
Osteopontin—A Master Regulator of Epithelial-Mesenchymal Transition

 ,
,
Abstract
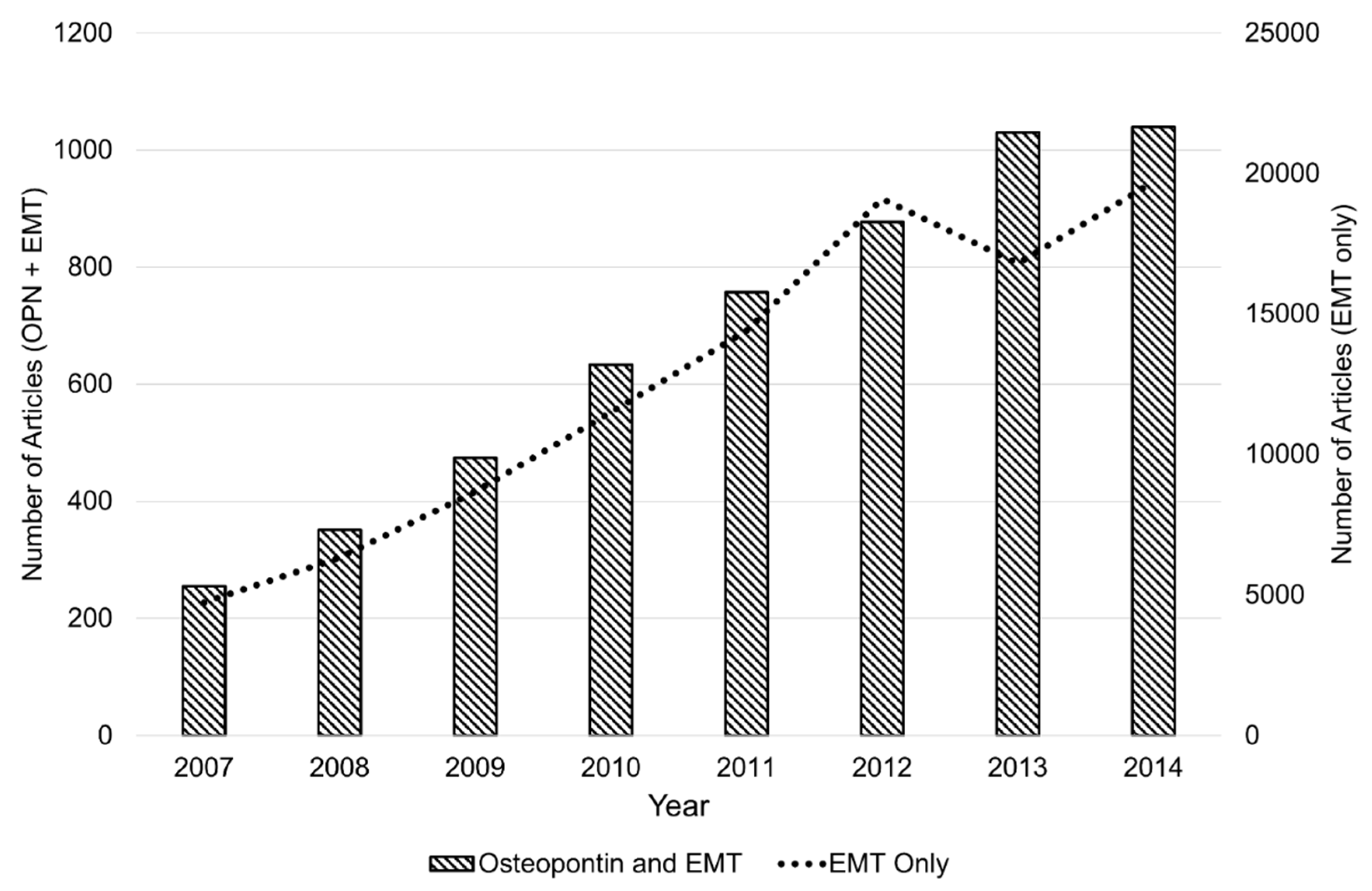
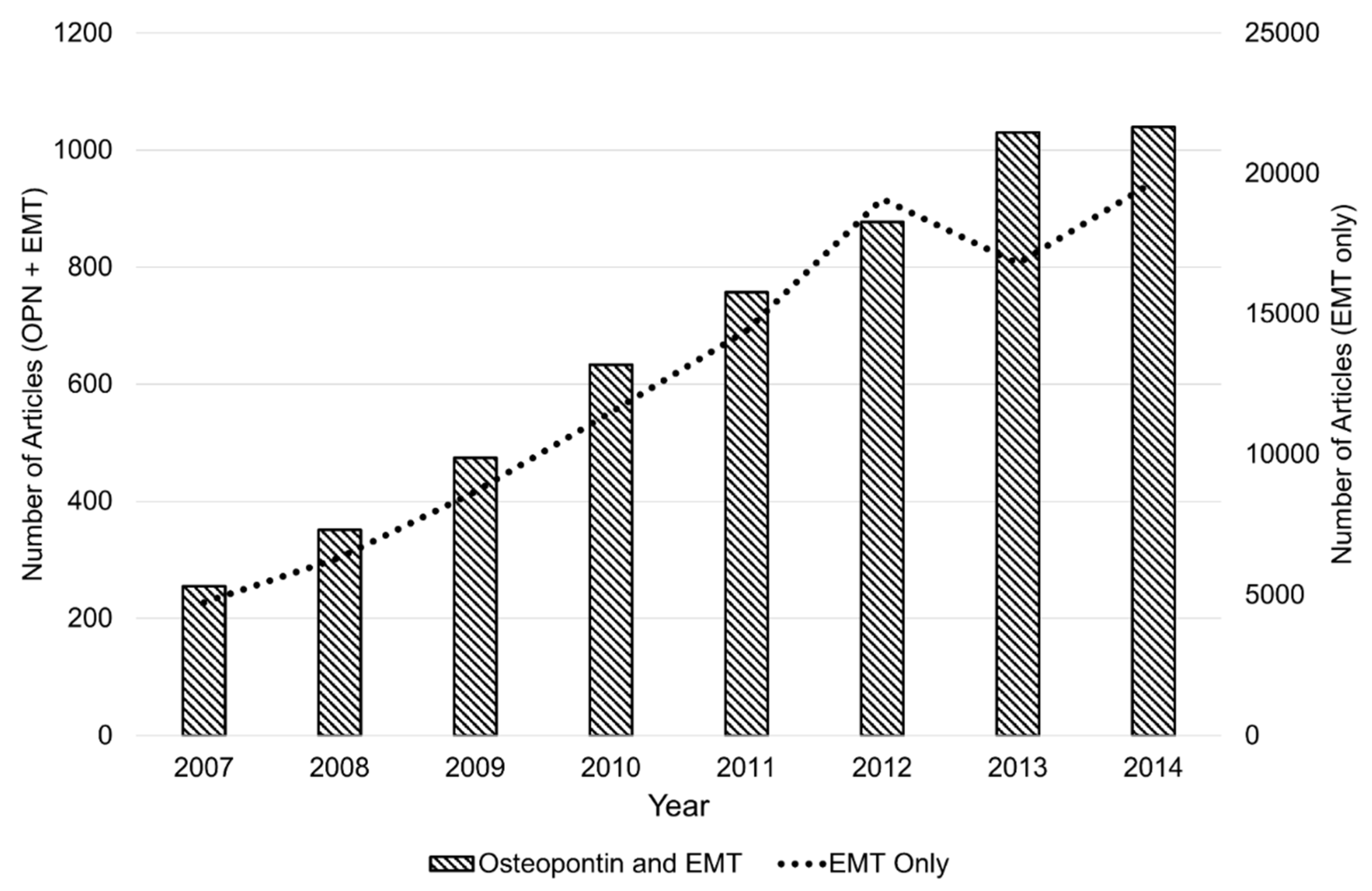
:1. Introduction
2. OPN Signaling and Transcriptional Regulation of EMT
2.1. Overview of Twist and EMT
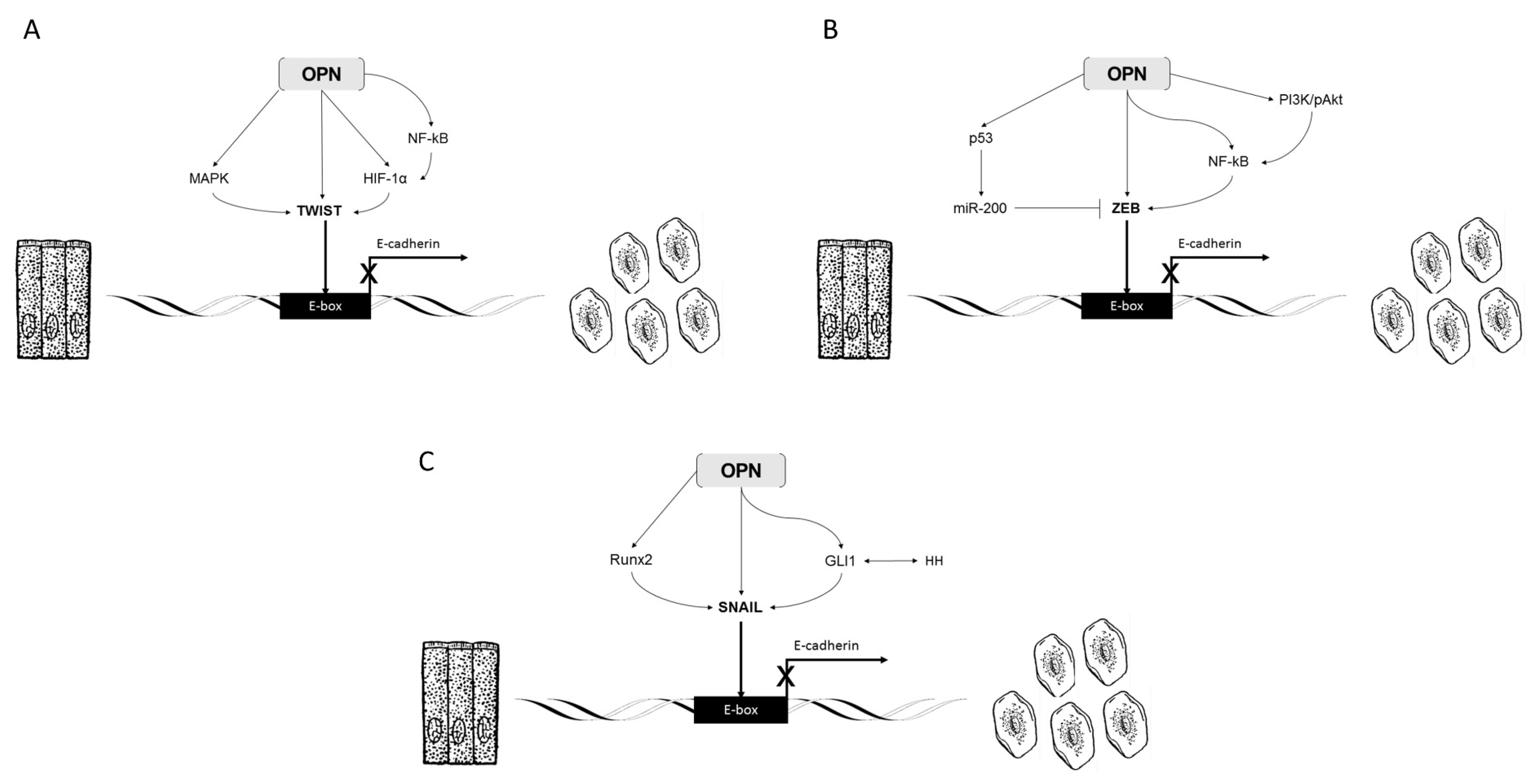
2.2. OPN, Twist, and EMT
2.3. Overview of ZEB and EMT
2.4. OPN, ZEB, and EMT
2.5. Overview of Snail and EMT
2.6. OPN, Snail, and EMT
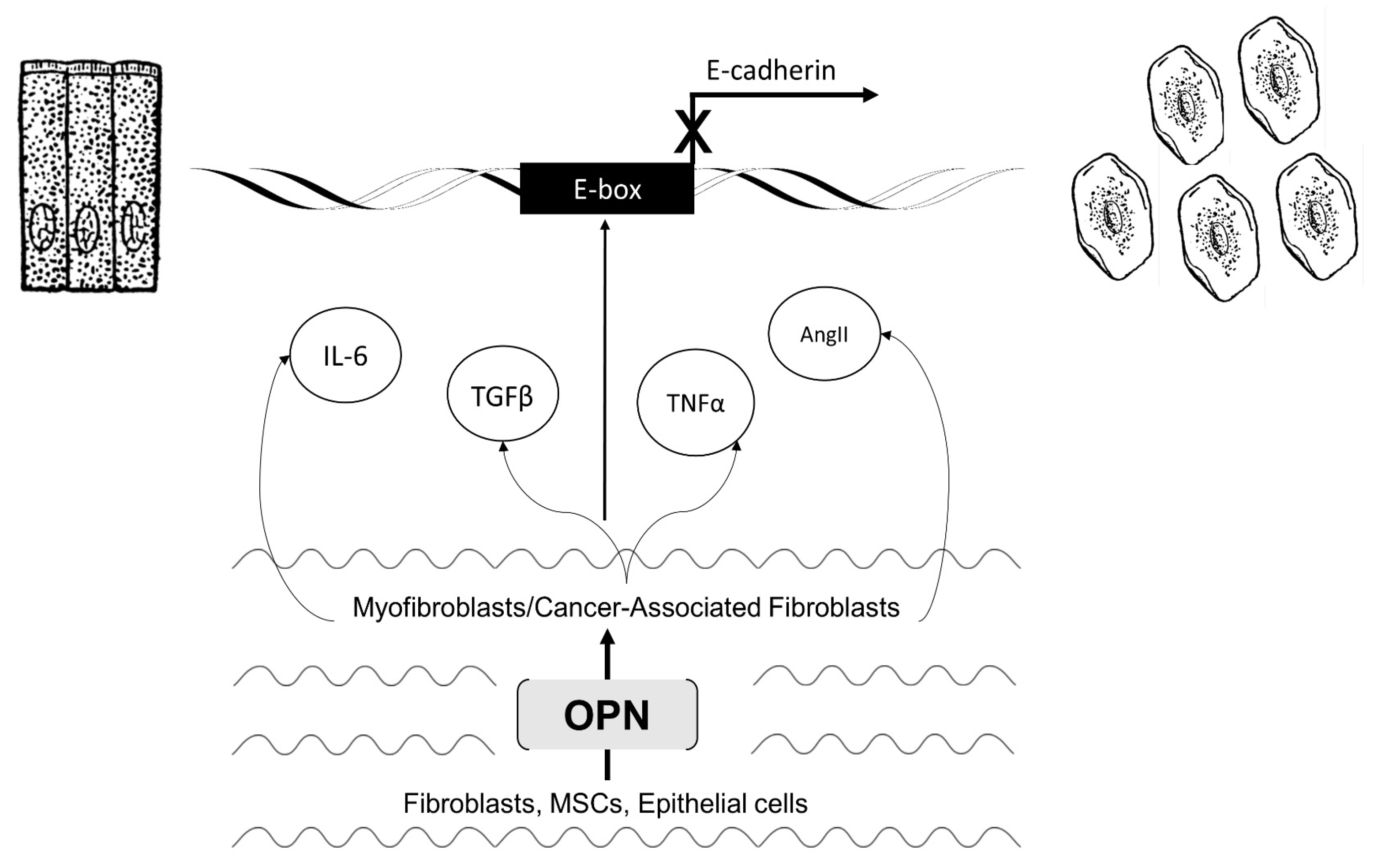
3. Section 2: OPN Modifies the Microenvironment to Regulate EMT
3.1. Myofibroblast Activation and Fibrosis
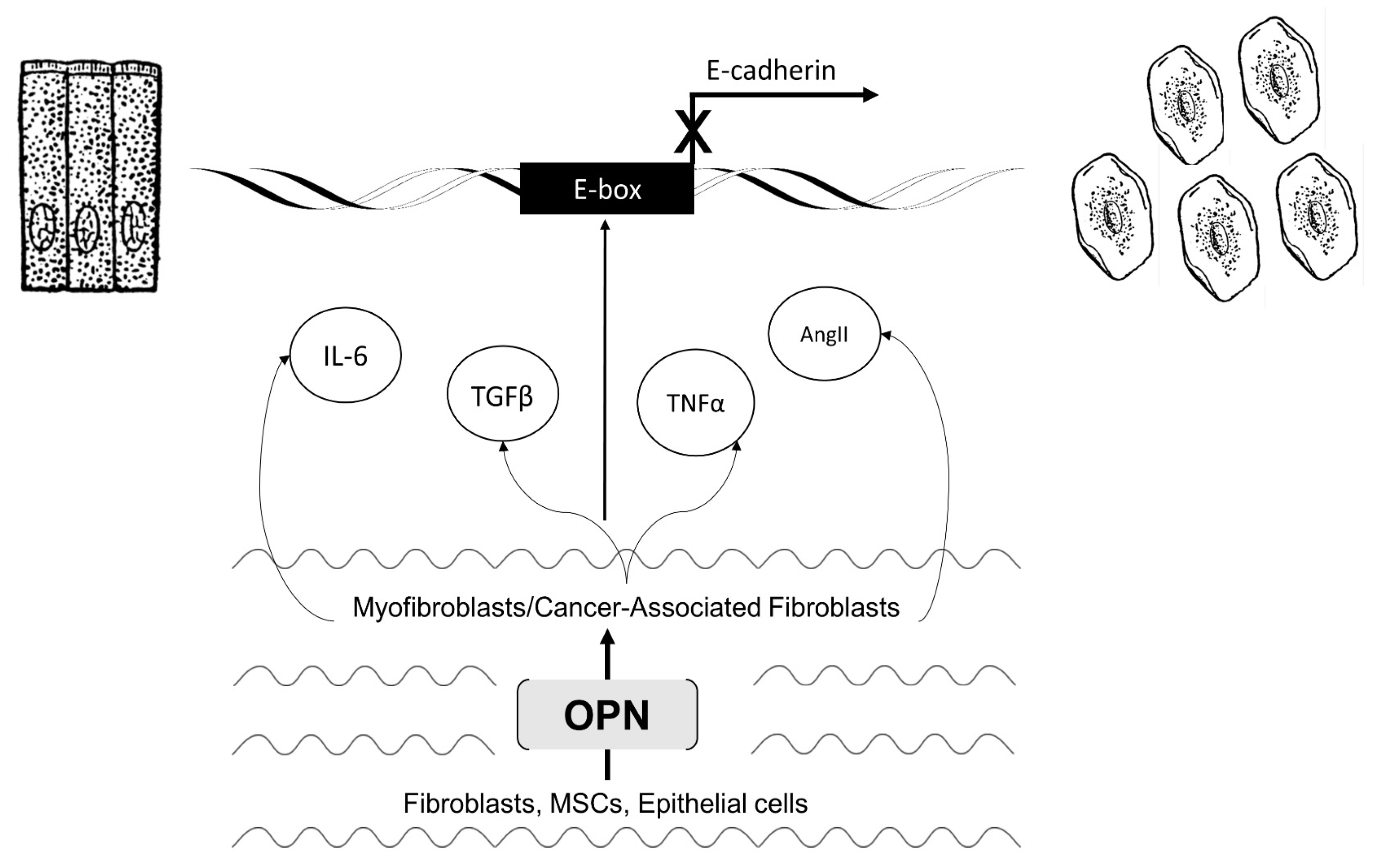
3.2. Activated Myofibroblasts Regulate EMT
3.3. Cancer-Associated Fibroblast Activation and Tumorigenicity
3.4. Activated CAFs Regulate EMT
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Senger, D.R.; Wirth, D.F.; Hynes, R.O. Transformed mammalian cells secrete specific proteins and phosphoproteins. Cell 1979, 16, 885–893. [Google Scholar] [CrossRef]
- Denhardt, D.T.; Guo, X. Osteopontin: A protein with diverse functions. FASEB J. 1993, 7, 1475–1482. [Google Scholar] [PubMed]
- Wai, P.Y.; Kuo, P.C. Osteopontin: Regulation in tumor metastasis. Cancer Metastasis Rev. 2008, 27, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009, 3, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Li, N.Y.; Wai, P.Y.; Kuo, P.C. Epithelial-mesenchymal transition, TGF-beta, and osteopontin in wound healing and tissue remodeling after injury. J. Burn Care Res. 2012, 33, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Wai, P.Y.; Kuo, P.C. The role of osteopontin in tumor metastasis. J. Surg. Res. 2004, 121, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, J.; Harvey, E.; Athwal, V.; Berry, A.; Rowe, C.; Oakley, F.; Moles, A.; Mann, D.A.; Bobola, N.; Sharrocks, A.D. Osteopontin is a novel downstream target of SOX9 with diagnostic implications for progression of liver fibrosis in humans. Hepatology 2012, 56, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.K.; Zheng, G.; Hsu, T.; Lee, S.R.; Zhang, J.; Zhao, Y.; Tian, X.; Wang, Y.; Wang, Y.M.; Cao, Q. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Laboratory Investig. 2013, 93, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell Cardiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Li, Z.; Jiang, X.; Lum, Y.L.; Khin, E.; Lee, N.P.; Wu, G.; Luk, J.M. Osteopontin as potential biomarker and therapeutic target in gastric and liver cancers. World J. Gastroenterol. 2012, 18, 3923. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Pan, C.; Hu, H.; Zheng, S.; Ding, L. Osteopontin-enhanced hepatic metastasis of colorectal cancer cells. PLoS ONE 2012, 7, e47901. [Google Scholar] [CrossRef] [PubMed]
- Hay, E. An overview of epithelio-mesenchymal transformation. Cells Tissues Organs (Print) 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. Leaving the neighborhood: Molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays 2001, 23, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Epithelial-mesenchymal transition: Untangling EMT's functions. Nat. Rev. Cancer 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Massagué, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Morel, A.; Hinkal, G.; Bastid, J.; Puisieux, A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene 2010, 29, 3173–3184. [Google Scholar] [CrossRef] [PubMed]
- Maestro, R.; Dei Tos, A.P.; Hamamori, Y.; Krasnokutsky, S.; Sartorelli, V.; Kedes, L.; Doglioni, C.; Beach, D.H.; Hannon, G.J. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999, 13, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Lapouge, G.; Rorive, S.; Drogat, B.; Desaedelaere, K.; Delafaille, S.; Dubois, C.; Salmon, I.; Willekens, K.; Marine, J. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 2015, 16, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.C.; Tozer, K.R.; Silber, J.R.; Mikheeva, S.; Deng, M.; Morrison, R.S.; Manning, T.C.; Silbergeld, D.L.; Glackin, C.A.; Reh, T.A. TWIST is expressed in human gliomas, promotes invasion. Neoplasia 2005, 7, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Hasselblatt, M.; Mertsch, S.; Koos, B.; Riesmeier, B.; Stegemann, H.; Jeibmann, A.; Tomm, M.; Schmitz, N.; Wrede, B.; Wolff, J.E.; et al. TWIST-1 is overexpressed in neoplastic choroid plexus epithelial cells and promotes proliferation and invasion. Cancer Res. 2009, 69, 2219–2223. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Poon, R.T.; Yuen, A.P.; Ling, M.T.; Kwok, W.K.; Wang, X.H.; Wong, Y.C.; Guan, X.Y.; Man, K.; Chau, K.L.; et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin. Cancer Res. 2006, 12, 5369–5376. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Yuen, H.F.; Chan, Y.P.; Wong, M.L.; Kwok, W.K.; Chan, K.K.; Lee, P.Y.; Srivastava, G.; Law, S.Y.; Wong, Y.C.; Wang, X.; et al. Upregulation of Twist in oesophageal squamous cell carcinoma is associated with neoplastic transformation and distant metastasis. J. Clin. Pathol. 2007, 60, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Li, P.; Song, H.; Song, F.; Qu, Y.; Ma, X.; Shi, R.; Wu, J. CXCL12/CXCR4 axis upregulates twist to induce EMT in human glioblastoma. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Chen, R.; Alvero, A.B.; Fu, H.; Holmberg, J.; Glackin, C.; Rutherford, T.; Mor, G. TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene 2010, 29, 3545–3553. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lowe, G.N.; Strong, D.D.; Wergedal, J.E.; Glackin, C.A. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. J. Cell. Biochem. 1999, 75, 566–577. [Google Scholar] [CrossRef]
- Li, N.Y.; Weber, C.E.; Mi, Z.; Wai, P.Y.; Cuevas, B.D.; Kuo, P.C. Osteopontin up-regulates critical epithelial-mesenchymal transition transcription factors to induce an aggressive breast cancer phenotype. J. Am. Coll Surg. 2013, 217, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Li, N.Y.; Weber, C.E.; Wai, P.Y.; Cuevas, B.D.; Zhang, J.; Kuo, P.C.; Mi, Z. An MAPK-dependent pathway induces epithelial-mesenchymal transition via Twist activation in human breast cancer cell lines. Surgery 2013, 154, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Giannini, E.; Antonacci, C.; Pistolese, C.A.; Spagnoli, L.G.; Bonanno, E. Microcalcifications in breast cancer: An active phenomenon mediated by epithelial cells with mesenchymal characteristics. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Hornik, C.; Brand-Saberi, B.; Rudloff, S.; Christ, B.; Füchtbauer, E. Twist is an integrator of SHH, FGF, and BMP signaling. Anat. Embryol. 2004, 209, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Zhu, X.; Dai, C.; Zhang, X.; Jia, H.; Ye, Q.; Qin, L. Osteopontin regulated epithelial-mesenchymal transition via PI3K/AKT signaling pathway in hepatocellular carcinoma. Cancer Res. 2013, 73, 2695–2695. [Google Scholar] [CrossRef]
- Yang, M.; Wu, M.; Chiou, S.; Chen, P.; Chang, S.; Liu, C.; Teng, S.; Wu, K. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Cai, Q.; Mao, Y.; Ming, Y.; Bao, S.; Ouyang, G. Osteopontin promotes ovarian cancer progression and cell survival and increases HIF-1α expression through the PI3-K/Akt pathway. Cancer Sci. 2008, 99, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Mao, Y.; Ming, Y.; Bao, S.; Hu, T. Osteopontin promotes gastric cancer metastasis by augmenting cell survival and invasion through Akt-mediated HIF-1α up-regulation and MMP9 activation. J. Cell. Mol. Med. 2009, 13, 1706–1718. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Fan, X.; Jing, W.; Liang, Y.; Chen, R.; Liu, Y.; Zhu, M.; Jia, R.; Wang, H.; Zhang, X.; et al. Osteopontin promotes a cancer stem cell-like phenotype in hepatocellular carcinoma cells via an integrin-NF-kappaB-HIF-1alpha pathway. Oncotarget 2015, 6, 6627–6640. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.; Wan, T.M.; Lam, C.S.; Chow, A.K.; Wong, S.K.; Man, J.H.; Li, H.; Cheng, N.S.; Pak, R.C.; Cheung, A.H. Post-operative plasma osteopontin predicts distant metastasis in human colorectal cancer. PLoS ONE 2015, 10, e0126219. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Boil. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Kang, Y. Multilayer control of the EMT master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A.; Depp, J.L.; Taylor, J.J.; Kroll, K.L. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Sharma, A.; Smith, J.J.; Krishnan, M.; Chen, X.; Eschrich, S.; Washington, M.K.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. Claudin-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells. Gastroenterology 2011, 141, 2140–2153. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Natsuizaka, M.; Wong, G.S.; Michaylira, C.Z.; Grugan, K.D.; Stairs, D.B.; Kalabis, J.; Vega, M.E.; Kalman, R.A.; Nakagawa, M.; et al. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res. 2010, 70, 4174–4184. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, S.; Ohashi, S.; Kimura, S.; Natsuizaka, M.; Gimotty, P.; Klein-Szanto, A.J.; Itoh, H.; Nakagawa, H. ZEB1 and ZEB2 promote EMT and invasion in esophageal squamous cell carcinoma. Cancer Res. 2010, 70, 2293–2293. [Google Scholar] [CrossRef]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, R.; Lopategi, A.; George, J.; Leung, T.; Lu, Y.; Wang, X.; Ge, X.; Fiel, M.I.; Nieto, N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin αVβ3 engagement and PI3K/pAkt/NFκB signaling. Hepatology 2012, 55, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Chua, H.; Bhat-Nakshatri, P.; Clare, S.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-κB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Baud, J.; Varon, C.; Chabas, S.; Chambonnier, L.; Darfeuille, F.; Staedel, C. Helicobacter pylori initiates a mesenchymal transition through ZEB1 in gastric epithelial cells. PLoS ONE 2013, 8, e60315. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Gnemmi, V.; Bouillez, A.; Gaudelot, K.; Hémon, B.; Ringot, B.; Pottier, N.; Glowacki, F.; Villers, A.; Vindrieux, D.; Cauffiez, C. MUC1 drives epithelial–mesenchymal transition in renal carcinoma through Wnt/β-catenin pathway and interaction with SNAIL promoter. Cancer Lett. 2014, 346, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Yin, F.; Fan, X.; Jing, W.; Chen, R.; Liu, L.; Zhang, L.; Liu, Y.; Liang, Y.; Bu, F. Decreased TIP30 promotes Snail-mediated epithelial–mesenchymal transition and tumor-initiating properties in hepatocellular carcinoma. Oncogene 2015, 34, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, ZEB and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.D.; Mi, Z.; Kim, V.M.; Guo, H.; Talbot, L.J.; Kuo, P.C. Osteopontin regulates epithelial mesenchymal transition-associated growth of hepatocellular cancer in a mouse xenograft model. Ann. Surg. 2012, 255, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; De Tribolet, N.; Radovanovic, I.; i Altaba, A.R. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Huang, S.; Yang, L.; Ma, X.; Xie, J.; Zhang, H. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J. Gastroenterol. 2007, 13, 1659. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Hernandez, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; i Altaba, A.R. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Lieu, A.; Wu, C.; Chang, L.; Loh, J.; Lin, R.; Chen, W.; Liao, H.; Fu, W.; Chang, C. Differential expression of hedgehog signaling components and Snail/E-cadherin in human brain tumors. Oncol. Rep. 2010, 24, 1225–1232. [Google Scholar] [PubMed]
- Li, X.; Deng, W.; Nail, C.D.; Bailey, S.K.; Kraus, M.H.; Ruppert, J.M.; Lobo-Ruppert, S.M. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 2006, 25, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Kita, Y.; Frank, D.J.; Majewski, R.R.; Konicek, B.A.; Nobrega, M.A.; Jacob, H.; Walterhouse, D.; Iannaccone, P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J. Biol. Chem. 2002, 277, 5548–5555. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Harris, L.G.; Metge, B.J.; Liu, S.; Riker, A.I.; Samant, R.S.; Shevde, L.A. The hedgehog pathway transcription factor GLI1 promotes malignant behavior of cancer cells by up-regulating osteopontin. J. Biol. Chem. 2009, 284, 22888–22897. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, B.; Liu, B.; Xie, Y.; Cao, X. Combined Runx2 and Snail overexpression is associated with a poor prognosis in breast cancer. Tumor. Biol. 2015, 36, 4565–4573. [Google Scholar] [CrossRef] [PubMed]
- Inman, C.K.; Shore, P. The osteoblast transcription factor Runx2 is expressed in mammary epithelial cells and mediates osteopontin expression. J. Biol. Chem. 2003, 278, 48684–48689. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Christakos, S. The vitamin D receptor, Runx2, and the Notch signaling pathway cooperate in the transcriptional regulation of osteopontin. J. Biol. Chem. 2005, 280, 40589–40598. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Claridge, L.C.; Jhaveri, R.; Swiderska-Syn, M.; Clark, P.; Suzuki, A.; Pereira, T.A.; Mi, Z.; Kuo, P.C.; Guy, C.D.; et al. Osteopontin is up-regulated in chronic hepatitis C and is associated with cellular permissiveness for hepatitis C virus replication. Clin. Sci. (Lond.) 2014, 126, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; McRae, S.; Mai, T.; Banaudha, K.; Sarkar-Dutta, M.; Waris, G. Role of hepatitis C virus induced osteopontin in epithelial to mesenchymal transition, migration and invasion of hepatocytes. PLoS ONE 2014, 9, e87464. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Y.; Xu, Y.; Voorhees, J.J.; Fisher, G.J. UV irradiation induces Snail expression by AP-1 dependent mechanism in human skin keratinocytes. J. Dermatol. Sci. 2010, 60, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.D.; Swiderska-Syn, M.; Dolle, L.; Reid, D.; Eksteen, B.; Claridge, L.; Briones-Orta, M.A.; Shetty, S.; Oo, Y.H.; Riva, A.; et al. Osteopontin neutralisation abrogates the liver progenitor cell response and fibrogenesis in mice. Gut 2015, 64, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Lenga, Y.; Koh, A.; Perera, A.S.; McCulloch, C.A.; Sodek, J.; Zohar, R. Osteopontin expression is required for myofibroblast differentiation. Circ. Res. 2008, 102, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Kohan, M.; Breuer, R.; Berkman, N. Osteopontin induces airway remodeling and lung fibroblast activation in a murine model of asthma. Am. J. Respir. Cell Mol. Biol. 2009, 41, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Seo, M.W.; Kim, Y.W.; Lee, D. Osteopontin Potentiates Pulmonary Inflammation and Fibrosis by Modulating IL-17/IFN-γ-secreting T-cell Ratios in Bleomycin-treated Mice. Immune Netw. 2015, 15, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med. 2005, 2. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis–from bench to bedside. J. Hepatol. 2003, 38, 38–53. [Google Scholar] [CrossRef]
- Ginès, P.; Cárdenas, A.; Arroyo, V.; Rodés, J. Management of cirrhosis and ascites. N. Engl. J. Med. 2004, 350, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.L.; Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.; Wang, X.; Kitamura, N.; Fiel, M.I.; Nieto, N. Osteopontin delays resolution of liver fibrosis. Lab. Investig. 2013, 93, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, Y.; Ikeda, H.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Sphingosine-1-phosphate as a mediator involved in development of fibrotic diseases. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Kilter, H.; Konkol, C.; Wassmann, S.; Bohm, M.; Nickenig, G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc. Res. 2002, 53, 911–920. [Google Scholar] [CrossRef]
- Guarino, M.; Tosoni, A.; Nebuloni, M. Direct contribution of epithelium to organ fibrosis: Epithelial-mesenchymal transition. Hum. Pathol. 2009, 40, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, M.; Fushida, S.; Harada, S.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Tajima, H.; Ninomiya, I.; Fujimura, T.; Ohta, T. The Angiotensin II type 1 receptor blocker candesartan suppresses proliferation and fibrosis in gastric cancer. Cancer Lett. 2014, 355, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Zhou, X.; Cui, J.; Zou, Z.; Xu, Y.; You, D. Role of complement 3 in TNF-α-induced mesenchymal transition of renal tubular epithelial cells in vitro. Mol. Biotechnol. 2013, 54, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Arito, M.; Suematsu, N.; Kamijo-Ikemori, A.; Omoteyama, K.; Sato, T.; Kurokawa, M.S.; Okamoto, K.; Kimura, K.; Shibagaki, Y. Roles of layilin in TNF-α-induced epithelial-mesenchymal transformation of renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 2015, 467, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Böttinger, E.P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Saika, S.; Kono-Saika, S.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Flanders, K.C.; Yoo, J.; Anzano, M.; Liu, C. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 2004, 164, 651–663. [Google Scholar] [CrossRef]
- Flanders, K.C.; Wakefield, L.M. Transforming growth factor-βs and mammary gland involution; functional roles and implications for cancer progression. J. Mammary Gland Biol. Neoplasia 2009, 14, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Zhang, J.; Peng, X.; Dong, Y.; Jia, L.; Li, H.; Du, J. The Notch γ-secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-β/Smad2/3 signaling pathway activation. Int. J. Biochem. Cell Biol. 2014, 55, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, S.; Qi, D.; Zheng, S.; Guo, J.; Zhang, S.; Weng, Z. Inhibition of Notch signaling by a γ-secretase inhibitor attenuates hepatic fibrosis in rats. PLoS ONE 2012, 7, e46512. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.; Deacon, K.; Pang, L. Activation Of Epidermal Growth Factor Receptor (EGFR) Is Required For Tgfβ1-Induced Epithelial-Mesenchymal Transition (EMT) In Idiopathic Pulmonary Fibrosis (IPF). Am. J. Respir. Crit. Care Med. 2013, 187. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Kothari, A.N.; Mi, Z.; Zapf, M.; Kuo, P.C. Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clin. Transl. Med. 2014, 3, 14–35. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Kothari, A.; Wai, P.; Li, N.; Driver, J.; Zapf, M.; Franzen, C.; Gupta, G.; Osipo, C.; Zlobin, A. Osteopontin mediates an MZF1–TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2014, 34, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.; Weber, C.E.; Callaci, J.J.; Kothari, A.N.; Zapf, M.A.; Roper, P.M.; Borys, D.; Franzen, C.A.; Gupta, G.N.; Wai, P.Y.; et al. Alcohol inhibits osteopontin-dependent transforming growth factor-beta1 expression in human mesenchymal stem cells. J. Biol. Chem. 2015, 290, 9959–9973. [Google Scholar] [CrossRef] [PubMed]
- Sharon, Y.; Raz, Y.; Cohen, N.; Ben-Shmuel, A.; Schwartz, H.; Geiger, T.; Erez, N. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. 2015, 75, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.; Tan, L.; Wang, Q.; Li, X.; Feng, Y. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Ariyama, H.; Tamura, S.; Isobe, T.; Miyawaki, K.; Okumura, Y.; Kusaba, H.; Ueki, T.; Baba, E.; Akashi, K. Plasticity of CD44 colorectal cancer stem cells depends on TGF-beta-induced epithelial mesenchymal transition (EMT): Evidences from ex vivo culture system. Cancer Res. 2015, 75. [Google Scholar] [CrossRef]
- Pore, M.M.; Buikema, L.; Hiltermann, T.; Kruyt, F. TGF beta-mediated epithelial to mesenchymal transition in non small cell lung cancer: Effects on stemness, invasiveness and chemotherapy sensitivity. Cancer Res. 2012, 72. [Google Scholar] [CrossRef]
- Liu, F.; Kong, X.; Lv, L.; Gao, J. TGF-β1 acts through miR-155 to down-regulate TP53INP1 in promoting epithelial–mesenchymal transition and cancer stem cell phenotypes. Cancer Lett. 2015, 359, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Wu, X.; Zhou, Q.; Wang, C.; Liu, B. Cancer-associated fibroblast promotes gastric cancer invasion and epithelial-mesenchymal transition via the IL-6/JAK/STAT3 signaling pathway. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Zhang, H.; Wen, Z.; Gu, Y.; Cheng, Y.; Sun, Y.; Zhang, T.; Jia, C.; Lu, Z.; Chen, J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014, 345, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Mao, Y.; Wang, J.; Zu, L.; Hao, M.; Cheng, G.; Qu, Q.; Cui, D.; Keller, E.; Chen, X. IL-6 secreted by cancer-associated fibroblasts induces tamoxifen resistance in luminal breast cancer. Oncogene 2014. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Pathway | Model | Mechanism |
|---|---|---|
| Twist | Osteoblast-like | Twist upregulation causes OPN mRNA upregulation |
| Breast Cancer | OPN/Twist/Bmi-1 pathway activates EMT programs | |
| Hepatocellular Carcinoma | OPN activates PI3K/AKT/Twist pathway leading to EMT | |
| Ovarian Cancer | OPN upregulates HIF-1α through PI3K/AKT pathway, upregulating Twist | |
| Gastric Cancer | OPN upregulates HIF-1 α through PI3K/AKT pathway, upregulating Twist | |
| Colorectal Cancer | High OPN secreting cell lines interact with Twist enhances metastasis | |
| ZEB | Breast Cancer | OPN activates NFκB and increases ZEB1 and 2 to induce EMT |
| Gastric Epithelial Cells | OPN activates NFκB, which transactivates ZEB1 to induce EMT in H. Pylori infection | |
| Liver Cells | OPN activates PI3K/pAkt/NFkB-signaling to cause liver fibrosis | |
| Hepatocellular Carcinoma | OPN interacts with p53, which upregulates miR-200 family to downregulate ZEB1 and ZEB2 to suppress metastasis | |
| Snail | Breast Cancer | OPN expression cause EMT through overexpression of Snail |
| Breast Cancer | OPN interacts with Runx2, Runx/Snail positive tumors exhibit EMT and increased malignancy | |
| Hepatocellular Carcinoma | OPN expression cause EMT through overexpression of Snail | |
| Skin Cancer | OPN and GLI1 are coregulated, GLI1 induces Snail1 and promotes EMT | |
| Brain Tumor | OPN and GLI1 are coregulated, GLI1 induces Snail1 and promotes EMT | |
| Nonalcoholic Steatohepatitis | OPN and GLI1 are coregulated, GLI1 induces Snail1 and promotes EMT | |
| Melanoma | OPN and GLI1 are coregulated, GLI1 induces Snail1 and promotes EMT |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kothari, A.N.; Arffa, M.L.; Chang, V.; Blackwell, R.H.; Syn, W.-K.; Zhang, J.; Mi, Z.; Kuo, P.C. Osteopontin—A Master Regulator of Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 39. https://doi.org/10.3390/jcm5040039
Kothari AN, Arffa ML, Chang V, Blackwell RH, Syn W-K, Zhang J, Mi Z, Kuo PC. Osteopontin—A Master Regulator of Epithelial-Mesenchymal Transition. Journal of Clinical Medicine. 2016; 5(4):39. https://doi.org/10.3390/jcm5040039
Chicago/Turabian StyleKothari, Anai N., Matthew L. Arffa, Victor Chang, Robert H. Blackwell, Wing-Kin Syn, Jiwang Zhang, Zhiyong Mi, and Paul C. Kuo. 2016. "Osteopontin—A Master Regulator of Epithelial-Mesenchymal Transition" Journal of Clinical Medicine 5, no. 4: 39. https://doi.org/10.3390/jcm5040039
APA StyleKothari, A. N., Arffa, M. L., Chang, V., Blackwell, R. H., Syn, W.-K., Zhang, J., Mi, Z., & Kuo, P. C. (2016). Osteopontin—A Master Regulator of Epithelial-Mesenchymal Transition. Journal of Clinical Medicine, 5(4), 39. https://doi.org/10.3390/jcm5040039