
Role of EMT in Metastasis and Therapy Resistance


{kind=link}
{kind=link}
Abstract
:1. EMT Classifications
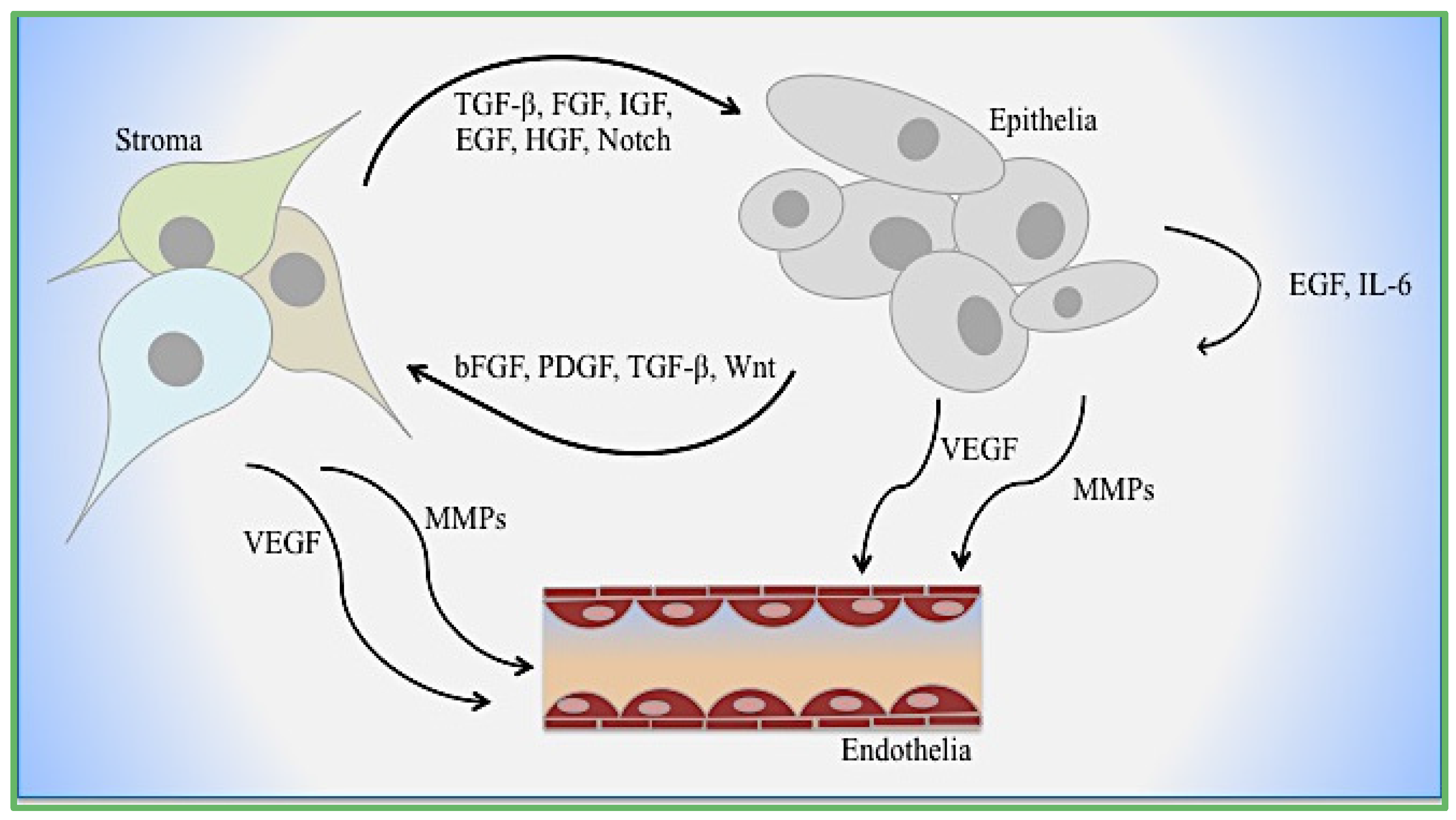
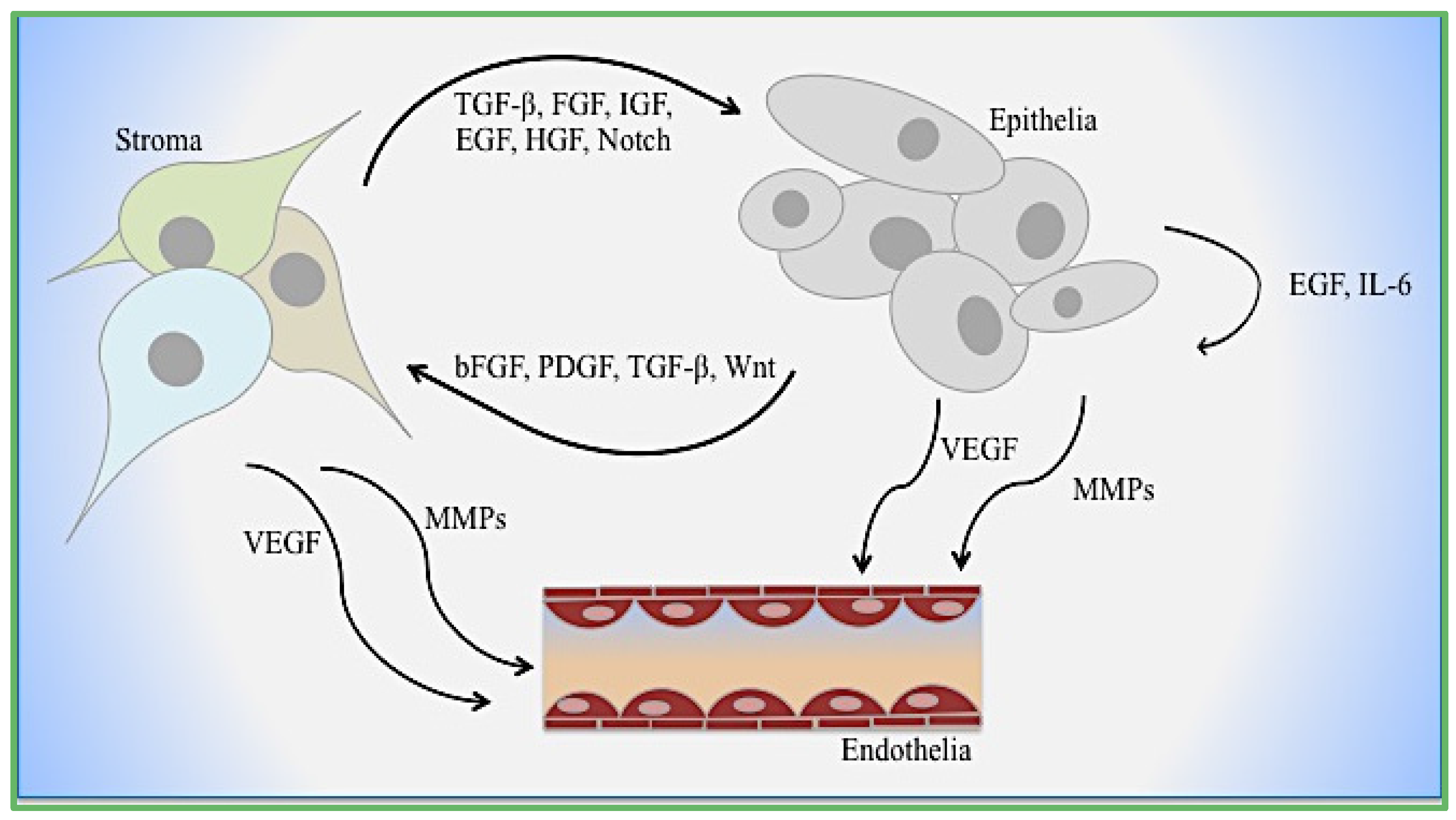
2. EMT Mediators: Tumor and Microenvironment Crosstalk
3. EMT and Tumor Metastasis
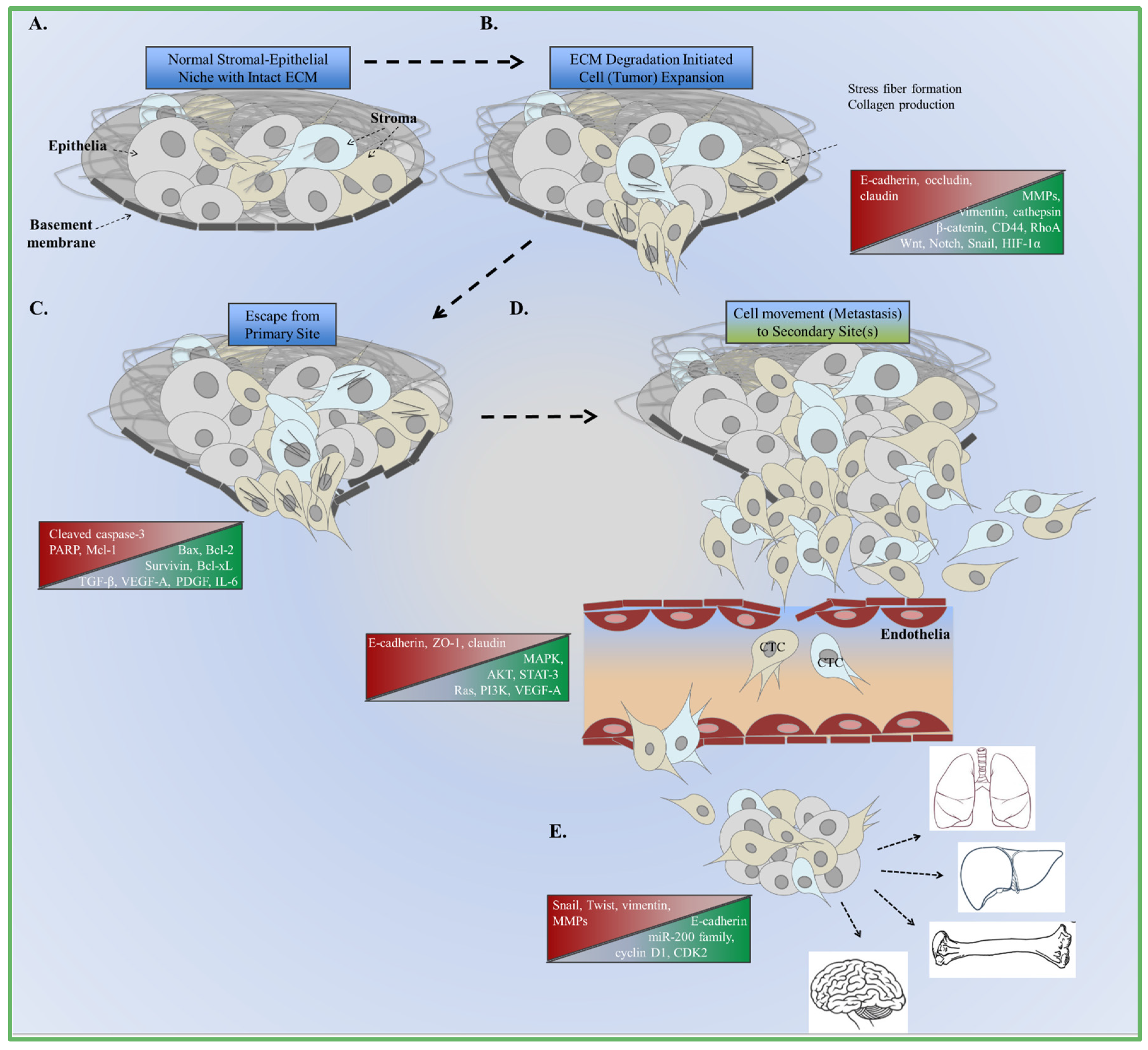
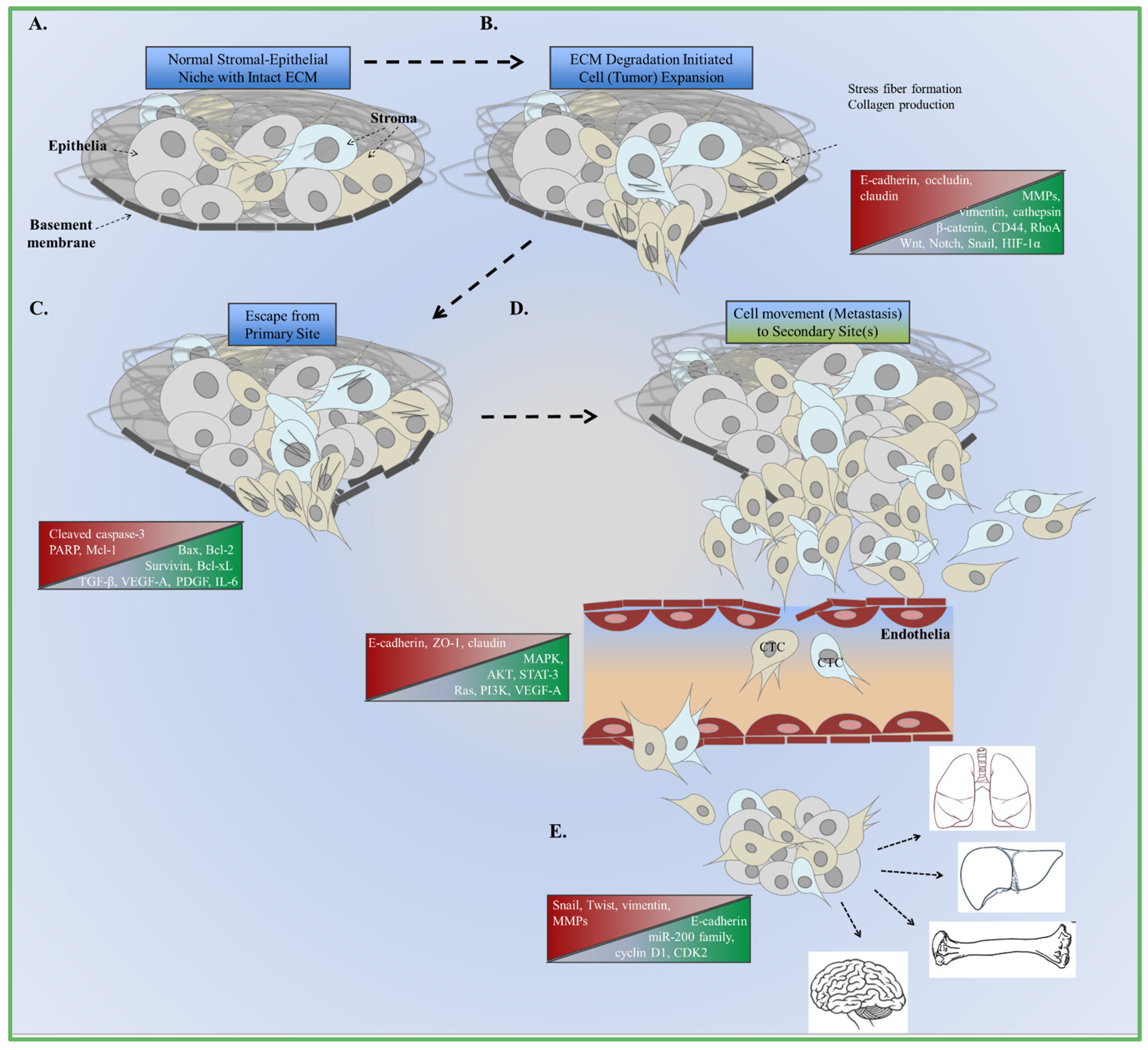
4. Cooperative EMT Signaling Mechanisms
5. Role of EMT in Therapeutic Resistance
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| EMT | Epithelial mesenchymal transition |
| TGF-β | Transforming growth factor β |
| EGF | Epidermal growth factor |
| MET | Mesenchymal-epithelial transition |
| MAPK | Mitogen activated protein kinase |
| ECM | Extracellular matrix |
| VEGF-A | Vascular endothelial growth factor-A |
| NED | Neuroendocrine differentiation |
| RANKL | Receptor activator of NFκB ligand |
| TME | Tumor microenvironment |
| PDGF | Platelet-derived growth factor |
| CAF | Cancer-associated fibroblast |
| NAF | Normal-associated fibroblast |
| FSP-1 | Fibroblast-specific protein-1 |
| LNCaP | Prostate cancer cells from left supraclavicular lymph node |
| PI3K | Phosphatidylinositol-3 kinase |
| IL-6 | Pro-inflammatory cytokine interleukin-6 |
| STC-1 | Stanniocalcin-1 |
| AR | Androgen receptor |
| ZEB-1 | Zinc finger E-box-binding homeobox 1 |
| ARCaPM | Androgen Repressed Metastatic Human Prostate Cancer Cell Line |
| UO126 | MEK1 and MEK2 selective inhibitor |
| MMP | Matrix metalloproteinase |
| BMP | Bone morphogenetic protein |
| HGF | Hepatocyte growth factor |
| FGF | Fibroblast growth factor |
| CTC | Circulating tumor cells |
| PAR1 | Protease-activated receptor-1 |
| α-SMA | α-smooth muscle actin |
| MSC | Mesenchymal stem cells |
| ROS | Reactive oxygen species |
| HNSCC | Head and neck squamous cell cancer |
| OSCC | Oral squamous cell cancer |
| FAK | Focal adhesion kinase |
| CSC | Cancer stem cells |
| ALDH1 | Aldehyde dehydrogenase 1 |
| CXCR4 | chemokine (C-X-C motif) receptor 4 |
| uPA | Urokinase plasminogen activator |
| IGF | Insulin growth factor |
| OC | Osteocalcin |
| BSP | Bone sialoprotein |
| OPN | Osteopontin |
| OPG | Osteoprotegrin |
| PTEN | Phosphatase and tensin homolog |
| NSCLC | Non-small-cell lung cancer |
References
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition. From: Cardiovascular Development to Disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Invest. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Die Cellular Pathologie in Ibrer Begruendung auf Physiologische und Pathologische Gewebelebre; August Hirschwald: Berlin, Germany, 1858. [Google Scholar]
- Arnoux, V.; Come, C.; Kusewitt, D.F.; Hudson, L.G.; Savagner, P. Cutaneous wound reepithelialization: A partial and reversible EMT. In Rise and Fall of Epithelial Phenotype: Concepts of Epithelial-Mesenchymal Transition; Savagner, P., Ed.; Springer: New York, NY, USA, 2005; pp. 111–134. [Google Scholar]
- Antsiferova, M.; Werner, S. The bright and dark sides of activin in wound healing and cancer. J. Cell Sci. 2012, 125, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, V.; Schoenwolf, G.C. Primitive-streak origin of the cardiovascular system in avian embryos. Dev. Biol. 1993, 159, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Kaissling, B.; le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Invest. 2011, 121, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.; Acloque, H.; Huang, R.Y.J.; Angela Nieto, M. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Neal, C.L.; Mckeithen, D.; Odero-Marah, V.A. Snail negatively regulates cell adhesion to extracellular matrix and integrin expression via the MAPK pathway in prostate cancer cells. Cell Adh. Migr. 2015, 5, 249–257. [Google Scholar] [CrossRef]
- Neal, C.L.; Henderson, V.; Smith, B.N.; Mckeithen, D.; Graham, T.; Vo, B.T.; Odero-Marah, V.A. Snail transcription factor negatively regulates maspin tumor suppressor in human prostate cancer cells. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Baala, L.; Brialt, S.; Etchevers, H.C.; Laumonnier, F.; Natiq, A.; Amiel, J.; Boddaert, N.; Picard, C.; Sbiti, A.; Asermouh, A.; et al. Homozygous silencing T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 2007, 39, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, X.-Y.; Willis, A.L.; Liu, C.; Chen, G.; Weiss, S.J. Snail1-dependent control of embryonic stem cell pluripotency and lineage commitment. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Sokol, S.Y. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development 2011, 138, 4341–4350. [Google Scholar] [CrossRef] [PubMed]
- Ten Berge, D.; Kurek, D.; Blauwkamp, T.; Koole, W.; Maas, A.; Eroglu, E.; Siu, R.K.; Nusse, R. Embryonic stem cells require Wnt proteins to prevent differentation to epiblast stem cells. Nat. Cell Biol. 2011, 13, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the microRNA-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriciton in differentiating ESCs. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Samatov, T.R.; Tonevitsky, A.G.; Schumacher, U. Epithelial-mesenchymal transition: Focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Duangkumpha, K.; Techasen, A.; Loilome, W.; Namwat, N.; Thanan, R.; Khuntikeo, N.; Yongvanit, P. BMP-7 blocks the effects of TGF-β-induced EMT in cholangiocarcinoma. Tumour Biol. 2014, 35, 9667–9676. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Oh, S.C.; Lee, D.H.; Kim, J.L.; Lee, S.Y.; Kang, M.H.; Lee, S.I.; Kang, S.; Joung, S.Y.; Min, B.W. BMP-2 induces motility and invasiveness by promoting colon cancer stemness through STAT3 activation. Tumour Biol. 2015, 36, 9475–9486. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.P.; Parameswaran, M.; Kinder, S.J.; Weinberger, R.P. The allocation of epiblast cells to the embryonic heart and other mesodermal lineages: The role of ingression and tissue movement during gastrulation. Development 1997, 124, 1631–1642. [Google Scholar] [PubMed]
- Kissa, K.; Herbomel, P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 2010, 464, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Rastaldi, M.P.; Ferrario, F.; Giardino, L.; Dell’Antonio, G.; Grillo, C.; Grillo, P.; Strutz, F.; Muller, G.A.; Colasanti, G.; D’Amico, G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002, 62, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Shah, A.A.; Kalluri, R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J. Biol. Chem. 2005, 280, 8094–8100. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Alpers, C.E.; Seifert, R.A.; Hudkins, K.L.; Johnson, R.J.; Bowen-Pope, D.F. PDGF-receptor localizes to mesangial, parietal epithelial, and interstitial cells in human and primate kidneys. Kidney Int. 1993, 43, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.; Skalli, O.; Gabbiani, G. α-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab. Invest. 1990, 63, 21–30. [Google Scholar] [PubMed]
- Dawson, T.P.; Gandhi, R.; Le Hir, M.; Kaissling, B. Ecto-5′-nucleotidase: Localization in rat kidney by light microscopic histochemical methods. J. Histochem. Cytochem. 1989, 37, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Liau, G.; Yamada, Y.; de Crombrugghe, B. Coordinate regulation of the levels of type III and type I collagen mRNA in most but not all mouse fibroblasts. J. Biol. Chem. 1985, 260, 531–536. [Google Scholar] [PubMed]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [PubMed]
- Venkov, C.; Link, A.J.; Jennings, J.L.; Plieth, D.; Inoue, T.; Nagai, K.; Xu, C.; Dimitrova, Y.N.; Rauscher, F.J.; Neilson, E.G. A proximal activator of transcription in epithelial-mesenchymal transition. J. Clin. Invest. 2007, 117, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Zent, R.; Ghiassi, M.; McDonnell, M.; Moses, H.L. Integrin beta 1 signaling is necessary for transforming growth factor-β activation of p38MAPK and epithelial plasticity. J. Biol. Chem. 2001, 276, 46707–46713. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Nabi, I.R. Actin cytoskeletal regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Burton, L.J.; Henderson, V.; Randle, D.D.; Morton, D.J.; Smith, B.A.; Taliaferro-Smith, L.; Nagappan, P.; Yates, C.; Zayzafoon, M.; et al. Snail promotes epithelial mesenchymal transition in breast cancer cells in part via activation of nuclear ERK2. PLoS ONE 2014, 9, e104987. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, M.; Pozzi, A.; Bhowmick, N.; Moses, H.L.; Zent, R. Transforming growth factor-beta (TGF-β) and TGF-β-associated kinase 1 are required for R-Ras-mediated transformation of mammary epithelial cells. Cancer Res. 2008, 68, 6224–6231. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Begai, S.; Jakkaraju, S.; Mattingly, R.R.; Pan, D.; Schuger, L. High RhoA activity maintains the undifferentiated mesenchymal cell phenotype, whereas RhoA down-regulation by laminin-2 induces smooth muscle myogenesis. JCB 2002, 156, 893–903. [Google Scholar]
- Yang, Y.; Yang, C.; Zhang, J. C23 protein meditates bone morphogenetic protein-2-mediated EMT via up-regulation of Erk1/2 and Akt in gastric cancer. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef] [PubMed]
- Liao, A.; Wang, W.; Sun, D.; Jiang, Y.; Tian, S.; Li, J.; Yang, X.; Shi, R. Bone morphogenetic protein 2 mediates epithelial-mesenchymal transition via AKT and ERK signaling pathways in gastric cancer. Tumour Biol. 2015, 36, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-α through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, A.; Riazi, G.H.; Azizi, E.; Amanpour, S.; Muhammadnejad, S.; Haddadi, M.; Zekri, A.; Shirkoohi, R. FGF10: Type III epithelial mesenchymal transition and invasion in breast cancer cell lines. J. Cancer 2014, 5, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wu, H.; Zhang, M.; Ding, L.; Meng, F.; Fan, X. Expression of the epithelial-mesenchymal transition-related proteins and their clinical significance in lung adenocarcinoma. Diagn. Pathol. 2013, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Uchikado, Y.; Okumura, H.; Ishigami, S.; Setoyama, T.; Matsumoto, M.; Owaki, T.; Kita, Y.; Natsugoe, S. Increased Slug and decreased E-cadherin expression is related to poor prognosis in patients with gastric cancer. Gastric Cancer. 2011, 14, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Jain, A.K.; Ramteke, A.; Ting, H.; Vijendra, K.C.; Gangar, S.C.; Agarwal, C.; Agarwal, R. Snai1 is critical for the aggressiveness of prostate cancer cells with low E-cadherin. Mol. Cancer. 2014, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tan, Y.; Ren, Y.; Dong, L.; Xie, Z.; Tang, L.; Cao, D.; Zhang, W.; Hu, H.; Wang, H. Zinc finger protein ZBTB20 expression is increased in hepatocellular carcinoma and associated with poor prognosis. BMC Cancer 2011, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, D.; Kajita, M.; Wade, P.A. The transcription factor Snail mediates epithelial to mesenchymal transition by repression of estrogen receptor-alpha. Mol. Endocrinol. 2007, 21, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, A.; Phadke, D.; Mav, D.; Shah, R.R.; Wade, P.A. The transcription factors of Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS ONE 2011, 6, e26514. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Q.; Fan, L.Q.; Wang, L.L.; Tan, B.B.; Leng, Y.L.; Liu, Y.; Wang, D. Zinc finger protein 139 expression in gastric cancer and its clinical significance. World J. Gastroenterol. 2014, 20, 18346–18353. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Jiang, H.; Ying, G.; Dong, Q.; Zhang, R.; Yu, J.; Fan, D.; Hao, X. Poor survival is associated with the methylated degree of zing-finger protein 545 (ZNF545) DNA promoter in gastric cancer. Oncotarget 2015, 6, 4482–4495. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Hirohashi, S.; Kanai, Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci. 2003, 94, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Pantel, K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastab. Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Valles, A.M.; Jouanneau, J.; Yamada, K.M.; Thiery, J.P. Alternative splicing fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol. Biol. Cell 1994, 5, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Nabi, I.R. Actin cytoskeletal regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar]
- Chaw, S.Y.; Majeed, A.; Dalley, A.J.; Chan, A.; Stein, S.; Farah, C.S. Epithelial to mesenchymal transition (EMT) biomarkers—E-cadherin, beta-catenin, APC and Vimentin—In oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012, 48, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, M.; Pozzi, A.; Bhowmick, N.; Moses, H.L.; Zent, R. Transforming growth factor-beta (TGF-β) and TGF-β-associated kinase I are required for R-Ras-mediated transformation of mammary epithelial cells. Cancer Res. 2008, 68, 6224–6231. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Sheng, T.; Stelter, A.A.; Li, C.; Zhang, X.; Sinha, M.; Luxon, B.A.; Xie, J. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J. Biol. Chem. 2006, 281, 35598–33602. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.; Cespedes, M.V.; Lindh, M.B.; Kiflemariam, S.; Mezheyeuski, A.; Edgvist, P.H.; Hägglöf, C.; Birgisson, H.; Bojmar, L.; Jirström, K.; et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013, 73, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Sneddon, J.B.; Alizadeh, A.A.; Sood, R.; West, R.B.; Montgomery, K.; Chi, J.-T.; van de Rijn, M.; Botstein, D.; Brown, P.O. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004, 2, E7. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression profiles clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, J.; Chanreshekar, C.; Radharkrishnan, R. Critical biomarkers of epithelial-mesenchymal transition in the head and neck cancers. J. Cancer Res. Ther. 2014, 10, 512–518. [Google Scholar] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Keirsebilck, A.; Bonne, S.; Bruyneel, E.; Vermassen, P.; Lukanidin, E.; Mareel, M.; van Roy, F. E-cadherin and metastasin (mts-1/S100A4) expression levels are inversely regulated in two tumor cell families. Cancer Res. 1998, 58, 4587–4591. [Google Scholar] [PubMed]
- Noe, V.; Fingleton, B.; Jacobs, K.; Crawford, H.C.; Vermeulen, S.; Steelant, W.; Bruyneel, E.; Matrisian, L.M.; Mareel, M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 2001, 114, 111–118. [Google Scholar] [PubMed]
- Ramos, D.M.; Dang, D.; Sadler, S. The role of the integrin αvβ6 in regulating the epithelial to mesenchymal transition in oral cancer. Anticancer Res. 2009, 29, 125–130. [Google Scholar] [PubMed]
- Dang, D.; Bamburg, J.R.; Ramos, D.M. αvβ3 integrin and cofilin molecule modulate K1735 melanoma cell invasion. Exp. Cell Res. 2006, 312, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Balasundaram, P.; Singh, M.K.; Dinda, A.K.; Thakar, A.; Yadav, R. Study of β-catenin, E-cadherin and vimentin in oral squamous cell carcinoma with and without lymph node metastases. Diagn. Pathol. 2014, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegrin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Odero-Marah, V.A.; Wang, R.; Chu, G.; Zayzafoon, M.; Xu, J.; Shi, C.; Marshall, F.F.; Zhau, H.E.; Chung, L.W. Receptor activator of NF-κB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008, 18, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [PubMed]
- Fizazi, K.; Yang, J.; Peleg, S.; Sikes, C.R.; Kreimann, E.L.; Daliani, D.; Olive, M.; Raymond, K.A.; Janus, T.J.; Logothetis, C.J.; et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegrin in osteoblasts. Clin. Cancer Res. 2003, 9, 2587–2597. [Google Scholar] [PubMed]
- Koeneman, K.S.; Yeung, F.; Chung, L.W. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate 1999, 39, 246–261. [Google Scholar] [CrossRef]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and Slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Oft, M.; Peli, J.; Rudaz, C.; Schwarz, H.; Beug, H.; Reichmann, E. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996, 10, 2462–2477. [Google Scholar] [CrossRef] [PubMed]
- Aluwihare, P.; Munger, J.S. What the lung has taught us about latent TGF-beta activation. Am. J. Respir. Cell Mol. Biol. 2008, 39, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Keshavarzian, A. Alcohol stimulates activation of Snail, epidermal growth factor receptor signaling, and biomarkers of epithelial-mesenchymal transition in colon and breast cancer cells. Alcohol. Clin. Exp. Res. 2010, 34, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Pasche, B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat. Rev. Rheumatol. 2009, 5, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.L. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol. 2009, 175, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Wu, Q.; Acquafondata, M.; Dhir, R.; Wells, A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 2012, 5, 19–28. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Jiborn, T.; Biartell, A.; Abrahamsson, P.A. Neuroendocrine differentiation in prostatic carcinoma during hormonal treatment. Urology 1998, 51, 585–589. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Chio, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Mckeithen, D.; Graham, T.; Chung, L.W.; Odero-Marah, V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate 2010, 70, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Bruchovsky, N.; Sadar, M.D. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J. Biol. Chem. 2002, 277, 7076–7085. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Murakami-Mori, K.; Bonavida, B. Interleukin-6 induces G1 arrest through induction of p27(Kip1), a cyclin-dependent kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem. Biophys. Res. Commun. 1999, 257, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, L.; Hu, J.; Li, F.; Shao, Y.; Kalvakolanu, D.V.; Kopecko, D.J.; Zhao, X.; Xu, D.-Q. Down-regulation of signal transducer and activator of transcription 3 expression using vector-based small interfering RNA suppressing growth of human prostate tumor in vivo. Clin. Cancer Res. 2005, 11, 6333–6341. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Yeung, K.; Palladino, M.; Berenson, J.; Bonavida, B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cells chemoimmunosensitization. Cancer Res. 2009, 69, 8376–8385. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; Lisok, A.; Kimble, B.; Domek, J.; Kato, Y.; van der Groep, P.; Artemov, D.; Kowalski, J.; Carraway, H.; van Diest, P.; et al. Twist contributes to hormone resistance in breast cancer by down-regulating estrogen receptor alpha. Oncogene 2012, 31, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour microenvironment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Cavaletti, G. Management of side effects in the personalized medicine era: Chemotherapy-induced peripheral neuropathy. Pharmacogn. Drug Discov. Dev. Methods Mol. Biol. 2014, 1175, 301–322. [Google Scholar]
- Hewitt, K.J.; Agarwal, R.; Morin, P.J. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer 2006, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005, 65, 9603–9606. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Adjei, A.A.; Lorusso, P.M.; Waterhouse, D.; Hecht, J.R.; Natale, R.B.; Hamid, O.; Varterasian, M.; Asbury, P.; Kaldjian, E.P.; et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 2004, 22, 4456–4462. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Maiello, M.R.; Campiglio, M.; Napolitano, M.; Mancino, M.; Carotenuto, A.; Viglietto, G.; Menard, S. The MEK/MAPK pathway is involved in the resistance of breast cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell. Physiol. 2006, 207, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Su, H.Y.; Lai, H.C.; Lin, Y.W.; Liu, C.Y.; Chen, C.K.; Chou, Y.C.; Lin, S.P.; Lin, W.C.; Lee, H.Y.; Yu, M.H. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through wnt signaling pathway. Int. J. Cancer 2010, 127, 555–567. [Google Scholar] [CrossRef] [PubMed]
- El Touny, L.H.; Banerjee, P.P. Akt GSK-3 pathway as a target in Genistein-induced inhibition of TRAMP prostate cancer progression toward a poorly differentiated phenotype. Carcinogenesis 2007, 28, 1710. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.N.; Singh, C.; Nall, D.; Meeker, D.; Shankar, S.; Srivastava, R.K. The dietary bioflavonoid Quercetin synergizes with epigallocathechin gallate (EGCG) to inhibit prostate cancer stem cell characteristics, invasion, migration and epithelial-mesenchymal transition. J. Mol. Signal. 2010, 5, 14. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. https://doi.org/10.3390/jcm5020017
Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. Journal of Clinical Medicine. 2016; 5(2):17. https://doi.org/10.3390/jcm5020017
Chicago/Turabian StyleSmith, Bethany N., and Neil A. Bhowmick. 2016. "Role of EMT in Metastasis and Therapy Resistance" Journal of Clinical Medicine 5, no. 2: 17. https://doi.org/10.3390/jcm5020017
APA StyleSmith, B. N., & Bhowmick, N. A. (2016). Role of EMT in Metastasis and Therapy Resistance. Journal of Clinical Medicine, 5(2), 17. https://doi.org/10.3390/jcm5020017