
Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer


Abstract
:1. Introduction of EMT
2. Effects of Cigarette Smoke on Cancer Development and Treatment Response
3. Molecular Mechanisms of Cigarette Smoke-Induced EMT
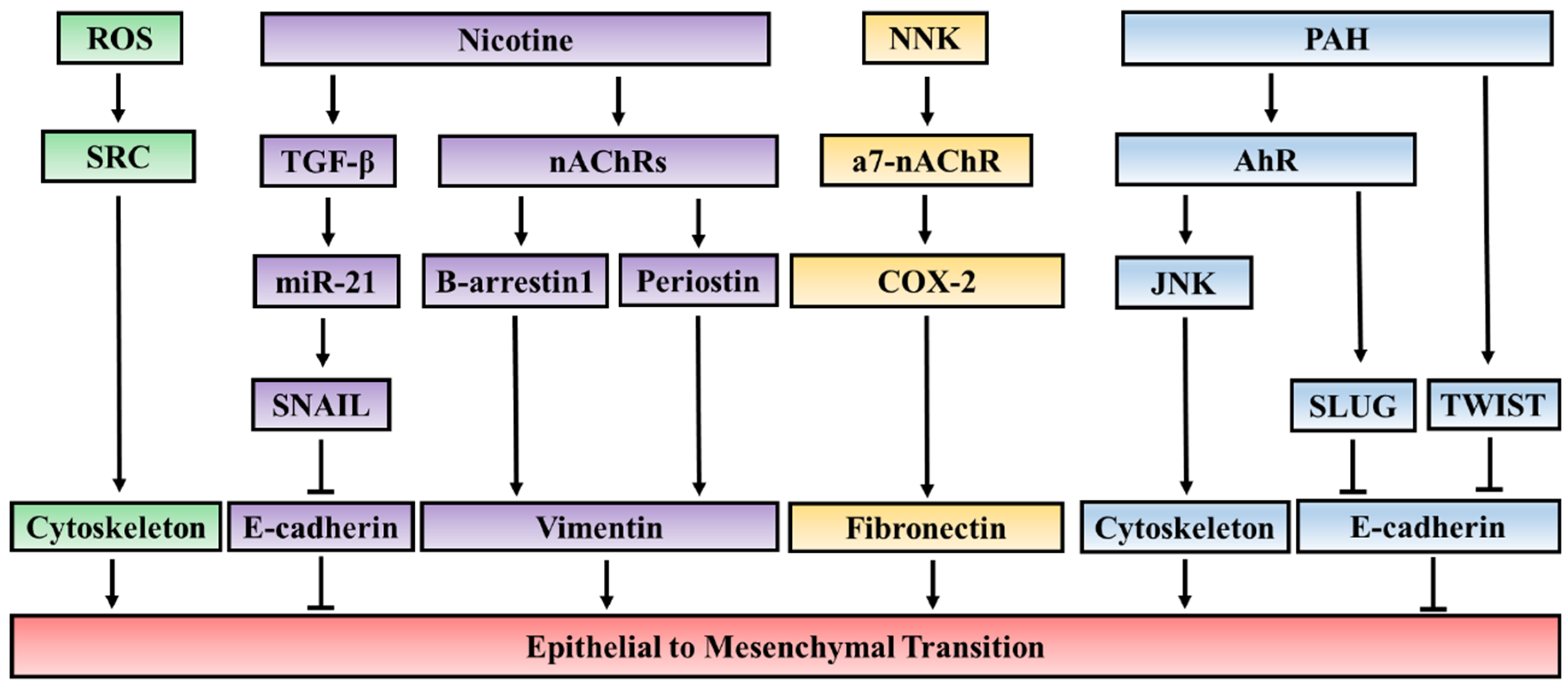
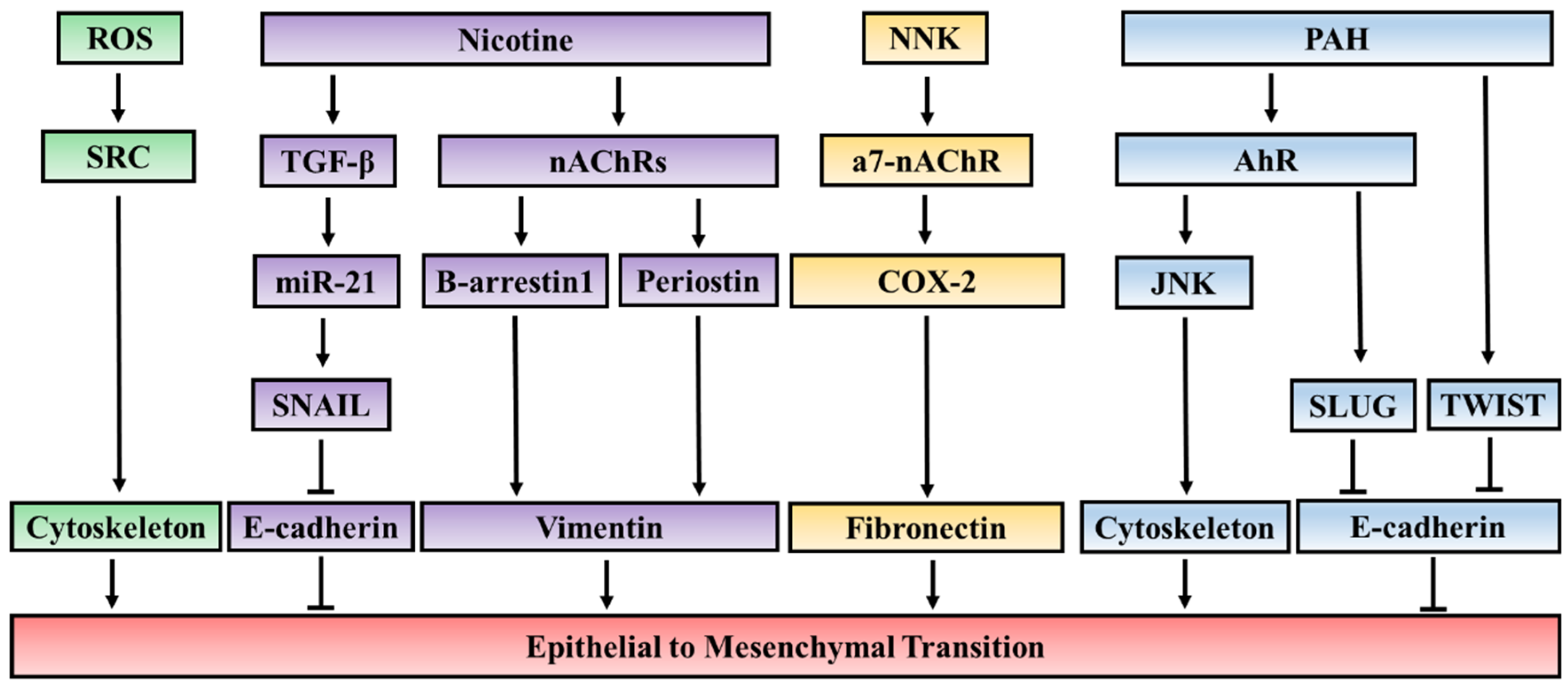
3.1. The EMT-Inducing Effect of Key Components of Cigarette Smoke
3.1.1. Nicotine
3.1.2. Polycyclic Aromatic Hydrocarbons (PAH)
3.1.3. Reactive Oxygen Species (ROS)
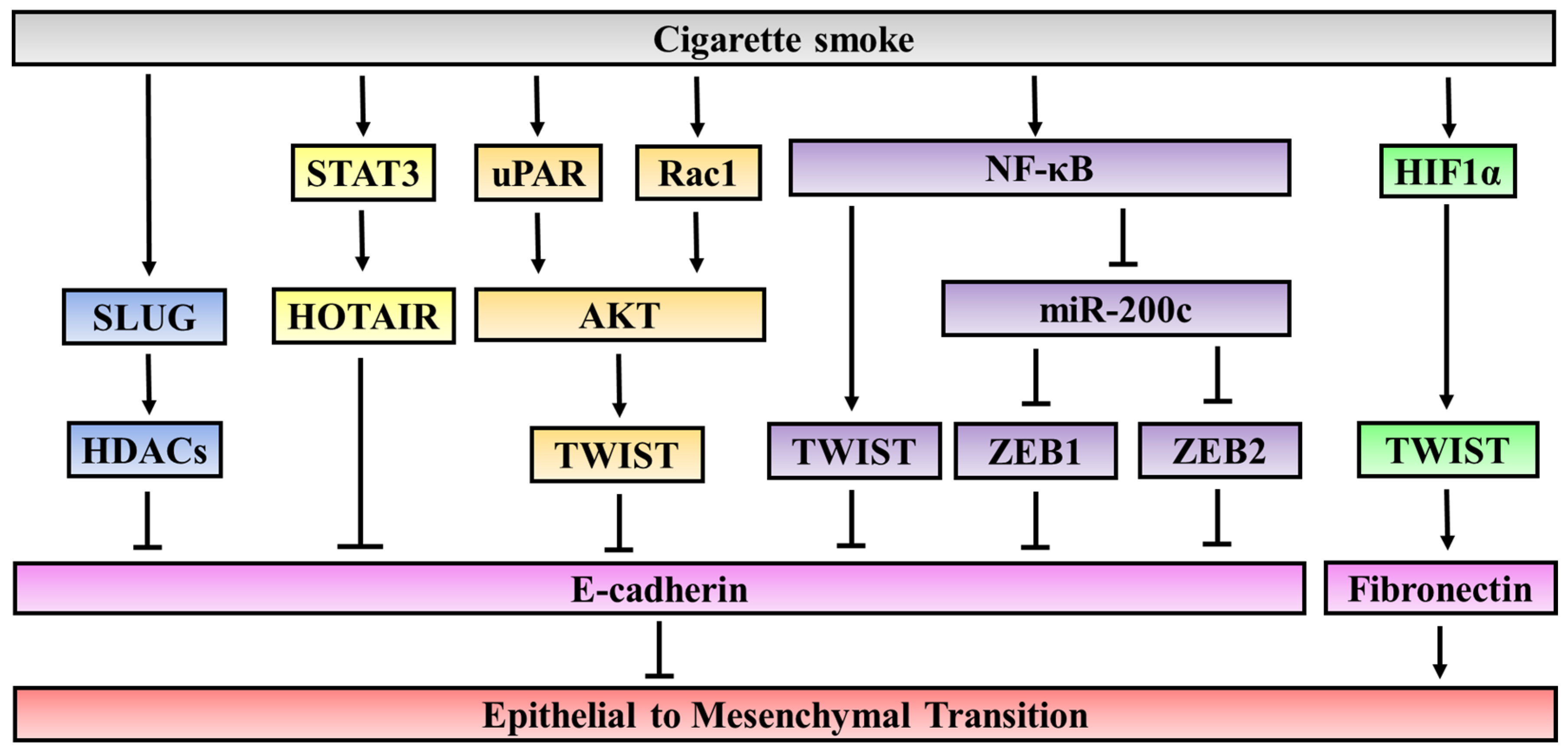
3.2. Cigarette Smoke Mediated Epigenetic Modifications and Oncogenic Pathways
3.2.1. Epigenetic Regulatory Mechanisms
3.2.2. Oncogenic Pathways
4. Conclusion and Perspectives
- (1)
- In addition to carcinogenic effects, various compounds found in cigarette smoke such as PAH, nicotine, and ROS can induce EMT through different signaling pathways. The effects can be mediated through specific receptors such as nAChR and AhR for nicotine and PAH, respectively or through other molecular factors. Despite the diversity in signaling pathways and molecules involved in cigarette smoke-induced EMT, the effects are mainly associated with upregulation and enhanced activation of EMT-inducing transcription factors. As these regulators are also found to be associated with poor prognosis in lung cancer, it is essential to get further insight into the molecular network regulated by EMT-inducing cigarette smoke components.
- (2)
- The induction in EMT in cancer by cigarette smoke can be mediated by both changes in epigenetic regulation and activities of a variety of oncogenic signaling pathways. Importantly, several modifications are also linked to biological responses to cigarette smoke, such as inflammation, hypoxia and oxidative stress. Therefore, to effectively study the complex network underlying the effects of cigarette smoke on EMT, it is more applicable to use cigarette smoke extracts to mimic the environmental stresses caused by long-term cigarette smoking. Furthermore, by using CSE, scientists can study the crosstalk between different signaling pathways activated by concerted effects of individual cigarette components in picogram concentrations.
- (3)
- The effect of cigarette smoke on EMT has not only been found in lung cancer, the cancer type directly related to smoking, but also in other cancer types. This is consistent with previous observations that smoking also has adverse effects on health outcomes in patients with different types of cancer. However, it is not well established how cigarette smoking affects cancers other than lung cancer in vivo.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lim, J.; Thiery, J.P. Epithelial to mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486. [Google Scholar] [PubMed]
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial to mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [PubMed]
- Kang, Y.; Massague, J. Epithelial to mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial to mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–219. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Li, C.H.; Peng, Y.J.; Cheng, Y.W.; Chen, H.W.; Liao, P.L.; Kang, J.J.; Yeng, M.H. SNAIL regulates Nanog status during the epithelial to mesenchymal transition via the Smad1/AKT/GSK3beta signaling pathway in non-small-cell lung cancer. Oncotarget 2014, 5, 3880–3894. [Google Scholar] [CrossRef] [PubMed]
- Merikallio, H.; Turpeenniemi-Hujanen, T.; Pääkkö, P.; Mäkitaro, R.; Riitta, K.; Salo, S.; Salo, T.; Harju, T.; Soini, Y. SNAIL promotes an invasive phenotype in lung carcinoma. Respir. Res. 2012, 13, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.J.; Yang, M.H.; Hsu, H.S.; Hsu, W.H.; Liu, J.S.; Wu, K.J. Prognostic significance of hypoxia-inducible factor-1alpha, TWIST1 and SNAIL expression in resectable non-small cell lung cancer. Thorax 2009, 64, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Merikallio, H.; Pääkkö, P.; Mäkitaro, R.; Kaarteenaho, R.; Lehtonen, S.; Salo, S.; Salo, T.; Harju, T.; Soini, Y.H. SLUG is associated with poor survival in squamous cell carcinoma of the lung. Int. J. Clin. Exp. Pathol. 2014, 7, 5846. [Google Scholar] [PubMed]
- Wushou, A.; Hou, J.; Zhao, Y.J.; Shao, Z.M. Twist-1 up-regulation in carcinoma correlates to poor survival. Int. J. Mol. Sci. 2014, 15, 21621–21630. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zhan, P.; Wu, G.; Yang, W.; Liang, W.; Lv, T.; Song, Y. Prognostic value of Twist in lung cancer: Systematic review and meta-analysis. Transl. Lung Cancer Res. 2015, 4, 236–241. [Google Scholar] [PubMed]
- Nakashima, H.; Hashimoto, N.; Aoyama, D.; Kohnoh, T.; Sakamoto, K.; Kusunose, M.; Imaizumi, K.; Takeyama, Y.; Sato, M.; Kawabe, T.; et al. Involvement of the transcription factor twist in phenotype alteration through epithelial to mesenchymal transition in lung cancer cells. Mol. Carcinog. 2012, 51, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Merikallio, H.; Kaarteenaho, R.; Pääkkö, P.; Lehtonen, S.; Hirvikoski, P.; Mäkitaro, R.; Harju, T.; Soini, Y. Zeb1 and twist are more commonly expressed in metastatic than primary lung tumours and show inverse associations with claudins. J. Clin. Pathol. 2011, 64, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, X.; Wang, Y.; Qu, Z.; Lu, Q.; Zhao, J.; Yan, X.; Zhang, H.; Zhou, Y. Notch3 is important for TGF-β-induced epithelial to mesenchymal transition in non-small cell lung cancer bone metastasis by regulating ZEB-1. Cancer Gene Ther. 2014, 21, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Pang, X.G.; Wang, Q.; Shen, Y.X.; Chen, X.K.; Xi, J.J. Prognostic role of Twist, SLUG, and Foxc2 expression in stage I non-small-cell lung cancer after curative resection. Clin. Lung Cancer 2012, 13, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, X.; Zhu, H.; Wang, X.; Ni, S.; Huang, J. FoxQ1 overexpression influences poor prognosis in non-small cell lung cancer, associates with the phenomenon of EMT. PLoS ONE 2012, 7, 39937–39947. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.X. High expression of FOXC1 is associated with poor clinical outcome in non-small cell lung cancer patients. Tumor Biol. 2013, 34, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Xu, N. FoxM1 is associated with poor prognosis of non-small cell lung cancer patients through promoting tumor metastasis. PLoS ONE 2013, 8, 59412–59420. [Google Scholar] [CrossRef] [PubMed]
- Kase, S.; Sugio, K.; Yamazaki, K.; Okamoto, T.; Yano, T.; Sugimachi, K. Expression of E-cadherin and beta-catenin in human non-small cell lung cancer and the clinical significance. Clin. Cancer Res. 2000, 6, 4789–4798. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, G.; Kang, Y.; Dong, Z.; Qian, Q.; Ma, X. N-cadherin expression is associated with acquisition of EMT phenotype and with enhanced invasion in erlotinib-resistant lung cancer cell lines. PLoS ONE 2013, 8, 57692–57700. [Google Scholar] [CrossRef] [PubMed]
- Anttonen, A.; Leppä, S.; Ruotsalainen, T.; Alfthan, H.; Mattson, K.; Joensuu, H. Pretreatment serum syndecan-1 levels and outcome in small cell lung cancer patients treated with platinum-based chemotherapy. Lung Cancer 2003, 41, 171–177. [Google Scholar] [CrossRef]
- Liu, X.G. High expression of serum miR-21 and tumor miR-200c associated with poor prognosis in patients with lung cancer. Med. Oncol. 2012, 29, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, Y.; Zhang, X.; Li, J.; Han, B.; Liu, S.; Wang, L.; Ling, Y.; Mao, S.; Wang, X. Overexpression of integrin-linked kinase correlates with malignant phenotype in non-small cell lung cancer and promotes lung cancer cell invasion and migration via regulating epithelial-mesenchymal transition (EMT)—Related genes. Acta Histochem. 2013, 115, 128–238. [Google Scholar] [CrossRef] [PubMed]
- Soltermann, A.; Tischler, V.; Arbogast, S.; Braun, J.; Probst-Hensch, N.; Weder, W.; Moch, H.; Kristiansen, G. Prognostic significance of epithelial-mesenchymal and mesenchymal-epithelial transition protein expression in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 7430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zou, L.; Zhang, Y.; Chen, Y.; Xing, P.; Yang, W.; Li, F.; Ji, X.; Liu, F.; Lu, X. Transforming growth factor-β1 and α-smooth muscle actin in stromal fibroblasts are associated with a poor prognosis in patients with clinical stage I–IIIA nonsmall cell lung cancer after curative resection. Tumor Biol. 2014, 35, 6707–6720. [Google Scholar] [CrossRef] [PubMed]
- Barrallo-Gimeno, A.; Nieto, M.A. The SNAIL genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. SNAIL mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Pasini, D.; Díaz, V.M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E-cadherin repression by the SNAIL1 transcription factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and SNAIL to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ponn, A.; Hu, X.; Law, B.K.; Lu, J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial to mesenchymal transition. Oncogene 2010, 29, 4896–4904. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with SNAIL and is critical for SNAIL-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is critical for SNAIL-mediated E-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Tan, E.J.; Peinado, H.; Cano, A.; Heldin, C.H.; Moustakas, A. HMGA2 and Smads coregulate SNAIL1 expression during induction of epithelial toto-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef] [PubMed]
- Smit, M.A.; Geiger, T.R.; Song, J.Y.; Gitelman, I.; Peeper, D.S. A Twist-SNAIL axis critical for TrkB-induced epithelial to mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol. Cell. Biol. 2009, 29, 3722–3737. [Google Scholar] [CrossRef] [PubMed]
- Guaita, S.; Puig, I.; Franci, C.; Garrido, M.; Dominguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; De Herreros, A.G.; Baulida, J. SNAIL induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39220. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.; Stanners, S.R.; Yong, R.; Tang, O.; Pollock, C.A. Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the SNAIL transcription factor. Int. J. Biochem. Cell Biol. 2010, 42, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L.; Huang, Q.; Xu, W.; Cai, X.; Zhang, J.; Yan, W.; Song, D.; Liu, T.; Zhou, W.; et al. Wnt signaling through SNAIL1 and Zeb1 regulates bone metastasis in lung cancer. Am. J. Cancer Res. 2015, 5, 748–755. [Google Scholar] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Sun, L.; Li, Q.; Han, X.; Lei, L.; Zhang, H.; Shang, Y. SET8 promotes epithelial to mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012, 31, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial to mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, K.J. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle 2008, 7, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Dean, D.C. ZEB represses transcription through interaction with the corepressor CtBP. Proc. Natl. Acad. Sci. USA 1999, 96, 6683–6699. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.E. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3510. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003, 22, 2453–2463. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Scully, K.; Zhu, X.; Cai, L.; Zhang, J.; Prefontaine, G.G.; Krones, A.; Ohgi, K.A.; Zhu, P.; Garcia-Bassets, I.; et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007, 446, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, À.; Beltran, M.; Peiró, S.; de Herreros, A.G. Functional cooperation between SNAIL1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12034. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial to mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Doll, R.; Hill, A.B. Smoking and carcinoma of the lung: Preliminary report. Br. Med. J. 1950, 2, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Kassie, F.; Hatsukami, D.K. Chemoprevention of lung carcinogenesis in addicted smokers and ex-smokers. Nat. Rev. Cancer 2009, 9, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.; Daley, A.; Begh, R.; Aveyard, P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: Systematic review of observational studies with meta-analysis. BMJ 2010, 340, 5569. [Google Scholar] [CrossRef] [PubMed]
- Kobrinsky, N.L.; Klug, M.G.; Hokanson, P.J.; Sjolander, D.E.; Burd, L. Impact of smoking on cancer stage at diagnosis. J. Clin. Oncol. 2003, 21, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Murin, S.; Inciardi, J. Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest 2001, 119, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Abrams, J.A.; Lee, P.C.; Port, J.L.; Altorki, N.K.; Neugut, A.I. Cigarette smoking and risk of lung metastasis from esophageal cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- McBride, S.M.; Ali, N.N.; Margalit, D.N.; Chan, A.W. Active tobacco smoking and distant metastasis in patients with oropharyngeal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.M.; Aronson, W.J.; Terris, M.K.; Kane, C.J.; Amling, C.L.; Cooperberg, M.R.; Boffetta, P.; Freedland, S.J. Cigarette smoking is associated with an increased risk of biochemical disease recurrence, metastasis, castration-resistant prostate cancer, and mortality after radical prostatectomy: Results from the SEARCH database. Cancer 2014, 120, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Iizasa, T.; Saitoh, Y.; Sekine, Y.; Motohashi, S.; Yasukawa, T.; Shibuya, K.; Hiroshima, K.; Ohwada, H. Smoking before surgery predicts poor long-term survival in patients with stage I non-small-cell lung carcinomas. J. Clin. Oncol. 1999, 17, 2086–2091. [Google Scholar] [PubMed]
- Fox, J.L.; Rosenzweig, K.E.; Ostroff, J.S. The effect of smoking status on survival following radiation therapy for non-small cell lung cancer. Lung Cancer 2004, 44, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kamdar, O.; Le, W.; Rosen, G.D.; Upadhyay, D. Nicotine induces resistance to chemotherapy by modulating mitochondrial signaling in lung cancer. Am. J. Respir. Cell Mol. Biol. 2009, 40, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Le, W.; Yee, A.; Kamdar, O.; Hwang, P.H.; Upadhyay, D. Nicotine induces resistance to chemotherapy in nasal epithelial cancer. Am. J. Rhinol. Allergy 2010, 24, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.W.; Singh, A.K. Nicotine and lung cancer. J. Carcinog. 2013, 12, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Videtic, G.M.; Stitt, L.W.; Dar, A.R.; Kocha, W.I.; Tomiak, A.T.; Truong, P.T.; Vincent, M.D.; Yu, E.W. Continued cigarette smoking by patients receiving concurrent chemoradiotherapy for limited-stage small-cell lung cancer is associated with decreased survival. J. Clin. Oncol. 2003, 21, 1544–1549. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Reid, D.; Soltani, A.; Ward, C.; Weston, S.; Muller, H.K.; Wood-Baker, R.; Walters, E.H. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology 2010, 15, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Peiró, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Mahmood, M.Q.; Walters, E.H. Clinical significance of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD): Potential target for prevention of airway fibrosis and lung cancer. Clin. Transl. Med. 2014, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, Y.; Li, Y.; Xu, W.; Luo, F.; Wang, B.; Pang, Y.; Xiang, Q.; Zhou, J.; Wang, X.; et al. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol. Sci. 2013, 135, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Y.; Lin, C.; Jia, J.; Tian, L.; Yang, K.; Zhao, L.; Lai, N.; Jiang, Q.; Sun, Y.; et al. Effects of chronic exposure to cigarette smoke on canonical transient receptor potential expression in rat pulmonary arterial smooth muscle. Am. J. Physiol. Cell Physiol. 2014, 306, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Walters, E.H. Role of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD). Respir. Res. 2013, 14, 120. [Google Scholar] [CrossRef] [PubMed]
- Siafakas, N.M.; Antoniou, K.M.; Tzortzaki, E.G. Role of angiogenesis and vascular remodeling in chronic obstructive pulmonary disease. Int. J. Chron. Obstr. Pulm. Dis. 2007, 2, 453–462. [Google Scholar]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Fowles, J.; Dybing, E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob. Control 2003, 12, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial to mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rastogi, S.; Pillai, S.; Ordonez-Ercan, D.; Morris, M.; Haura, E.; Chellappan, S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Investig. 2006, 116, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Plummer, H.K., 3rd; Jull, B.A. Receptor-mediated effects of nicotine and its nitrosated derivative NNK on pulmonary neuroendocrine cells. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2003, 270, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.; Rizwani, W.; Banerjee, S.; Kovacs, M.; Haura, E.; Coppola, D.; Chellappan, S. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS ONE 2009, 4, 7524. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Shenoy, S.K. Arrestins and protein ubiquitination. Prog. Mol. Biol. Transl. Sci. 2013, 118, 175–204. [Google Scholar] [PubMed]
- Pillai, S.; Trevino, J.; Rawal, B.; Singh, S.; Kovacs, M.; Li, X.; Schell, M.; Haura, E.; Bepler, G.; Chellappan, S. Beta-arrestin-1 mediates nicotine-induced metastasis through E2F1 target genes that modulate epithelial to mesenchymal transition. Cancer Res. 2015, 75, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zou, Y.; Zhao, Z.; Li, B.; Ran, P. Nicotine-induced epithelial to mesenchymal transition via Wnt/beta-catenin signaling in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Li, X.Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates SLUG activity and links epithelial to mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Shao, R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial to mesenchymal transformation. J. Biol. Chem. 2006, 281, 19700–19708. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Q.; Lv, Y.E.; Lin, B.H.; Luo, L.M.; Lv, S.L.; Bi, A.H.; Jia, Y.S. Silencing of periostin inhibits nicotine-mediated tumor cell growth and epithelial to mesenchymal transition in lung cancer cells. Mol. Med. Rep. 2013, 7, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, I.; Kometani, T.; Shoji, F.; Osoegawa, A.; Ohba, T.; Kouso, H.; Takenaka, T.; Yohena, T.; Maehara, Y. Induction of epithelial to mesenchymal transition-related genes by benzo[a]pyrene in lung cancer cells. Cancer 2007, 110, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, T.; Kawajiri, K. Zinc finger transcription factor SLUG is a novel target gene of aryl hydrocarbon receptor. Exp. Cell Res. 2006, 312, 3585–3594. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Chang, H.; Ho, W.L.; Wu, M.H.; Su, J.M. Association of aryl hydrocarbon receptor and cytochrome P4501B1 expressions in human non-small cell lung cancers. Lung Cancer 2003, 42, 255–261. [Google Scholar] [CrossRef]
- Diry, M.; Tomkiewicz, C.; Koehle, C.; Coumoul, X.; Bock, K.W.; Barouki, R.; Transy, C. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene 2006, 25, 5570–5574. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [PubMed]
- Mallozzi, C.; Di Stasi, A.M.; Minetti, M. Activation of src tyrosine kinases by peroxynitrite. FEBS Lett. 1999, 456, 201–206. [Google Scholar] [CrossRef]
- Mehdi, M.Z.; Pandey, N.R.; Pandey, S.K.; Srivastava, A.K. H2O2-induced phosphorylation of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c-Src. Antioxid. Redox Signal. 2005, 7, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, H.; Borok, Z.; Davies, K.J.; Ursini, F.; Forman, H.J. Cigarette smoke extract stimulates epithelial to mesenchymal transition through Src activation. Free Radic. Biol. Med. 2012, 52, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta Biochim. Biophys. Sin. 2010, 42, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Massion, P.P.; Gonzalez, A.L.; Lu, P.; Datta, P.K. Smoking induces epithelial to-mesenchymal transition in non-small cell lung cancer through HDAC-mediated downregulation of E-cadherin. Mol. Cancer Ther. 2012, 11, 2362–2372. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, F.; Xu, Y.; Wang, B.; Zhao, Y.; Xu, W.; Shi, L.; Lu, X.; Liu, Q. Epithelial to mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol. Appl. Pharmacol. 2015, 282, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; An, Y.; Liang, Y.; Xie, X.W. Role of HOTAIR long noncoding RNA in metastatic progression of lung cancer. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1930–1936. [Google Scholar] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q. Activation of uPAR is required for cigarette smoke extract-induced epithelial to mesenchymal transition in lung epithelial cells. Oncol. Res. 2013, 21, 295. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.R.; Ellis, V.; Rønne, E.; Pyke, C.; Danø, K. Transcriptional and post-transcriptional regulation of the receptor for urokinase-type plasminogen activator by cytokines and tumour promoters in the human lung carcinoma cell line A549. Biochem. J. 1995, 310, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Duggan, C. The urokinase plasminogen activator system: A rich source of tumour markers for the individualised management of patients with cancer. Clin. Biochem. 2004, 37, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Nieder, C.; Bremnes, R.M. Effects of smoking cessation on hypoxia and its potential impact on radiation treatment effects in lung cancer patients. Strahlenther. Onkol. 2008, 184, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Sagone, A.L., Jr.; Lawrence, T.; Balcerzak, S.P. Effect of smoking on tissue oxygen supply. Blood 1973, 41, 845–850. [Google Scholar] [PubMed]
- Nurwidya, F.; Takahashi, F.; Kobayashi, I.; Murakami, A.; Kato, M.; Minakata, K.; Nara, T.; Hashimoto, M.; Yagishita, S.; Baskoro, H.; et al. Treatment with insulin-like growth factor 1 receptor inhibitor reverses hypoxia-induced epithelial to mesenchymal transition in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2014, 455, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, D.; Zhou, Q.; Chen, T.; Ibe, J.C.; Raj, J.U.; Zhou, G. cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial to mesenchymal transition, migration, and invasion in lung cancer cells. Cell. Signal. 2012, 24, 2396–2400. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Eurlings, I.M.; Reynaert, N.L.; Van den Beucken, T.; Gosker, H.R.; de Theije, C.C.; Verhamme, F.M.; Bracke, K.R.; Wouters, E.F.; Dentener, M.A. Cigarette smoke extract induces a phenotypic shift in epithelial cells; involvement of HIF1alpha in mesenchymal transition. PLoS ONE 2014, 9, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.J.; Sun, Y.H.; Zhang, S.J.; Jiang, J.X.; Dong, X.W.; Jia, Y.L.; Shen, J.; Guan, Y.; Zhang, L.H.; Li, F.F.; et al. Cigarette smoke-induced alveolar epithelial to mesenchymal transition is mediated by Rac1 activation. Biochim. Biophys. Acta 2014, 1840, 1838–1849. [Google Scholar] [CrossRef] [PubMed]
- Price, L.S.; Collard, J.G. Regulation of the cytoskeleton by Rho-family GTPases: Implications for tumour cell invasion. Semin. Cancer Biol. 2001, 11, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Lan, H.Y.; Huang, C.H.; Tai, S.K.; Tzeng, C.H.; Kao, S.Y.; Wu, K.J.; Hung, M.C.; Yang, M.H. RAC1 activation mediates Twist1-induced cancer cell migration. Nat. Cell Boil. 2012, 14, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Xie, W.; Wu, R.; Geng, H.; Zhao, L.; Xie, C.; Li, X.; Huang, C.; Zhu, J.; Zhu, M.; et al. ERK5 negatively regulates tobacco smoke-induced pulmonary epithelial to mesenchymal transition. Oncotarget 2015, 6, 19605–19618. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Nishida, E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006, 7, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Wu, Y.; Wehrli, B.; Chakrabarti, S.; Chakraborty, C. Modulation of ERK5 is a novel mechanism by which Cdc42 regulates migration of breast cancer cells. J. Cell. Biochem. 2015, 116, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Nitschke, A.M.; Xiong, W.; Zhang, Q.; Tang, Y.; Bloch, M.; Elliott, S.; Zhu, Y.; Bazzone, L.; Yu, D.; et al. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial to mesenchymal transition phenotype. Breast Cancer Res. 2008, 10, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Schnoll, R.; Rothman, R.; Newman, H.; Lerman, C.; Miller, S.M.; Movsas, B.; Sherman, E.; Ridge, J.A.; Unger, M.; Langer, C.; et al. Characteristics of cancer patients entering a smoking cessation program and correlates of quit motivation: Implications for the development of tobacco control programs for cancer patients. Psycho-Oncol. 2004, 13, 346–358. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Factors | Description | Relevance to Lung Cancer | Refs |
|---|---|---|---|
| Transcription factors | |||
| SNAIL1 | Zinc-finger protein, E-box transcriptional repressor | Positive expression associated with poor survival in squamous cell and adenocarcinomas | [8,9,10] |
| SLUG | Zinc-finger protein, E-box transcriptional repressor | Upregulation associated with poor survival in squamous cell carcinoma | [11] |
| TWIST1 | bHLH factor | Overexpression in primary NSCLCs associated with a shorter overall survival | [12,13,14] |
| ZEB1 | Zinc-finger protein, E-box transcriptional repressor | Higher expression found in metastatic lung tumors compared to primary tumors | [15,16] |
| FOXC2 | Forkhead box transcription factor | Overexpression associated with a worse overall survival and correlated with a shorter recurrence-free survival in patients with stage-I non-small cell lung cancer | [17] |
| FOXQ1 | Forkhead box transcription factor | Upregulation in NSCLC resulting in poor prognosis | [18] |
| FOXC1 | Forkhead box transcription factor | Upregulation correlated with poor tumor differentiation, tumor-node-metastasis stage, and lymph node metastasis in NSCLC patients | [19] |
| FOXM1 | Forkhead box transcription factor | Overexpression associated with poor prognosis of NSCLC patients and tumor metastasis | [20] |
| Factors directly associated with EMT | |||
| E-cadherin | Adhesion glycoprotein | Reduced E-cadherin expression significantly correlated with lymph node metastasis | [21] |
| N-cadherin | Adhesion glycoprotein | Overexpression associated with a shorter overall survival | [22] |
| Syndecan-1 | Transmembrane (type I) heparan sulfate proteoglycan | High pretreatment serum syndecan-1 level associated with poor prognosis in SCLC treated with platinum-based chemotherapy | [23] |
| miR-21 | Non-coding RNA | Upregulation of serum miR-21 strongly associated with lymph node metastasis and advanced clinical stage of NSCLC | [24] |
| α-SMA | α-Smooth muscle actin | Overexpression associated with a poor prognosis in patients with clinical stage I-IIIA NSCLC after curative resection | [27] |
| Vimentin | Member of the intermediate filament family | Upregulation correlated with lymph node metastasis in squamous cell lung carcinoma | [25] |
| Periostin | Osteoblast Specific Factor | Overexpression associated with decreased progression-free survival | [26] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, T.; Jin, L.; Datta, P.K. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. J. Clin. Med. 2016, 5, 44. https://doi.org/10.3390/jcm5040044
Vu T, Jin L, Datta PK. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. Journal of Clinical Medicine. 2016; 5(4):44. https://doi.org/10.3390/jcm5040044
Chicago/Turabian StyleVu, Trung, Lin Jin, and Pran K. Datta. 2016. "Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer" Journal of Clinical Medicine 5, no. 4: 44. https://doi.org/10.3390/jcm5040044
APA StyleVu, T., Jin, L., & Datta, P. K. (2016). Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. Journal of Clinical Medicine, 5(4), 44. https://doi.org/10.3390/jcm5040044