mendoza et al. [64] |
| Immunofluorescence | |||
| Gene Expression | |||
| Radiation-induced Pulmonary Fibrosis | Lung tissues (Surgery) | Immunofluorescence | Choi et al. [129] |
| SSc-associated Pulmonary Hypertension | Lung biopsies | Immunofluorescence | Good et al. [66] |
| Idiopathic Pulmonary Hypertension | Lung transplants | Immunofluorescence | Ranchoux et al. [65] |
| Transmission Electron Microscopy | |||
| Immunoelectron Microscopy | |||
| Cardiac Fibrosis | Heart transplants | Gene Expression | Xu et al. [170] |
| Chronic kidney disease-associated cardiac fibrosis | Heart tissue (Autopsies and cardiac surgery) | Immunohistochemistry | Charytan et al. [171] |
| Gene Expression | |||
| Diabetic kidney disease-associated renal fibrosis | Kidney biopsies | Immunohistochemistry | Li et al. [106] |
| Idiopathic Portal Hypertension | Liver biopsies | Immunohistochemistry | Kitao et al. [62] |
| Intestinal Fibrosis | Colonic mucosa | Immunohistochemistry | Reider et al. [59] |
| Radiation-induced Rectal Fibrosis | Rectal tissues (Surgery) | Immunofluorescence | Mintet et al. [63] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J. Clin. Med. 2016, 5, 45. https://doi.org/10.3390/jcm5040045
Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. Journal of Clinical Medicine. 2016; 5(4):45. https://doi.org/10.3390/jcm5040045
Chicago/Turabian StylePiera-Velazquez, Sonsoles, Fabian A. Mendoza, and Sergio A. Jimenez. 2016. "Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases" Journal of Clinical Medicine 5, no. 4: 45. https://doi.org/10.3390/jcm5040045
APA StylePiera-Velazquez, S., Mendoza, F. A., & Jimenez, S. A. (2016). Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. Journal of Clinical Medicine, 5(4), 45. https://doi.org/10.3390/jcm5040045