
Immunotherapy for Gastroesophageal Cancer

Abstract
:
1. Introduction
2. Gastroesophageal (OG) Cancer: Epidemiology and Current Treatment Patterns
3. Immunotherapy–Basic Premises
4. Interaction between Immune Status and OG Cancer/Outcomes
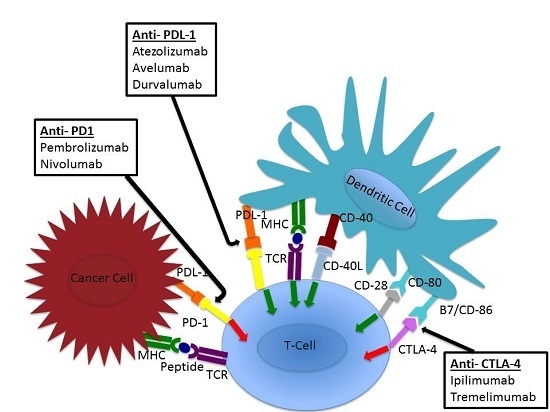
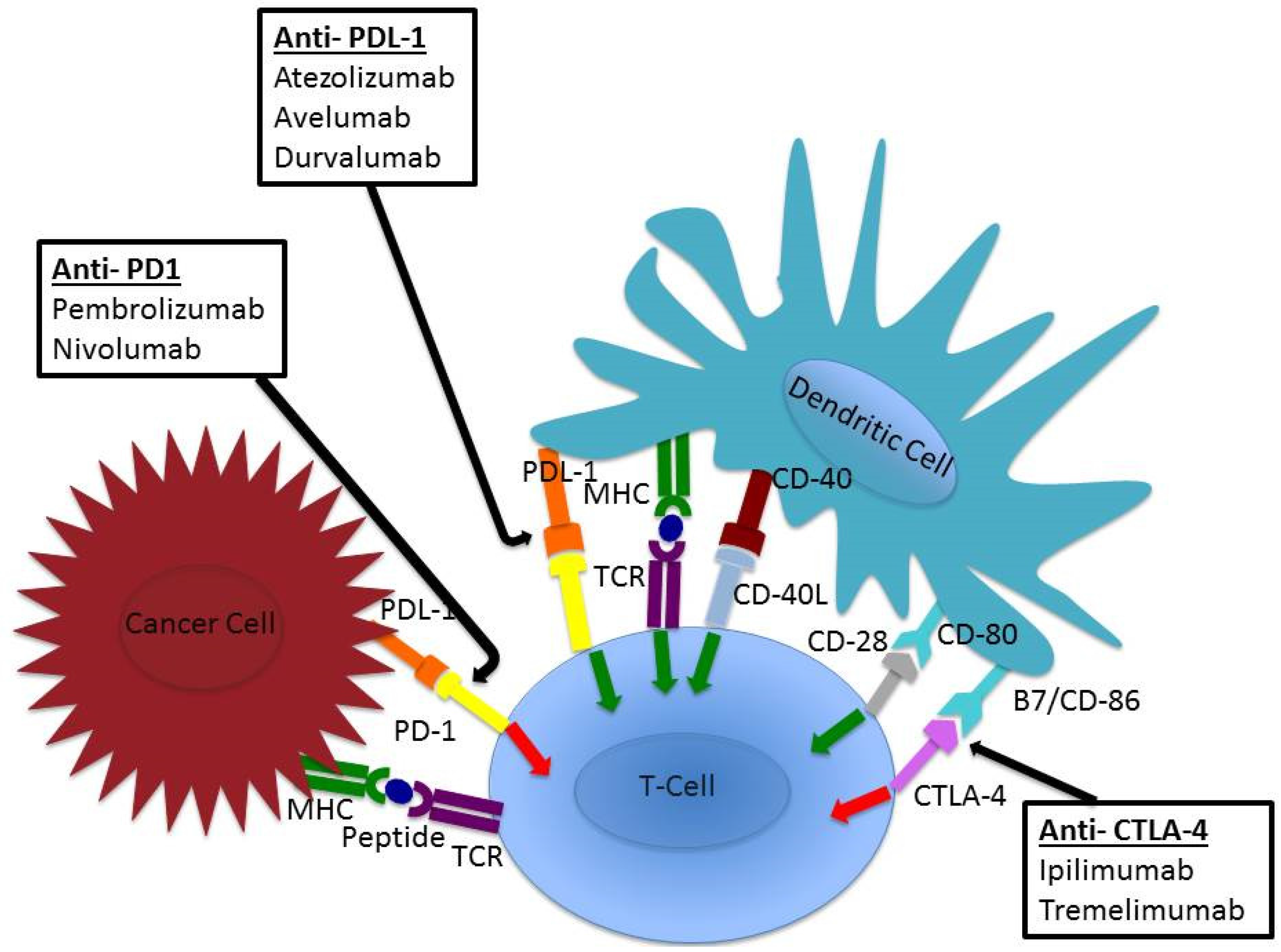
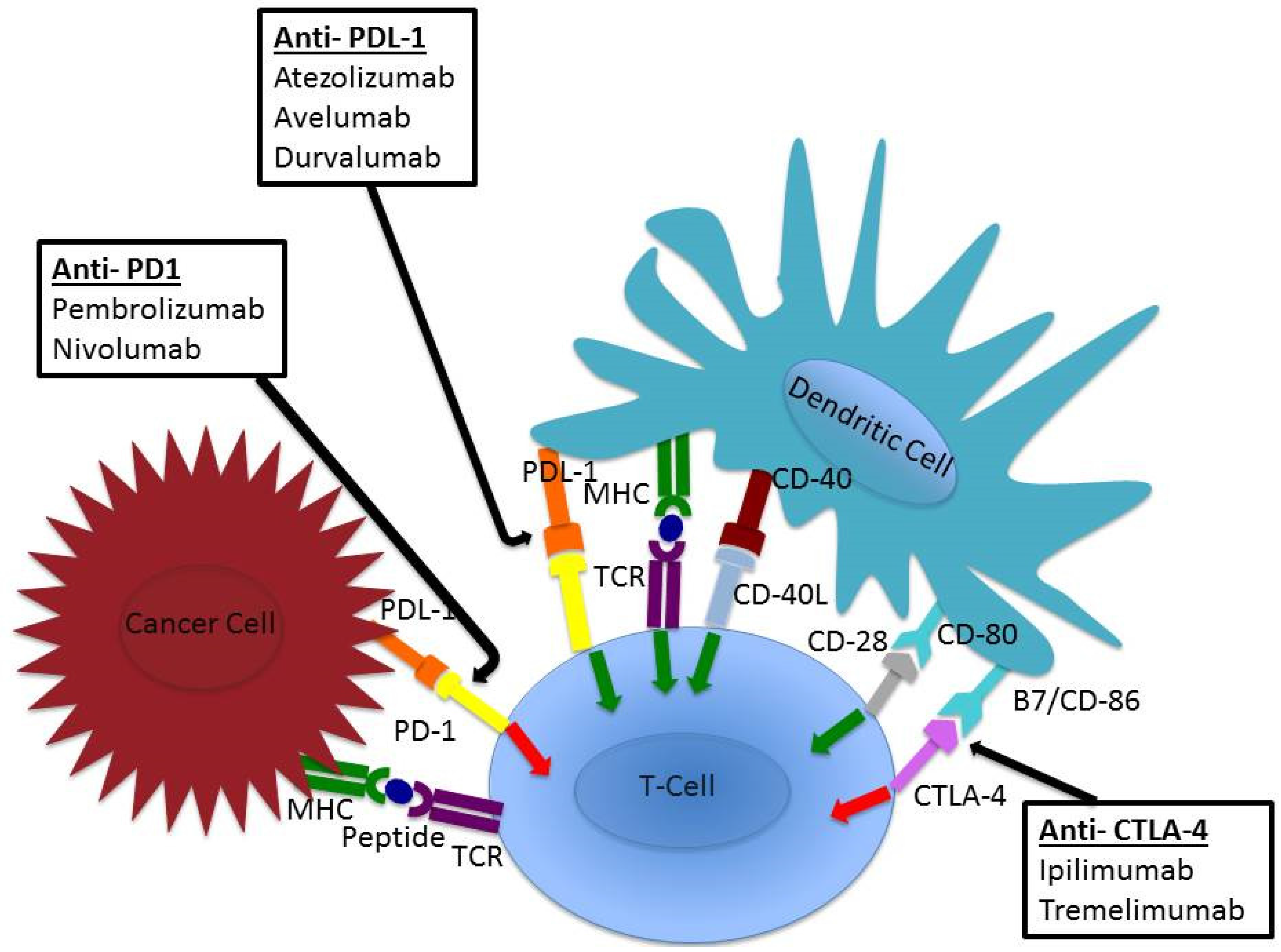
5. Checkpoint Inhibition
6. Anti-PD-L1 (Atezolizumab, Avelumab, Durvalumab)
7. Anti-PD-1 and PD-L1 Therapy in Esophageal Adenocarcinoma
8. Biomarkers Associated with Response to Checkpoint Inhibitor Therapy in Gastroesophageal Cancer
9. Vaccines, Adoptive T Cell Transfer, CAR T Cells
10. Impact of Next Generation Sequencing and Computational Biology on Immunotherapy
11. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; Burley, V.J. Gastric cancer: Global pattern of the disease and an overview of environmental risk factors. Best. Pract. Res. Clin. Gastroenterol 2006, 20, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Kelsen, D.P. Gastric cancer: A primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J. Natl. Compr. Cancer Netw. 2010, 8, 437–447. [Google Scholar]
- Cunningham, D.; Starling, N.; Rao, S.; Iveson, T.; Nicolson, M.; Coxon, F.; Middleton, G.; Daniel, F.; Oates, J.; Norman, A.R. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N. Engl. J. Med. 2008, 358, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Moiseyenko, V.M.; Tjulandin, S.; Majlis, A.; Constenla, M.; Boni, C.; Rodrigues, A.; Fodor, M.; Chao, Y.; Voznyi, E.; et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: A report of the V325 study group. J. Clin. Oncol. 2006, 24, 4991–4997. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Ford, H.E.; Marshall, A.; Bridgewater, J.A.; Janowitz, T.; Coxon, F.Y.; Wadsley, J.; Mansoor, W.; Fyfe, D.; Madhusudan, S.; Middleton, G.W.; et al. Docetaxel versus active symptom control for refractory oesophagogastric adenocarcinoma (COUGAR-02): An open-label, phase 3 randomised controlled trial. Lancet Oncol. 2014, 15, 78–86. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, S.I.; Lim, D.H.; Park, K.W.; Oh, S.Y.; Kwon, H.C.; Hwang, I.G.; Lee, S.C.; Nam, E.; Shin, D.B.; et al. Salvage chemotherapy for pretreated gastric cancer: A randomized phase III trial comparing chemotherapy plus best supportive care with best supportive care alone. J. Clin. Oncol. 2012, 30, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Fontana, E.; Smyth, E.C. Novel targets in the treatment of advanced gastric cancer: A perspective review. Ther. Adv. Med. Oncol. 2016, 8, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Drake, C.G. Primer on tumor immunology and cancer immunotherapy. J. Immunother. Cancer 2013. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Gao, Z.; Cai, Z.; Wang, M.; He, J. Clinicopathological and prognostic significance of FoxP3+ tumor infiltrating lymphocytes in patients with breast cancer: A meta-analysis. BMC Cancer 2015, 15, 727. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Park, S.; Lee, W.Y.; Yun, S.H.; Chun, H.K. Clinical impact of tumor-infiltrating lymphocytes for survival in stage II colon cancer. Cancer 2010, 116, 5188–5199. [Google Scholar] [CrossRef] [PubMed]
- Bogunovic, D.; O’Neill, D.W.; Belitskaya-Levy, I.; Vacic, V.; Yu, Y.L.; Adams, S.; Darvishian, F.; Berman, R.; Shapiro, R.; Pavlick, A.C.; et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc. Natl. Acad. Sci. USA 2009, 106, 20429–20434. [Google Scholar] [CrossRef] [PubMed]
- Grogg, K.L.; Lohse, C.M.; Pankratz, V.S.; Halling, K.C.; Smyrk, T.C. Lymphocyte-rich gastric cancer: Associations with epstein-barr virus, microsatellite instability, histology, and survival. Mod. Pathol. 2003, 16, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, K.S.; Cho, H.J.; Kim, Y.H.; Yang, H.K.; Kim, W.H.; Kang, G.H. Prognostic implications of tumor-infiltrating FoxP3+ regulatory T cells and CD8+ cytotoxic T cells in microsatellite-unstable gastric cancers. Hum. Pathol. 2014, 45, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y.; Kono, K.; Kawaguchi, Y.; Akaike, H.; Kamimura, K.; Sugai, H.; Fujii, H. Localisation pattern of FoxP3+ regulatory T cells is associated with clinical behaviour in gastric cancer. Br. J. Cancer 2008, 98, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Chae, S.W.; Lee, Y.J.; Kim, M.A.; Lee, H.S.; Lee, B.L.; Kim, W.H. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. Br. J. Cancer 2008, 99, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.D.; Zahurak, M.; Murphy, A.; Cornish, T.; Cuka, N.; Abdelfatah, E.; Yang, S.; Duncan, M.; Ahuja, N.; Taube, J.M.; et al. Patterns of PD-L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut 2016. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Ralph, C.; Elkord, E.; Burt, D.J.; O’Dwyer, J.F.; Austin, E.B.; Stern, P.L.; Hawkins, R.E.; Thistlethwaite, F.C. Modulation of lymphocyte regulation for cancer therapy: A phase II trial of tremelimumab in advanced gastric and esophageal adenocarcinoma. Clin. Cancer Res. 2010, 16, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.H.; Cho, J.Y.; Kim, Y.H.; Kim, J.W.; Di Bartolomeo, M.; Ajani, J.A.; Yamaguchi, K.; Balogh, A.; Kong-Sanchez, M.T.; Bang, Y.-J. A randomized, open-label, two-arm phase II trial comparing the efficacy of sequential ipilimumab (ipi) versus best supportive care (BSC) following first-line (1L) chemotherapy in patients with unresectable, locally advanced/metastatic (A/M) gastric or gastro-esophageal junction (G/GEJ) cancer. ASCO Meet. Abstr. 2016, 34, 4011. [Google Scholar]
- Smyth, E.C.; Cunningham, D. Encouraging results for PD-1 inhibition in gastric cancer. Lancet Oncol. 2016, 17, 682–683. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Bendell, J.C.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Ott, P.A.; Bono, P.; Jaeger, D.; Evans, T.R.J.; et al. Checkmate-032: Phase I/II, open-label study of safety and activity of nivolumab (nivo) alone or with ipilimumab (ipi) in advanced and metastatic (A/M) gastric cancer (GC). ASCO Meet. Abstr. 2016, 34, 4010. [Google Scholar]
- Chung, H.C.; Arkenau, H.-T.; Wyrwicz, L.; Oh, D.-Y.; Lee, K.-W.; Infante, J.R.; Lee, S.S.; Lee, J.; Keilholz, U.; Mita, A.C.; et al. Avelumab (MSB0010718C; anti-PD-L1) in patients with advanced gastric or gastroesophageal junction cancer from JAVELIN solid tumor phase Ib trial: Analysis of safety and clinical activity. ASCO Meet. Abstr. 2016, 34, 4009. [Google Scholar]
- Lutzky, J.; Antonia, S.J.; Blake-Haskins, A.; Li, X.; Robbins, P.B.; Shalabi, A.M.; Vasselli, J.; Ibrahim, R.A.; Khleif, S.; Segal, N.H. A phase 1 study of medi4736, an anti-PD-L1 antibody, in patients with advanced solid tumors. ASCO Meet. Abstr. 2014, 32, 3001. [Google Scholar]
- Bendell, J.C.; Kim, T.W.; Goh, B.C.; Wallin, J.; Oh, D.-Y.; Han, S.-W.; Lee, C.B.; Hellmann, M.D.; Desai, J.; Lewin, J.H.; et al. Clinical activity and safety of cobimetinib (COBI) and atezolizumab in colorectal cancer (CRC). ASCO Meet. Abstr. 2016, 34, 3502. [Google Scholar]
- Doi, T.; Piha-Paul, S.A.; Jalal, S.I.; Mai-Dang, H.; Saraf, S.; Koshiji, M.; Csiki, I.; Bennouna, J. Updated results for the advanced esophageal carcinoma cohort of the phase Ib KEYNOTE-028 study of pembrolizumab (MK-3475). ASCO Meet. Abstr. 2016, 34, 4046. [Google Scholar]
- Spencer, K.R.; Wang, J.; Silk, A.W.; Ganesan, S.; Kaufman, H.L.; Mehnert, J.M. Biomarkers for immunotherapy: Current developments and challenges. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.A.; Lee, H.J.; Yang, H.K.; Bang, Y.J.; Kim, W.H. Heterogeneous amplification of ERBB2 in primary lesions is responsible for the discordant ERBB2 status of primary and metastatic lesions in gastric carcinoma. Histopathology 2011, 59, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Robert, C.; Hodi, F.S.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.-J.; Weber, J.S.; Zarour, H.M.; Kefford, R.; Loboda, A.; et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. ASCO Meet. Abstr. 2015, 33, 3001. [Google Scholar]
- Shankaran, V.; Muro, K.; Bang, Y.-J.; Geva, R.; Catenacci, D.V.T.; Gupta, S.; Eder, J.P.; Berger, R.; Loboda, A.; Albright, A.; et al. Correlation of gene expression signatures and clinical outcomes in patients with advanced gastric cancer treated with pembrolizumab (MK-3475). ASCO Meet. Abstr. 2015, 33, 3026. [Google Scholar]
- Lin, S.J.; Gagnon-Bartsch, J.A.; Tan, I.B.; Earle, S.; Ruff, L.; Pettinger, K.; Ylstra, B.; van Grieken, N.; Rha, S.Y.; Chung, H.C.; et al. Signatures of tumour immunity distinguish asian and non-asian gastric adenocarcinomas. Gut 2015, 64, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.; Li, G.M.; Longley, M.; Modrich, P.; Liu, B.; Berk, T.; Hamilton, S.R.; Kinzler, K.W.; Vogelstein, B. Mismatch repair deficiency in phenotypically normal human cells. Science 1995, 268, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Choi, S.I.; Lee, H.K.; Kim, H.S.; Yang, H.K.; Kang, G.H.; Kim, Y.I.; Lee, B.L.; Kim, W.H. Distinct clinical features and outcomes of gastric cancers with microsatellite instability. Mod. Pathol. 2002, 15, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Beghelli, S.; de Manzoni, G.; Barbi, S.; Tomezzoli, A.; Roviello, F.; Di Gregorio, C.; Vindigni, C.; Bortesi, L.; Parisi, A.; Saragoni, L.; et al. Microsatellite instability in gastric cancer is associated with better prognosis in only stage ii cancers. Surgery 2006, 139, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Falchetti, M.; Saieva, C.; Lupi, R.; Masala, G.; Rizzolo, P.; Zanna, I.; Ceccarelli, K.; Sera, F.; Mariani-Costantini, R.; Nesi, G.; et al. Gastric cancer with high-level microsatellite instability: Target gene mutations, clinicopathologic features, and long-term survival. Hum. Pathol. 2008, 39, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Oki, E.; Kakeji, Y.; Zhao, Y.; Yoshida, R.; Ando, K.; Masuda, T.; Ohgaki, K.; Morita, M.; Maehara, Y. Chemosensitivity and survival in gastric cancer patients with microsatellite instability. Ann. Surg. Oncol. 2009, 16, 2510–2515. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; An, J.Y.; Noh, S.H.; Shin, S.K.; Lee, Y.C.; Kim, H. High microsatellite instability predicts good prognosis in intestinal-type gastric cancers. J. Gastroenterol. Hepatol. 2011, 26, 585–592. [Google Scholar] [CrossRef] [PubMed]
- An, J.Y.; Kim, H.; Cheong, J.H.; Hyung, W.J.; Noh, S.H. Microsatellite instability in sporadic gastric cancer: Its prognostic role and guidance for 5-FU based chemotherapy after R0 resection. Int. J. Cancer 2012, 131, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Shin, N.R.; Kim, A.; Lee, H.J.; Park, W.Y.; Lee, C.H.; Huh, G.Y.; Park, D.Y. Microsatellite instability status in gastric cancer: A reappraisal of its clinical significance and relationship with mucin phenotypes. Korean J. Pathol. 2013, 47, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Turner, N.C.; Pearson, A.; Peckitt, C.; Chau, I.; Watkins, D.J.; Starling, N.; Rao, S.; Gillbanks, A.; Kilgour, E.; et al. Phase II study of AZD4547 in FGFR amplified tumours: Gastroesophageal cancer (GC) cohort pharmacodynamic and biomarker results. ASCO Meet. Abstr. 2016, 34, 154. [Google Scholar]
- Le, D.T.; Yoshino, T.; Jager, D.; Andre, T.; Bendell, J.C.; Wang, R.; Kang, S.P.; Koshiji, M.; Diaz, L.A. Keynote-164: Phase II study of pembrolizumab (MK-3475) for patients with previously treated, microsatellite instability-high advanced colorectal carcinoma. ASCO Meet. Abstr. 2016, 34, TPS787. [Google Scholar]
- Polk, D.B.; Peek, R.M. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Llosa, A.E.; Friedman, G.D.; Vogelman, J.H.; Orentreich, N.; Stolzenberg-Solomon, R.Z.; Parsonnet, J. Helicobacter pylori infection and development of pancreatic cancer. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Tsukamoto, T.; Toyoda, T.; Mori, A.; Tanaka, H.; Maekita, T.; Ichinose, M.; Tatematsu, M.; Ushijima, T. Inflammatory processes triggered by helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010, 70, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Mao, X.H.; Li, J.X.; Tong, W.D.; Wang, B.; Zhang, Y.J.; Guo, G.; Zhao, Z.J.; Li, L.; Wu, D.L.; et al. Efficacy, safety, and immunogenicity of an oral recombinant helicobacter pylori vaccine in children in china: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1457–1464. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Camargo, M.C.; Kim, W.H.; Chiaravalli, A.M.; Kim, K.M.; Corvalan, A.H.; Matsuo, K.; Yu, J.; Sung, J.J.; Herrera-Goepfert, R.; Meneses-Gonzalez, F.; et al. Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: An international pooled analysis. Gut 2014, 63, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Mori, M.; Honda, M.; Li, J.; Shibuta, K.; Mimori, K.; Ueo, H.; Akiyoshi, T. The expression of tumor-rejection antigen “mage” genes in human gastric carcinoma. Gastroenterology 1995, 109, 1522–1525. [Google Scholar] [CrossRef]
- Mashino, K.; Sadanaga, N.; Tanaka, F.; Yamaguchi, H.; Nagashima, H.; Inoue, H.; Sugimachi, K.; Mori, M. Expression of multiple cancer-testis antigen genes in gastrointestinal and breast carcinomas. Br. J. Cancer 2001, 85, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Ueda, S.; Nishikawa, H.; Kitano, S.; Hirayama, M.; Ikeda, H.; Toyoda, H.; Tanaka, K.; Kanai, M.; Takabayashi, A.; et al. Antibody responses against NY-ESO-1 and HER2 antigens in patients vaccinated with combinations of cholesteryl pullulan (CHP)-NY-ESO-1 and CHP-HER2 with OK-432. Vaccine 2009, 27, 6854–6861. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, S.; Wada, H.; Muro, K.; Niwa, Y.; Ueda, S.; Miyata, H.; Takiguchi, S.; Sugino, S.H.; Miyahara, Y.; Ikeda, H.; et al. Dose-dependent effects of NY-ESO-1 protein vaccine complexed with cholesteryl pullulan (CHP-NY-ESO-1) on immune responses and survival benefits of esophageal cancer patients. J. Transl. Med. 2013, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Kawada, J.; Wada, H.; Isobe, M.; Gnjatic, S.; Nishikawa, H.; Jungbluth, A.A.; Okazaki, N.; Uenaka, A.; Nakamura, Y.; Fujiwara, S.; et al. Heteroclitic serological response in esophageal and prostate cancer patients after NY-ESO-1 protein vaccination. Int. J. Cancer 2012, 130, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Uenaka, A.; Wada, H.; Isobe, M.; Saika, T.; Tsuji, K.; Sato, E.; Sato, S.; Noguchi, Y.; Kawabata, R.; Yasuda, T.; et al. T cell immunomonitoring and tumor responses in patients immunized with a complex of cholesterol-bearing hydrophobized pullulan (CHP) and NY-ESO-1 protein. Cancer immunity 2007, 7, 9. [Google Scholar] [PubMed]
- Wada, H.; Sato, E.; Uenaka, A.; Isobe, M.; Kawabata, R.; Nakamura, Y.; Iwae, S.; Yonezawa, K.; Yamasaki, M.; Miyata, H.; et al. Analysis of peripheral and local anti-tumor immune response in esophageal cancer patients after NY-ESO-1 protein vaccination. Int. J. Cancer 2008, 123, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Hecht, J.R.; Ho, L.; Baker, J.; Oortgiesen, M.; Eduljee, A.; Michaeli, D. An open-label, multinational, multicenter study of G17DT vaccination combined with cisplatin and 5-fluorouracil in patients with untreated, advanced gastric or gastroesophageal cancer: The GC4 study. Cancer 2006, 106, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, T.; Fujiwara, Y.; Okada, K.; Nakamura, A.; Takiguchi, S.; Nakajima, K.; Miyata, H.; Yamasaki, M.; Kurokawa, Y.; Osawa, R.; et al. Phase I/II study of S-1 plus cisplatin combined with peptide vaccines for human vascular endothelial growth factor receptor 1 and 2 in patients with advanced gastric cancer. Int. J. Oncol. 2012, 41, 1297–1304. [Google Scholar] [PubMed]
- Sadanaga, N.; Nagashima, H.; Mashino, K.; Tahara, K.; Yamaguchi, H.; Ohta, M.; Fujie, T.; Tanaka, F.; Inoue, H.; Takesako, K.; et al. Dendritic cell vaccination with mage peptide is a novel therapeutic approach for gastrointestinal carcinomas. Clin. Cancer Res. 2001, 7, 2277–2284. [Google Scholar] [PubMed]
- Kono, K.; Takahashi, A.; Sugai, H.; Fujii, H.; Choudhury, A.R.; Kiessling, R.; Matsumoto, Y. Dendritic cells pulsed with HER-2/neu-derived peptides can induce specific T-cell responses in patients with gastric cancer. Clin. Cancer Res. 2002, 8, 3394–3400. [Google Scholar] [PubMed]
- Kono, K.; Takahashi, A.; Ichihara, F.; Amemiya, H.; Iizuka, H.; Fujii, H.; Sekikawa, T.; Matsumoto, Y. Prognostic significance of adoptive immunotherapy with tumor-associated lymphocytes in patients with advanced gastric cancer: A randomized trial. Clin. Cancer Res. 2002, 8, 1767–1771. [Google Scholar] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Popplewell, L.L.; Wagner, J.R.; Naranjo, A.; Blanchard, M.S.; Mott, M.R.; Norris, A.P.; Wong, C.W.; Urak, R.Z.; Chang, W.C.; et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 2016, 127, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Heslop, H.E.; Brenner, M.K. Car-T cell therapy for lymphoma. Annu. Rev. Med. 2016, 67, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.; de Visser, K.E.; Tirion, F.H.; Coccoris, M.; Kruisbeek, A.M.; Schumacher, T.N. The impact of self-tolerance on the polyclonal CD8+ T cell repertoire. J. Immunol. 2004, 172, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; Marquardt, K.L.; Cheung, J.; Sherman, L.A. Functional differences between low- and high-affinity CD8(+) T cells in the tumor environment. Oncoimmunology 2012, 1, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.L.; Ansari, H.R.; Bradley, P.; Cawley, G.C.; Hertz, T.; Hu, X.; Jojic, N.; Kim, Y.; Kohlbacher, O.; Lund, O.; et al. Machine learning competition in immunology-prediction of HLA class I binding peptides. J. Immunol. Methods 2011, 374, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Lower, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, T.; Pignon, J.C.; Wang, B.; Wang, J.; Shukla, S.A.; Dou, R.; Chen, Q.; Hodi, F.S.; Choueiri, T.K.; et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 2016, 48, 725–732. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Trial Phase | Drugs Tested | N | Median PFS (m) | ORR | Median OS (m) | 1 Year OS |
|---|---|---|---|---|---|---|
| Phase II (28) | Tremilimumab | 18 | 2.8 | 6% | 4.8 | 33% |
| Phase II (29) | Ipilimumab | 57 | 2.7 | NR | 12.7 | NR |
| Phase Ib (3) | Pembrolizumab * | 39 | 1.9 | 22% | 11.4 | NR |
| Phase I/II(31) | Nivolumab | 59 | NR | 14% | 5.0 | 36 |
| Nivolumab 1 mg/kg, Ipilimumab 3 mg/kg | 49 | NR | 26% | 6.9 | 34 | |
| Nivolumab 3 mg/kg, Ipilimumab 1 mg/kg | 52 | NR | 10% | 4.8 | NR | |
| Phase Ib (32) | Avelumab 1st line maintenance | 89 | 12 weeks | 9% | NR | NR |
| Avelumab 2nd line | 62 | 6 weeks | 9.7% | NR | NR |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goode, E.F.; Smyth, E.C. Immunotherapy for Gastroesophageal Cancer. J. Clin. Med. 2016, 5, 84. https://doi.org/10.3390/jcm5100084
Goode EF, Smyth EC. Immunotherapy for Gastroesophageal Cancer. Journal of Clinical Medicine. 2016; 5(10):84. https://doi.org/10.3390/jcm5100084
Chicago/Turabian StyleGoode, Emily F., and Elizabeth C. Smyth. 2016. "Immunotherapy for Gastroesophageal Cancer" Journal of Clinical Medicine 5, no. 10: 84. https://doi.org/10.3390/jcm5100084
APA StyleGoode, E. F., & Smyth, E. C. (2016). Immunotherapy for Gastroesophageal Cancer. Journal of Clinical Medicine, 5(10), 84. https://doi.org/10.3390/jcm5100084