
Advances in the Microbiome: Applications to Clostridium difficile Infection



{kind=link}
Abstract
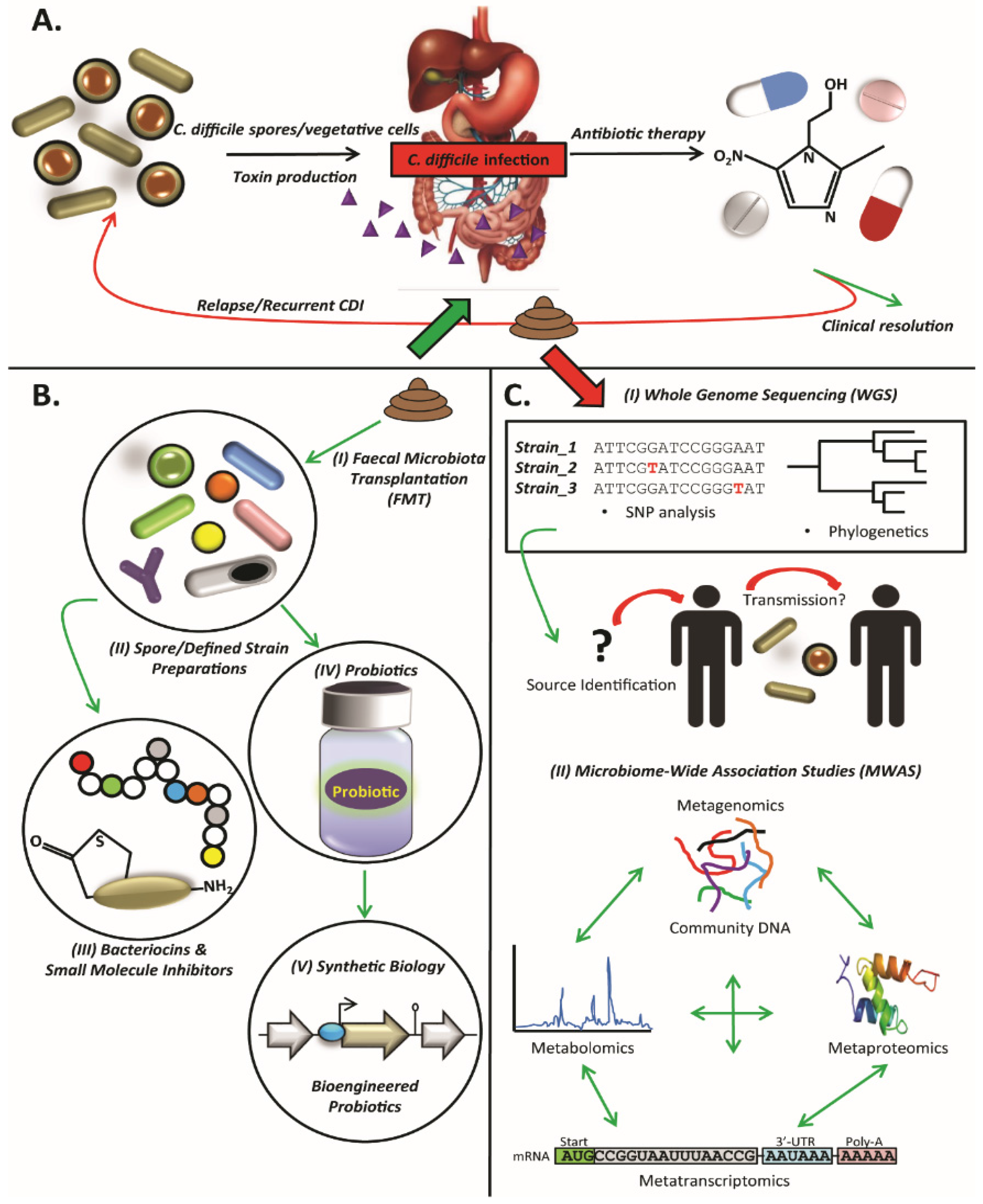
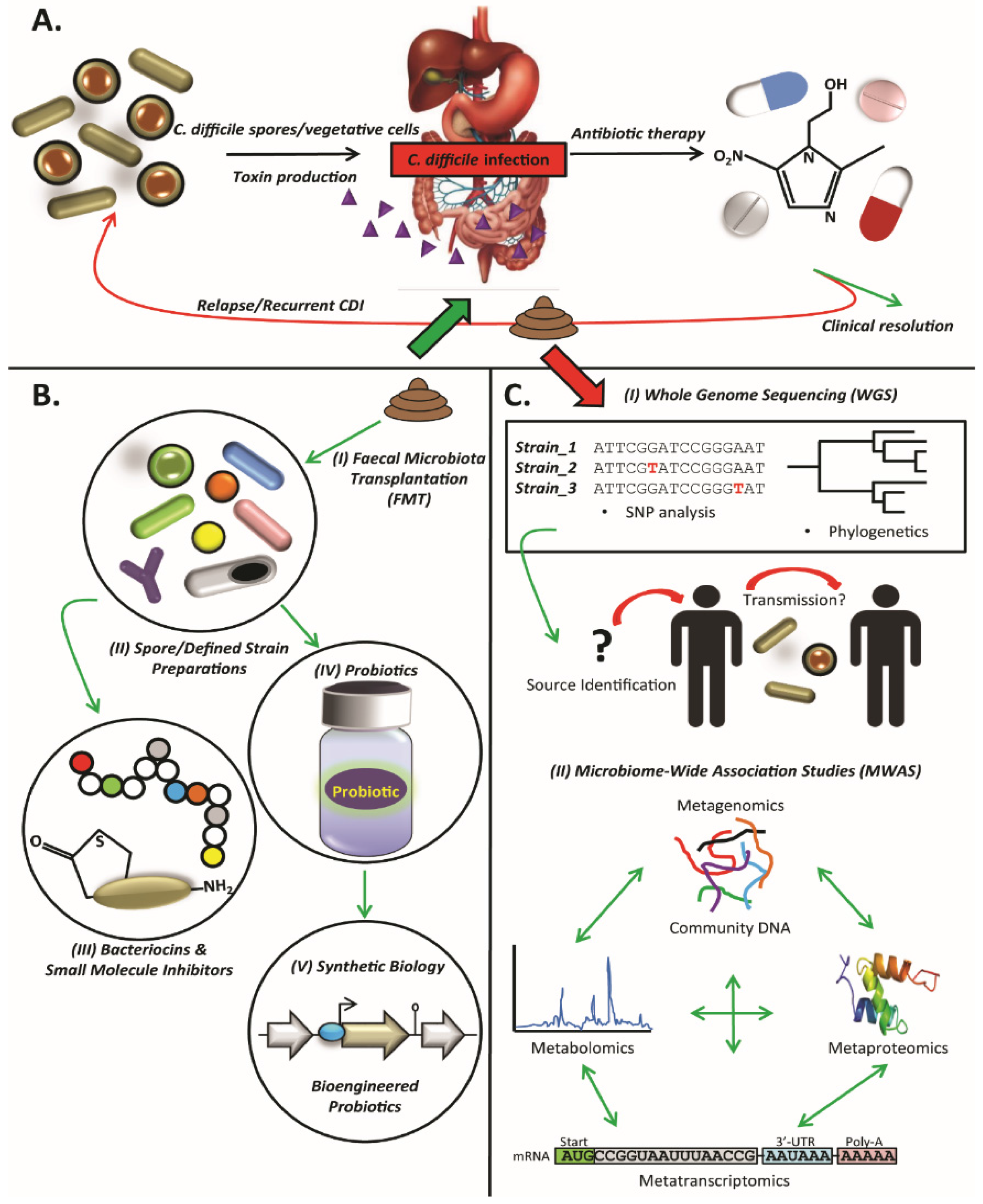
:1. Introduction
2. Clostridium difficile
3. Faecal Microbiota Transplantation
4. Defined Strain or Spore Formulations
5. Microbiome-Wide Association Studies (MWAS)
6. Whole-Genome Sequencing
7. Probiotics
8. Small Molecule Inhibitors
9. Bacteriocins
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Itoh, T.; Kuwahara, T.; Oshima, K.; Toh, H.; Toyoda, A.; Takami, H.; Morita, H.; Sharma, V.K.; Srivastava, T.P.; et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007, 14, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Gordon, J.I. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc. Natl. Acad. Sci. USA 2006, 103, 10011–10016. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.P.; Antoine, J.M.; Midtvedt, T.; van Hemert, S. Manipulating the gut microbiota to maintain health and treat disease. Microb. Ecol. Health Dis. 2015, 26, 25877. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Clostridium difficile infection: Epidemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, S.; Ascenzi, P.; Siarakas, S.; Petrosillo, N.; di Masi, A. Clostridium difficile toxins A and B: Insights into pathogenic properties and extraintestinal effects. Toxins 2016, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Wilm, M.; Selzer, J.; Rex, G.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. The enterotoxin from Clostridium difficile (toxa) monoglucosylates the rho proteins. J. Biol. Chem. 1995, 270, 13932–13936. [Google Scholar] [CrossRef] [PubMed]
- Lyerly, D.M.; Krivan, H.C.; Wilkins, T.D. Clostridium difficile: Its disease and toxins. Clin. Microbiol. Rev. 1988, 1, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lyras, D.; O’Connor, J.R.; Howarth, P.M.; Sambol, S.P.; Carter, G.P.; Phumoonna, T.; Poon, R.; Adams, V.; Vedantam, G.; Johnson, S.; et al. Toxin B is essential for virulence of Clostridium difficile. Nature 2009, 458, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010, 467, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, S.A.; Cartman, S.T.; Minton, N.P. Both, toxin A and toxin B, are important in Clostridium difficile infection. Gut Microbes 2011, 2, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.; Squire, M.M.; Thean, S.; Chang, B.J.; Brazier, J.S.; Rupnik, M.; Riley, T.V. New types of toxin A-negative, toxin B-positive strains among clinical isolates of Clostridium difficile in Australia. J. Med. Microbiol. 2011, 60, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Janezic, S.; Marin, M.; Martin, A.; Rupnik, M. A new type of toxin A-negative, toxin B-positive Clostridium difficile strain lacking a complete tcdA gene. J. Clin. Microbiol. 2015, 53, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.S.; Roberts, A.P.; Hussain, H.; Williams, R.J.; Allan, E.; Mullany, P. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat. Commun. 2013, 4, 2601. [Google Scholar] [CrossRef] [PubMed]
- Crobach, M.J.; Dekkers, O.M.; Wilcox, M.H.; Kuijper, E.J. European society of clinical microbiology and infectious diseases (ESCMID): Data review and recommendations for diagnosing Clostridium difficile-infection (CDI). Clin. Microbiol. Infect. 2009, 15, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Crogan, N.L.; Evans, B.C. Clostridium difficile: An emerging epidemic in nursing homes. Geriatr. Nurs. 2007, 28, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Severe Clostridium difficile-associated disease in populations previously at low risk—Four states, 2005. MMWR Morb. Mortal. Wkly. Rep. 2005, 54, 1201–1205. [Google Scholar]
- Cecil, J.A. Clostridium difficile: Changing epidemiology, treatment and infection prevention measures. Curr. Infect. Dis. Rep. 2012, 14, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Kutty, P.K.; Woods, C.W.; Sena, A.C.; Benoit, S.R.; Naggie, S.; Frederick, J.; Evans, S.; Engel, J.; McDonald, L.C. Risk factors for and estimated incidence of community-associated Clostridium difficile infection, North Carolina, USA. Emerg. Infect. Dis. 2010, 16, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Ghantoji, S.S.; Sail, K.; Lairson, D.R.; DuPont, H.L.; Garey, K.W. Economic healthcare costs of Clostridium difficile infection: A systematic review. J. Hosp. Infect. 2010, 74, 309–318. [Google Scholar] [CrossRef] [PubMed]
- McGlone, S.M.; Bailey, R.R.; Zimmer, S.M.; Popovich, M.J.; Tian, Y.; Ufberg, P.; Muder, R.R.; Lee, B.Y. The economic burden of Clostridium difficile. Clin. Microbiol. Infect. 2012, 18, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.H.; Cunniffe, J.G.; Trundle, C.; Redpath, C. Financial burden of hospital-acquired Clostridium difficile infection. J. Hosp. Infect. 1996, 34, 23–30. [Google Scholar] [CrossRef]
- Kuijper, E.J.; Coignard, B.; Tull, P. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin. Microbiol. Infect. 2006, 12 (Suppl. 6), 2–18. [Google Scholar] [CrossRef] [PubMed]
- Rupnik, M.; Wilcox, M.H.; Gerding, D.N. Clostridium difficile infection: New developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Aldeyab, M.A.; Kearney, M.P.; Scott, M.G.; Aldiab, M.A.; Alahmadi, Y.M.; Darwish Elhajji, F.W.; Magee, F.A.; McElnay, J.C. An evaluation of the impact of antibiotic stewardship on reducing the use of high-risk antibiotics and its effect on the incidence of Clostridium difficile infection in hospital settings. J. Antimicrob. Chemother. 2012, 67, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Hensgens, M.P.; Goorhuis, A.; Dekkers, O.M.; Kuijper, E.J. Time interval of increased risk for Clostridium difficile infection after exposure to antibiotics. J. Antimicrob. Chemother. 2012, 67, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Talpaert, M.J.; Gopal Rao, G.; Cooper, B.S.; Wade, P. Impact of guidelines and enhanced antibiotic stewardship on reducing broad-spectrum antibiotic usage and its effect on incidence of Clostridium difficile infection. J. Antimicrob. Chemother. 2011, 66, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.H.; Gerding, D.N.; Johnson, S.; Kelly, C.P.; Loo, V.G.; McDonald, L.C.; Pepin, J.; Wilcox, M.H. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control Hosp. Epidemiol. 2010, 31, 431–455. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V. Alternative treatments for Clostridium difficile disease: What really works? J. Med. Microbiol. 2005, 54, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Kuijper, E.J.; Coignard, B.; Brazier, J.S.; Suetens, C.; Drudy, D.; Wiuff, C.; Pituch, H.; Reichert, P.; Schneider, F.; Widmer, A.F.; et al. Update of Clostridium difficile-associated disease due to PCR ribotype 027 in Europe. Euro Surveill. 2007, 12, E1–E2. [Google Scholar] [PubMed]
- Surawicz, C.M.; Brandt, L.J.; Binion, D.G.; Ananthakrishnan, A.N.; Curry, S.R.; Gilligan, P.H.; McFarland, L.V.; Mellow, M.; Zuckerbraun, B.S. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 2013, 108, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, G.; Masucci, L.; Ianiro, G.; Bibbo, S.; Dinoi, G.; Costamagna, G.; Sanguinetti, M.; Gasbarrini, A. Randomised clinical trial: Faecal microbiota transplantation by colonoscopy vs. Vancomycin for the treatment of recurrent Clostridium difficile infection. Aliment. Pharmacol. Ther. 2015, 41, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Gough, E.; Shaikh, H.; Manges, A.R. Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin. Infect. Dis. 2011, 53, 994–1002. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Cai, H.F.; Wang, Z.H.; Xu, J.; Fang, J.Y. Systematic review with meta-analysis: Long-term outcomes of faecal microbiota transplantation for Clostridium difficile infection. Aliment. Pharmacol. Ther. 2016, 43, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Brandt, L.J.; Aroniadis, O.C.; Mellow, M.; Kanatzar, A.; Kelly, C.; Park, T.; Stollman, N.; Rohlke, F.; Surawicz, C. Long-term follow-up of colonoscopic fecal microbiota transplant for recurrent Clostridium difficile infection. Am. J. Gastroenterol. 2012, 107, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Mattila, E.; Uusitalo-Seppala, R.; Wuorela, M.; Lehtola, L.; Nurmi, H.; Ristikankare, M.; Moilanen, V.; Salminen, K.; Seppala, M.; Mattila, P.S.; et al. Fecal transplantation, through colonoscopy, is effective therapy for recurrent Clostridium difficile infection. Gastroenterology 2012, 142, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Aroniadis, O.C.; Brandt, L.J.; Kelly, C.; Freeman, S.; Surawicz, C.; Broussard, E.; Stollman, N.; Giovanelli, A.; Smith, B.; et al. The long-term efficacy and safety of fecal microbiota transplant for recurrent, severe, and complicated Clostridium difficile infection in 146 elderly individuals. J. Clin. Gastroenterol. 2016, 50, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.; Colville, A. Adverse events in faecal microbiota transplant: A review of the literature. J. Hosp. Infect. 2016, 92, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Bakken, J.S.; Borody, T.; Brandt, L.J.; Brill, J.V.; Demarco, D.C.; Franzos, M.A.; Kelly, C.; Khoruts, A.; Louie, T.; Martinelli, L.P.; et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin. Gastroenterol. Hepatol. 2011, 9, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Bojanova, D.P.; Bordenstein, S.R. Fecal transplants: What is being transferred? PLoS Biol. 2016, 14, e1002503. [Google Scholar] [CrossRef] [PubMed]
- Sachs, R.; Edelstein, C. Ensuring the safe and effective fda regulation of fecal microbiota transplantation. J. Law Biosci. 2015, 2. [Google Scholar] [CrossRef]
- Smith, M.B.; Kelly, C.; Alm, E.J. Policy: How to regulate faecal transplants. Nature 2014, 506, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Pardi, D.S.; Kelly, C.R.; Kraft, C.S.; Dhere, T.; Henn, M.R.; Lombardo, M.J.; Vulic, M.; Ohsumi, T.; Winkler, J.; et al. A novel microbiome therapeutic increases gut microbial diversity and prevents recurrent Clostridium difficile infection. J. Infect. Dis. 2016, 214, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Gerding, D.N.; Meyer, T.; Lee, C.; Cohen, S.H.; Murthy, U.K.; Poirier, A.; Van Schooneveld, T.C.; Pardi, D.S.; Ramos, A.; Barron, M.A.; et al. Administration of spores of nontoxigenic Clostridium difficile strain M3 for prevention of recurrent C difficile infection: A randomized clinical trial. JAMA J. Am. Med. Assoc. 2015, 313, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Dingle, K.E.; Griffiths, D.; Didelot, X.; Evans, J.; Vaughan, A.; Kachrimanidou, M.; Stoesser, N.; Jolley, K.A.; Golubchik, T.; Harding, R.M.; et al. Clinical Clostridium difficile: Clonality and pathogenicity locus diversity. PLoS ONE 2011, 6, e19993. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.P.; DeMaere, M.; Chapman, T.; Worden, P.; Charles, I.G.; Darling, A.E.; Djordjevic, S.P. Comparative genomic analysis of toxin-negative strains of Clostridium difficile from humans and animals with symptoms of gastrointestinal disease. BMC Microbiol. 2016, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Petrof, E.O.; Gloor, G.B.; Vanner, S.J.; Weese, S.J.; Carter, D.; Daigneault, M.C.; Brown, E.M.; Schroeter, K.; Allen-Vercoe, E. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Stares, M.D.; Connor, T.R.; Raisen, C.; Goulding, D.; Rad, R.; Schreiber, F.; Brandt, C.; et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012, 8, e1002995. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Quinn, R.A.; Debelius, J.; Xu, Z.Z.; Morton, J.; Garg, N.; Jansson, J.K.; Dorrestein, P.C.; Knight, R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016, 535, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht, M.J.; Theriot, C.M.; Bergin, I.L.; Schumacher, C.A.; Schloss, P.D.; Young, V.B. Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect. Immun. 2015, 83, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Kearney, S.; Li, N.; Bogart, E.; Bullock, K.; Gerber, G.K.; Bry, L.; Clish, C.B.; Alm, E.; Korzenik, J.R. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment. Pharmacol. Ther. 2016, 43, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Ashton, P.M.; Peters, T.; Ameh, L.; McAleer, R.; Petrie, S.; Nair, S.; Muscat, I.; de Pinna, E.; Dallman, T. Whole genome sequencing for the retrospective investigation of an outbreak of salmonella typhimurium DT 8. PLoS Curr. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.R.; Tarr, C.; Strain, E.; Jackson, K.A.; Conrad, A.; Carleton, H.; Katz, L.S.; Stroika, S.; Gould, L.H.; Mody, R.K.; et al. Implementation of nationwide real-time whole-genome sequencing to enhance listeriosis outbreak detection and investigation. Clin. Infect. Dis. 2016, 63, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, S.R.; Anderson, L.J.; Kotter, C.V.; Littlehorn, C.A.; Arms, L.E.; Dowell, E.; Todd, J.K.; Frank, D.N. Comparison of whole-genome sequencing and molecular-epidemiological techniques for Clostridium difficile strain typing. J. Pediatr. Infect. Dis. Soc. 2015. [Google Scholar] [CrossRef]
- Harris, S.R.; Cartwright, E.J.; Torok, M.E.; Holden, M.T.; Brown, N.M.; Ogilvy-Stuart, A.L.; Ellington, M.J.; Quail, M.A.; Bentley, S.D.; Parkhill, J.; et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant staphylococcus aureus: A descriptive study. Lancet Infect. Dis. 2013, 13, 130–136. [Google Scholar] [CrossRef]
- Lewis, T.; Loman, N.J.; Bingle, L.; Jumaa, P.; Weinstock, G.M.; Mortiboy, D.; Pallen, M.J. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J. Hosp. Infect. 2010, 75, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Cumley, N.; Wearn, C.M.; Niebel, M.; Constantinidou, C.; Thomas, C.M.; Pallen, M.J.; Moiemen, N.S.; Bamford, A.; Oppenheim, B.; et al. Seeking the source of Pseudomonas aeruginosa infections in a recently opened hospital: An observational study using whole-genome sequencing. BMJ Open 2014, 4, e006278. [Google Scholar] [CrossRef] [PubMed]
- Snitkin, E.S.; Zelazny, A.M.; Thomas, P.J.; Stock, F.; Henderson, D.K.; Palmore, T.N.; Segre, J.A. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 148ra116. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Miyajima, F.; He, M.; Roberts, P.; Swale, A.; Ellison, L.; Pickard, D.; Smith, G.; Molyneux, R.; Dougan, G.; et al. Genome-based infection tracking reveals dynamics of Clostridium difficile transmission and disease recurrence. Clin. Infect. Dis. 2016, 62, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Mac Aogain, M.; Moloney, G.; Kilkenny, S.; Kelleher, M.; Kelleghan, M.; Boyle, B.; Rogers, T.R. Whole-genome sequencing improves discrimination of relapse from reinfection and identifies transmission events among patients with recurrent Clostridium difficile infections. J. Hosp. Infect. 2015, 90, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Du, P.; Yang, H.; Zhang, Y.; Wang, J.; Zhang, W.; Han, G.; Han, N.; Yao, Z.; Wang, H.; et al. Nosocomial transmission of Clostridium difficile ribotype 027 in a chinese hospital, 2012–2014, traced by whole genome sequencing. BMC Genom. 2016, 17, 405. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D.W.; Tracey, L.; Elliott, B.; Slimings, C.; Huntington, P.G.; Stuart, R.L.; Korman, T.M.; Kotsiou, G.; McCann, R.; Griffiths, D.; et al. Emergence and spread of predominantly community-onset Clostridium difficile PCR ribotype 244 infection in Australia, 2010 to 2012. Euro Surveill. 2015, 20, 21059. [Google Scholar] [CrossRef] [PubMed]
- Steglich, M.; Nitsche, A.; von Muller, L.; Herrmann, M.; Kohl, T.A.; Niemann, S.; Nubel, U. Tracing the spread of Clostridium difficile ribotype 027 in germany based on bacterial genome sequences. PLoS ONE 2015, 10, e0139811. [Google Scholar] [CrossRef]
- Knetsch, C.W.; Connor, T.R.; Mutreja, A.; van Dorp, S.M.; Sanders, I.M.; Browne, H.P.; Harris, D.; Lipman, L.; Keessen, E.C.; Corver, J.; et al. Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the netherlands, 2002 to 2011. Euro Surveill. 2014, 19, 20954. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Ashton, P.; Calus, S.; Chatt, C.; Gossain, S.; Hawker, J.; Nair, S.; Neal, K.; Nye, K.; Peters, T.; et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of salmonella. Genome Biol. 2015, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Loose, M.; Malla, S.; Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 2016. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.C.; McCallum, N.; Sintchenko, V.; Howden, B.P. Whole genome sequencing in clinical and public health microbiology. Pathology 2015, 47, 199–210. [Google Scholar] [CrossRef]
- Allard, M.W. The future of whole-genome sequencing for public health and the clinic. J. Clin. Microbiol. 2016, 54, 1946–1948. [Google Scholar] [CrossRef] [PubMed]
- Fricke, W.F.; Rasko, D.A. Bacterial genome sequencing in the clinic: Bioinformatic challenges and solutions. Nat. Rev. Genet. 2014, 15, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Bletz, S.; Boking, T.; Kipp, F.; Becker, K.; Schultes, A.; Prior, K.; Harmsen, D. Real-time genome sequencing of resistant bacteria provides precision infection control in an institutional setting. J. Clin. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Culligan, E.P.; Hill, C.; Sleator, R.D. Probiotics and gastrointestinal disease: Successes, problems and future prospects. Gut Pathog. 2009, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D. Probiotic therapy—Recruiting old friends to fight new foes. Gut Pathog. 2010, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Maziade, P.J.; Pereira, P.; Goldstein, E.J. A decade of experience in primary prevention of Clostridium difficile infection at a community hospital using the probiotic combination Lactobacillus acidophilus CL1285, Lactobacillus casei LBC80R, and Lactobacillus rhamnosus CLR2 (Bio-K+). Clin. Infect. Dis. 2015, 60 (Suppl. 2), S144–S147. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.W.; Mubasher, M.; Fang, C.Y.; Reifer, C.; Miller, L.E. Dose-response efficacy of a proprietary probiotic formula of Lactobacillus acidophilus CL1285 and Lactobacillus casei LBC80R for antibiotic-associated diarrhea and Clostridium difficile-associated diarrhea prophylaxis in adult patients. Am. J. Gastroenterol. 2010, 105, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, J.Z.; Ma, S.S.; Saxton, J.D.; Martzen, M.R.; Vandvik, P.O.; Thorlund, K.; Guyatt, G.H.; Johnston, B.C. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Johnson, S.; Maziade, P.J.; McFarland, L.V.; Trick, W.; Donskey, C.; Currie, B.; Low, D.E.; Goldstein, E.J. Is primary prevention of Clostridium difficile infection possible with specific probiotics? Int. J. Infect. Dis. 2012, 16, e786–e792. [Google Scholar] [CrossRef] [PubMed]
- Johnston, B.C.; Ma, S.S.; Goldenberg, J.Z.; Thorlund, K.; Vandvik, P.O.; Loeb, M.; Guyatt, G.H. Probiotics for the prevention of Clostridium difficile-associated diarrhea: A systematic review and meta-analysis. Ann. Int. Med. 2012, 157, 878–888. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V.; Surawicz, C.M.; Greenberg, R.N.; Fekety, R.; Elmer, G.W.; Moyer, K.A.; Melcher, S.A.; Bowen, K.E.; Cox, J.L.; Noorani, Z.; et al. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. J. Am. Med.Assoc. 1994, 271, 1913–1918. [Google Scholar] [CrossRef]
- Surawicz, C.M.; McFarland, L.V.; Greenberg, R.N.; Rubin, M.; Fekety, R.; Mulligan, M.E.; Garcia, R.J.; Brandmarker, S.; Bowen, K.; Borjal, D.; et al. The search for a better treatment for recurrent Clostridium difficile disease: Use of high-dose vancomycin combined with Saccharomyces boulardii. Clin. Infect. Dis. 2000, 31, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Pozzoni, P.; Riva, A.; Bellatorre, A.G.; Amigoni, M.; Redaelli, E.; Ronchetti, A.; Stefani, M.; Tironi, R.; Molteni, E.E.; Conte, D.; et al. Saccharomyces boulardii for the prevention of antibiotic-associated diarrhea in adult hospitalized patients: A single-center, randomized, double-blind, placebo-controlled trial. Am. J. Gastroenterol. 2012, 107, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Pochapin, M. The effect of probiotics on Clostridium difficile diarrhea. Am. J. Gastroenterol. 2000, 95, S11–S13. [Google Scholar] [CrossRef]
- Wullt, M.; Hagslatt, M.L.; Odenholt, I. Lactobacillus plantarum 299v for the treatment of recurrent Clostridium difficile-associated diarrhoea: A double-blind, placebo-controlled trial. Scand. J. Infect. Dis. 2003, 35, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Wareham, K.; Wang, D.; Bradley, C.; Hutchings, H.; Harris, W.; Dhar, A.; Brown, H.; Foden, A.; Gravenor, M.B.; et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2013, 382, 1249–1257. [Google Scholar] [CrossRef]
- Allen, S.J.; Wareham, K.; Wang, D.; Bradley, C.; Sewell, B.; Hutchings, H.; Harris, W.; Dhar, A.; Brown, H.; Foden, A.; et al. A high-dose preparation of lactobacilli and bifidobacteria in the prevention of antibiotic-associated and Clostridium difficile diarrhoea in older people admitted to hospital: A multicentre, randomised, double-blind, placebo-controlled, parallel arm trial (placide). Health Technol. Assess. 2013, 17, 1–140. [Google Scholar] [PubMed]
- Evans, C.T.; Johnson, S. Prevention of Clostridium difficile infection with probiotics. Clin. Infect. Dis. 2015, 60 (Suppl. 2), S122–S128. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D. Digital biology: A new era has begun. Bioengineered 2012, 3, 311–312. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D. The synthetic biology future. Bioengineered 2014, 5, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, V.M.; Sleator, R.D.; Fitzgerald, G.F.; Hill, C. Heterologous expression of Betl, a betaine uptake system, enhances the stress tolerance of Lactobacillus salivarius UCC118. Appl. Environ. Microbiol. 2006, 72, 2170–2177. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, V.M.; Sleator, R.D.; Hill, C.; Fitzgerald, G.F. Improving gastric transit, gastrointestinal persistence and therapeutic efficacy of the probiotic strain Bifidobacterium breve UCC2003. Microbiology 2007, 153, 3563–3571. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D. Designer probiotics: Development and applications in gastrointestinal health. World J. Gastrointest. Pathophysiol. 2015, 6, 73–78. [Google Scholar] [PubMed]
- Sleator, R.D.; Hill, C. Patho-biotechnology: Using bad bugs to do good things. Curr. Opin. Biotechnol. 2006, 17, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D.; Hill, C. Patho-biotechnology; using bad bugs to make good bugs better. Sci. Prog. 2007, 90, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.; Sleator, R.D.; Hill, C.; Gahan, C.G. Enhancing bile tolerance improves survival and persistence of Bifidobacterium and Lactococcus in the murine gastrointestinal tract. BMC Microbiol. 2008, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.Y.; Tan, M.H.; Koh, E.; Ho, C.L.; Poh, C.L.; Chang, M.W. Reprogramming microbes to be pathogen-seeking killers. ACS Synth. Biol. 2014, 3, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, N.; Wong, C.; Lo, T.; Nguyen, H.; Ling, H.; Leong, S.; Poh, C.; Chang, M. Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Mol. Syst. Biol. 2011, 7, 521. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.W.; Morona, R.; Paton, J.C. A new biological agent for treatment of shiga toxigenic Escherichia coli infections and dysentery in humans. Nat. Med. 2000, 6, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Focareta, A.; Paton, J.C.; Morona, R.; Cook, J.; Paton, A.W. A recombinant probiotic for treatment and prevention of cholera. Gastroenterology 2006, 130, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.W.; Jennings, M.P.; Morona, R.; Wang, H.; Focareta, A.; Roddam, L.F.; Paton, J.C. Recombinant probiotics for treatment and prevention of enterotoxigenic Escherichia coli diarrhea. Gastroenterology 2005, 128, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Din, M.O.; Danino, T.; Prindle, A.; Skalak, M.; Selimkhanov, J.; Allen, K.; Julio, E.; Atolia, E.; Tsimring, L.S.; Bhatia, S.N.; et al. Synchronized cycles of bacterial lysis forin vivo delivery. Nature 2016, 536, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D.; Hill, C. Designer probiotics: A potential therapeutic for Clostridium difficile? J. Med. Microbiol. 2008, 57, 793–794. [Google Scholar] [CrossRef] [PubMed]
- Antunes, L.C.; Han, J.; Ferreira, R.B.; Lolic, P.; Borchers, C.H.; Finlay, B.B. Effect of antibiotic treatment on the intestinal metabolome. Antimicrob. Agents Chemother. 2011, 55, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Antunes, L.C.; McDonald, J.A.; Schroeter, K.; Carlucci, C.; Ferreira, R.B.; Wang, M.; Yurist-Doutsch, S.; Hira, G.; Jacobson, K.; Davies, J.; et al. Antivirulence activity of the human gut metabolome. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Darkoh, C.; DuPont, H.L.; Norris, S.J.; Kaplan, H.B. Toxin synthesis by Clostridium difficile is regulated through quorum signaling. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.O.; Garland, M.; Ferreyra, J.A.; Hryckowian, A.J.; Child, M.A.; Puri, A.W.; Solow-Cordero, D.E.; Higginbottom, S.K.; Segal, E.; Banaei, N.; et al. A small-molecule antivirulence agent for treating Clostridium difficile infection. Sci. Transl. Med. 2015, 7, 306ra148. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Ebselen study group. Stroke J. Cereb. Circ. 1998, 29, 12–17. [Google Scholar] [CrossRef]
- Tam, J.; Beilhartz, G.L.; Auger, A.; Gupta, P.; Therien, A.G.; Melnyk, R.A. Small molecule inhibitors of Clostridium difficile toxin B-induced cellular damage. Chem. Biol. 2015, 22, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Ripert, G.; Racedo, S.M.; Elie, A.M.; Jacquot, C.; Bressollier, P.; Urdaci, M.C. Secreted compounds of the probiotic bacillus clausii strain O/C inhibit the cytotoxic effects induced by Clostridium difficile and Bacillus cereus toxins. Antimicrob. Agents Chemother. 2016, 60, 3445–3454. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; Dreger, S.C.; Gerhard, R.; Fritz, G.; Just, I.; Genth, H. Upregulation of the immediate early gene product rhob by exoenzyme C3 from clostridium limosum and toxin B from Clostridium difficile. Biochemistry 2007, 46, 4923–4931. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Donia, M.S.; Cimermancic, P.; Schulze, C.J.; Wieland Brown, L.C.; Martin, J.; Mitreva, M.; Clardy, J.; Linington, R.G.; Fischbach, M.A. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 2014, 158, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.T.; Freed, S.D.; Lee, S.W.; Friedberg, I. A large scale prediction of bacteriocin gene blocks suggests a wide functional spectrum for bacteriocins. BMC Bioinform. 2015, 16, 381. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.J.; Guinane, C.M.; Hill, C.; Ross, R.P.; O’Toole, P.W.; Cotter, P.D. In silico identification of bacteriocin gene clusters in the gastrointestinal tract, based on the human microbiome project’s reference genome database. BMC Microbiol. 2015, 15, 183. [Google Scholar] [CrossRef] [PubMed]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Kommineni, S.; Bretl, D.J.; Lam, V.; Chakraborty, R.; Hayward, M.; Simpson, P.; Cao, Y.; Bousounis, P.; Kristich, C.J.; Salzman, N.H. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 2015, 526, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.C.; Sit, C.S.; Clayton, E.; O’Connor, P.M.; Whittal, R.M.; Zheng, J.; Vederas, J.C.; Ross, R.P.; Hill, C. Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc. Natl. Acad. Sci. USA 2010, 107, 9352–9357. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.C.; Dobson, A.; O’Sullivan, O.; Crispie, F.; Fouhy, F.; Cotter, P.D.; Shanahan, F.; Kiely, B.; Hill, C.; Ross, R.P. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4639–4644. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.C.; Alemayehu, D.; Casey, P.G.; O’Connor, P.M.; Lawlor, P.G.; Walsh, M.; Shanahan, F.; Kiely, B.; Ross, R.P.; Hill, C. Bioavailability of the anti-clostridial bacteriocin thuricin CD in gastrointestinal tract. Microbiology 2014, 160, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; O’Sullivan, O.; Rea, M.C.; Cotter, P.D.; Ross, R.P.; Hill, C. Genome mining for radical sam protein determinants reveals multiple sactibiotic-like gene clusters. PLoS ONE 2011, 6, e20852. [Google Scholar] [CrossRef] [PubMed]
- Bartoloni, A.; Mantella, A.; Goldstein, B.P.; Dei, R.; Benedetti, M.; Sbaragli, S.; Paradisi, F. In vitro activity of nisin against clinical isolates of Clostridium difficile. J. Chemother. 2004, 16, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, C.; Fernandez, B.; Hammami, R.; Ouellette, M.; Fliss, I. On lactococcus lactis ul719 competitivity and nisin (Nisaplin®) capacity to inhibit Clostridium difficile in a model of human colon. Front. Microbiol. 2015, 6, 1020. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.P.; Rea, M.C.; Hill, C.; Ross, R.P. An application in cheddar cheese manufacture for a strain of Lactococcus lactis producing a novel broad-spectrum bacteriocin, lacticin 3147. Appl. Environ. Microbiol. 1996, 62, 612–619. [Google Scholar] [PubMed]
- Rea, M.C.; Clayton, E.; O’Connor, P.M.; Shanahan, F.; Kiely, B.; Ross, R.P.; Hill, C. Antimicrobial activity of lacticin 3147 against clinical Clostridium difficile strains. J. Med. Microbiol. 2007, 56, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Gebhart, D.; Williams, S.R.; Bishop-Lilly, K.A.; Govoni, G.R.; Willner, K.M.; Butani, A.; Sozhamannan, S.; Martin, D.; Fortier, L.C.; Scholl, D. Novel high-molecular-weight, R-type bacteriocins of Clostridium difficile. J. Bacteriol. 2012, 194, 6240–6247. [Google Scholar] [CrossRef] [PubMed]
- Gebhart, D.; Lok, S.; Clare, S.; Tomas, M.; Stares, M.; Scholl, D.; Donskey, C.J.; Lawley, T.D.; Govoni, G.R. A modified R-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Boakes, S.; Ayala, T.; Herman, M.; Appleyard, A.N.; Dawson, M.J.; Cortes, J. Generation of an actagardine a variant library through saturation mutagenesis. Appl. Microbiol. Biotechnol. 2012, 95, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wilson-Stanford, S.; Cromwell, W.; Hillman, J.D.; Guerrero, A.; Allen, C.A.; Sorg, J.A.; Smith, L. Site-directed mutations in the lanthipeptide mutacin 1140. Appl. Environ. Microbiol. 2013, 79, 4015–4023. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.S.; Baines, S.D.; Todhunter, S.L.; Freeman, J.; Chilton, C.H.; Wilcox, M.H. Evaluation of NVB302 versus vancomycin activity in an in vitro human gut model of Clostridium difficile infection. J. Antimicrob. Chemother. 2013, 68, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Field, D.; Quigley, L.; O’Connor, P.M.; Rea, M.C.; Daly, K.; Cotter, P.D.; Hill, C.; Ross, R.P. Studies with bioengineered nisin peptides highlight the broad-spectrum potency of Nisin V. Microb. Biotechnol. 2010, 3, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Jarrad, A.M.; Karoli, T.; Blaskovich, M.A.; Lyras, D.; Cooper, M.A. Clostridium difficile drug pipeline: Challenges in discovery and development of new agents. J. Med. Chem. 2015, 58, 5164–5185. [Google Scholar] [CrossRef] [PubMed]
- Mathur, H.; Rea, M.C.; Cotter, P.D.; Ross, R.P.; Hill, C. The potential for emerging therapeutic options for Clostridium difficile infection. Gut Microbes 2014, 5, 696–710. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Culligan, E.P.; Sleator, R.D. Advances in the Microbiome: Applications to Clostridium difficile Infection. J. Clin. Med. 2016, 5, 83. https://doi.org/10.3390/jcm5090083
Culligan EP, Sleator RD. Advances in the Microbiome: Applications to Clostridium difficile Infection. Journal of Clinical Medicine. 2016; 5(9):83. https://doi.org/10.3390/jcm5090083
Chicago/Turabian StyleCulligan, Eamonn P., and Roy D. Sleator. 2016. "Advances in the Microbiome: Applications to Clostridium difficile Infection" Journal of Clinical Medicine 5, no. 9: 83. https://doi.org/10.3390/jcm5090083
APA StyleCulligan, E. P., & Sleator, R. D. (2016). Advances in the Microbiome: Applications to Clostridium difficile Infection. Journal of Clinical Medicine, 5(9), 83. https://doi.org/10.3390/jcm5090083