
Emerging Transcriptional Mechanisms in the Regulation of Epithelial to Mesenchymal Transition and Cellular Plasticity in the Kidney

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
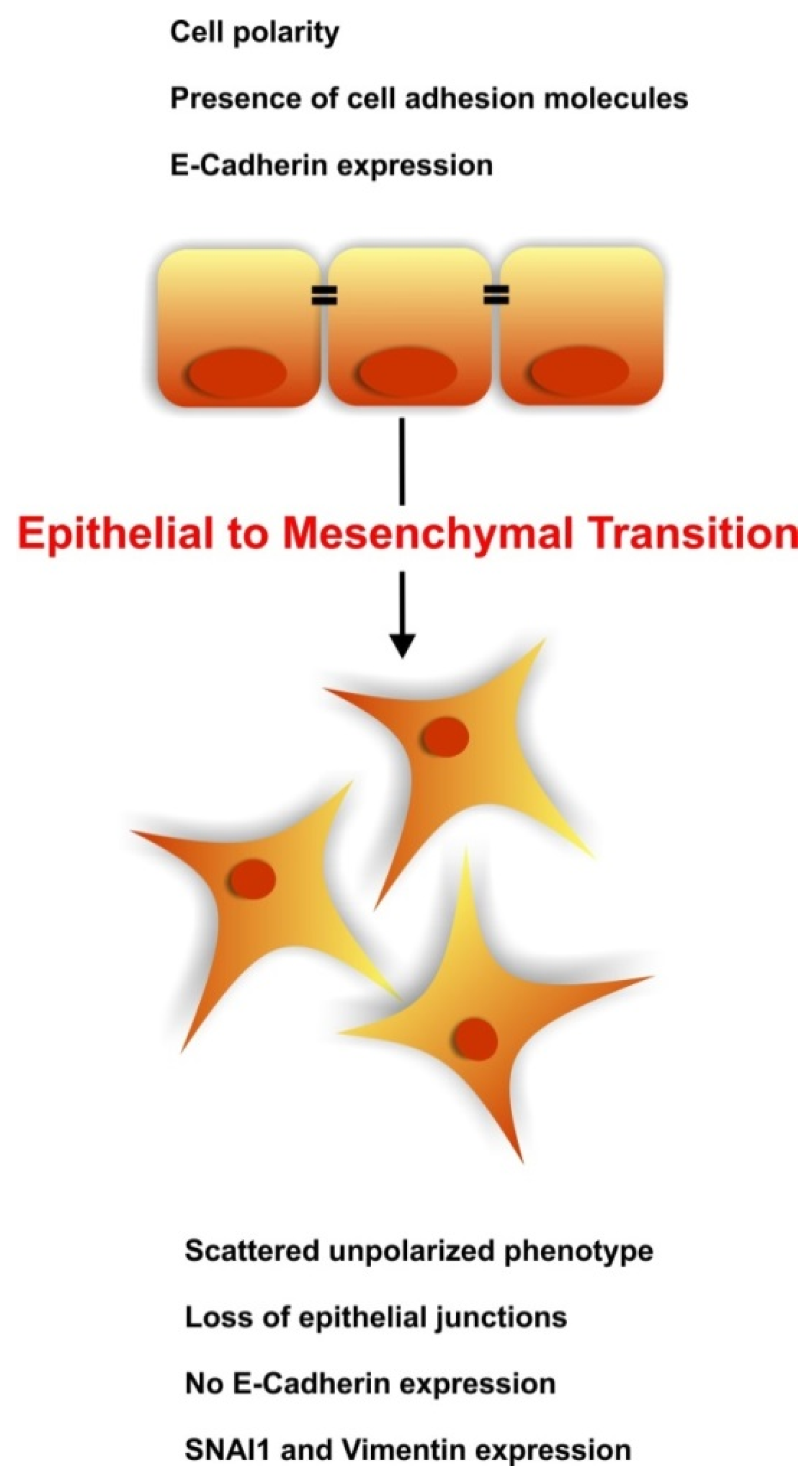
:1. Introduction
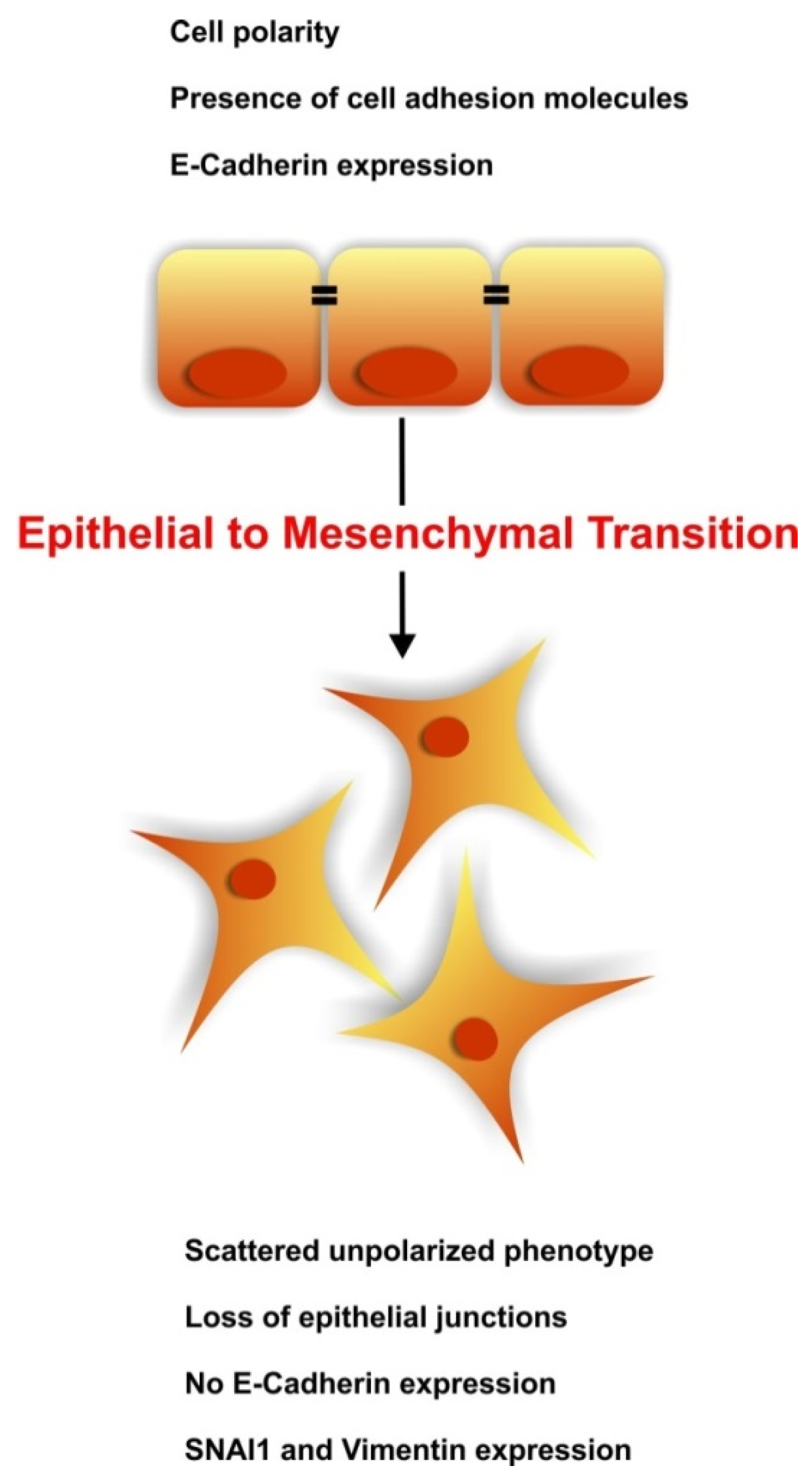
2. The Kidney at a Glance
Renal Development
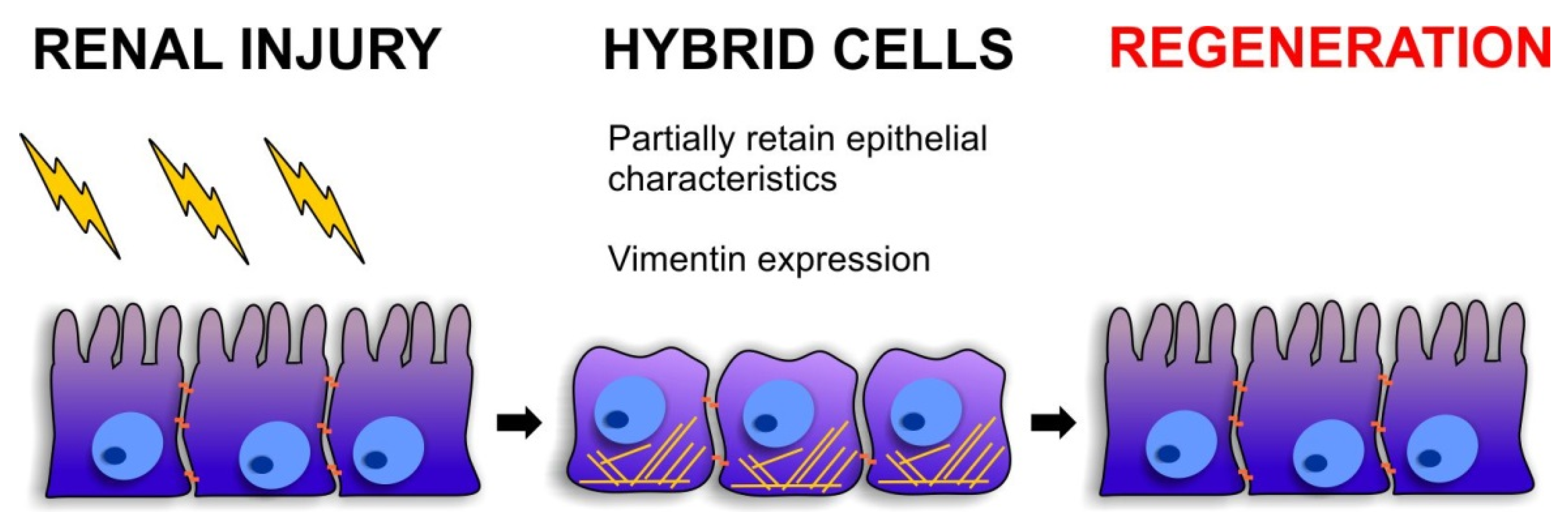
3. The Regenerative Potential of EMT
3.1. The Origin of the Myofibroblasts during Renal Disease
3.2. Cellular Plasticity during Renal Repair
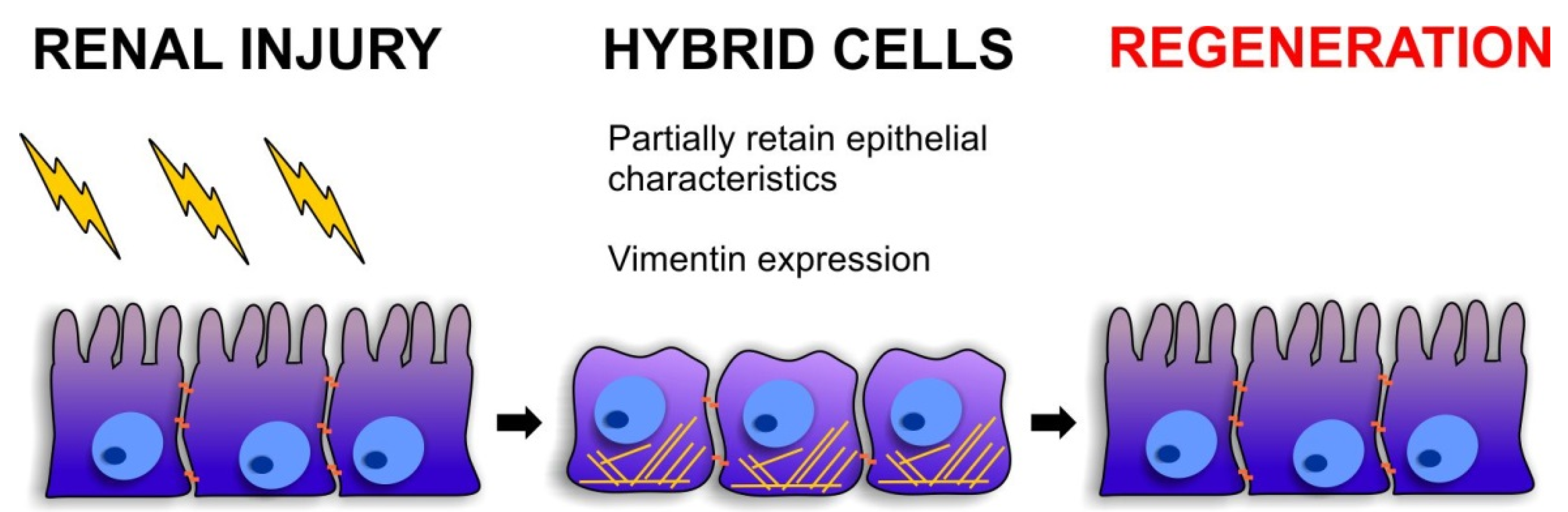
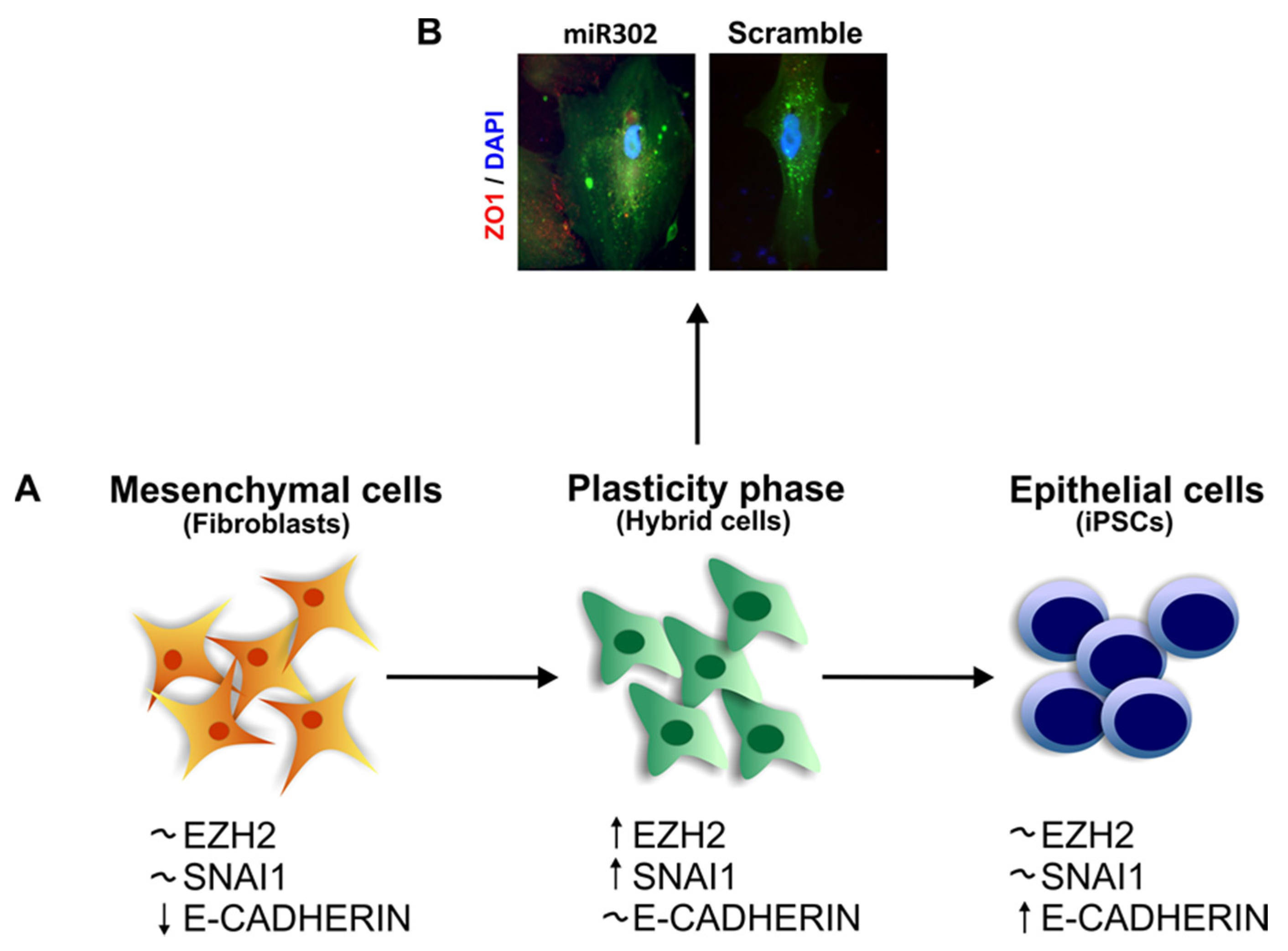
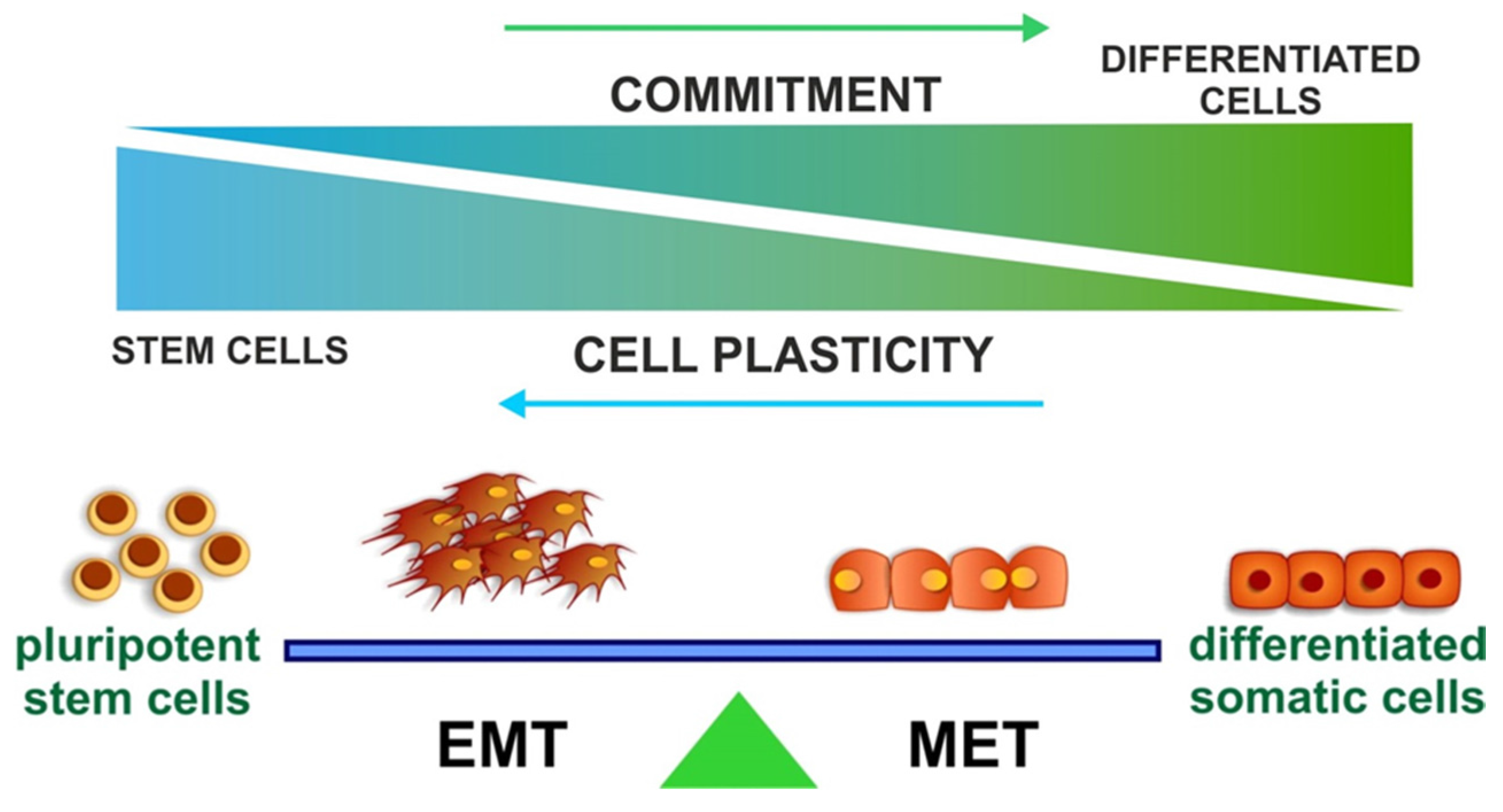
4. EMT and Stemness
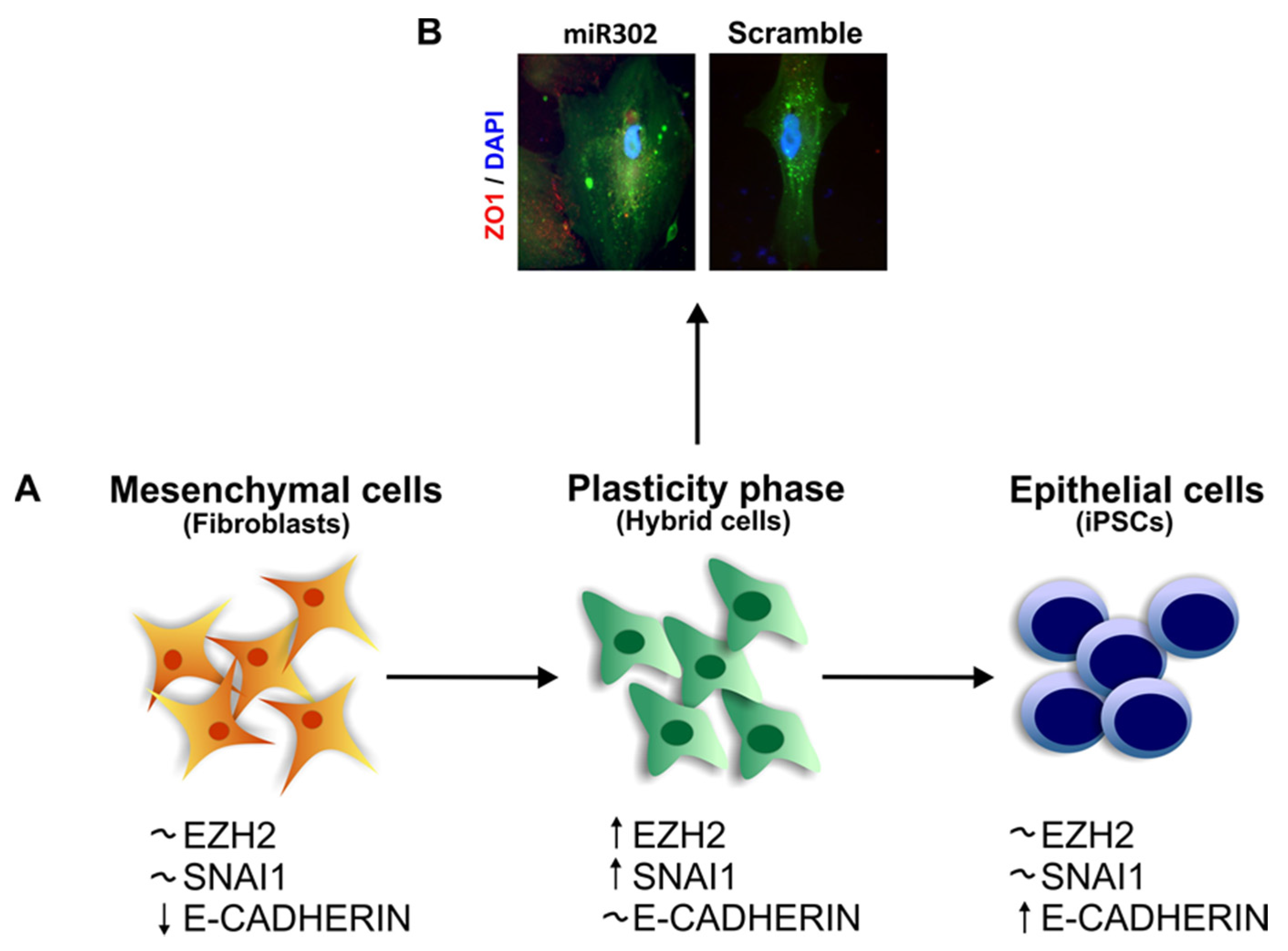
The Role of SNAI1 during Pluripotency and Plasticity Acquisition
5. Chromatin Remodeling during EMT
5.1. The PRC2 Axis
5.2. The S”MAD” Idea
6. Conclusions
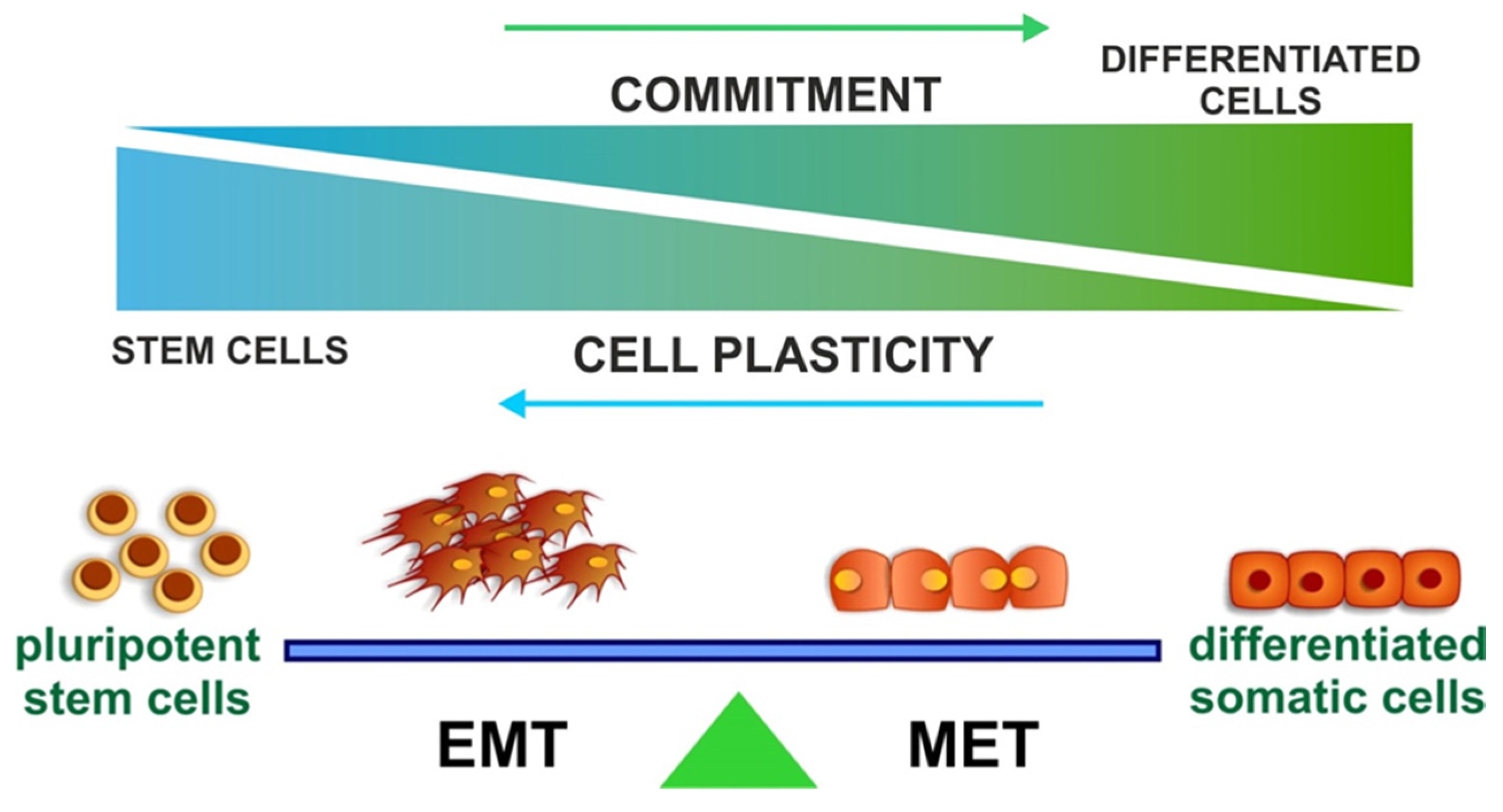
Acknowledgments
Conflicts of Interest
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [PubMed]
- Burns, W.C.; Thomas, M.C. The molecular mediators of type 2 epithelial to mesenchymal transition (EMT) and their role in renal pathophysiology. Expert Rev. Mol. Med. 2010, 12, e17. [Google Scholar] [CrossRef] [PubMed]
- Ombrato, L.; Malanchi, I. The EMT universe: Space between cancer cell dissemination and metastasis initiation. Crit. Rev. Oncog. 2014, 19, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, H.; Qi, J.; Wang, L.; He, S.; Liu, J.; Feng, C.; Chen, C.; Li, W.; Guo, Y.; et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat. Cell Biol. 2013, 15, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Rony, I.K.; Baten, A.; Bloomfield, J.A.; Islam, M.E.; Billah, M.M.; Islam, K.D. Inducing pluripotency in vitro: Recent advances and highlights in induced pluripotent stem cells generation and pluripotency reprogramming. Cell Prolif. 2015, 48, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, i37–i45. [Google Scholar] [CrossRef] [PubMed]
- Castro, N.E.; Kato, M.; Park, J.T.; Natarajan, R. Transforming growth factor β1 (TGF-β1) enhances expression of profibrotic genes through a novel signaling cascade and microRNAs in renal mesangial cells. J. Biol. Chem. 2014, 289, 29001–29013. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [PubMed]
- Zambon, J.P.; Magalhaes, R.S.; Ko, I.; Ross, C.L.; Orlando, G.; Peloso, A.; Atala, A.; Yoo, J.J. Kidney regeneration: Where we are and future perspectives. World J. Nephrol. 2014, 3, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Little, M.H.; McMahon, A.P. Mammalian kidney development: Principles, progress, and projections. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Kusaba, T.; Humphreys, B.D. Who regenerates the kidney tubule? Nephrol. Dial. Transplant. 2015, 30, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol. JASN 2003, 14, S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2014, 111, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Smeets, B.; Boor, P.; Dijkman, H.; Sharma, S.V.; Jirak, P.; Mooren, F.; Berger, K.; Bornemann, J.; Gelman, I.H.; Floege, J.; et al. Proximal tubular cells contain a phenotypically distinct, scattered cell population involved in tubular regeneration. J. Pathol. 2013, 229, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Sagrinati, C.; Netti, G.S.; Mazzinghi, B.; Lazzeri, E.; Liotta, F.; Frosali, F.; Ronconi, E.; Meini, C.; Gacci, M.; Squecco, R.; et al. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J. Am. Soc. Nephrol. JASN 2006, 17, 2443–2456. [Google Scholar] [CrossRef] [PubMed]
- Angelotti, M.L.; Ronconi, E.; Ballerini, L.; Peired, A.; Mazzinghi, B.; Sagrinati, C.; Parente, E.; Gacci, M.; Carini, M.; Rotondi, M.; et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells 2012, 30, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G. Renal progenitors in non-diabetic and diabetic nephropathies. Trends Endocrinol. Metab. TEM 2013, 24, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Crescioli, C.; Ronconi, E.; Mazzinghi, B.; Sagrinati, C.; Netti, G.S.; Angelotti, M.L.; Parente, E.; Ballerini, L.; Cosmi, L.; et al. Regenerative potential of embryonic renal multipotent progenitors in acute renal failure. J. Am. Soc. Nephrol. JASN 2007, 18, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Grobstein, C. Inductive epitheliomesenchymal interaction in cultured organ rudiments of the mouse. Science 1953, 118, 52–55. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.L.; McMahon, A.P. Induction and patterning of the metanephric nephron. Semin. Cell Dev. Biol. 2014, 36, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef]
- Torres, M.; Gomez-Pardo, E.; Dressler, G.R.; Gruss, P. Pax-2 controls multiple steps of urogenital development. Development 1995, 121, 4057–4065. [Google Scholar] [PubMed]
- Self, M.; Lagutin, O.V.; Bowling, B.; Hendrix, J.; Cai, Y.; Dressler, G.R.; Olivier, G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006, 25, 5214–5228. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Valerius, M.T.; Mugford, J.W.; Carroll, T.J.; Self, M.; Oliver, G.; McMahon, A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 2008, 3, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Imgrund, M.; Grone, E.; Grone, H.J.; Kretzler, M.; Holzman, L.; Schlondorff, D.; Rothenpieler, U.W. Re-expression of the developmental gene Pax-2 during experimental acute tubular necrosis in mice 1. Kidney Int. 1999, 56, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Loutochin, O.; Amin, M.; Capolicchio, J.P.; Goodyer, P.; Jednak, R. PAX2 is reactivated in urinary tract obstruction and partially protects collecting duct cells from programmed cell death. Am. J. Physiol. Ren. Physiol. 2007, 292, F1267–F1273. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Doerner, A.M.; Zuraw, B.L. TGF-β1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1β but not abrogated by corticosteroids. Respir. Res. 2009, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Masszi, A.; di Ciano, C.; Sirokmany, G.; Arthur, W.T.; Rotstein, O.D.; Wang, J.; McCulloch, C.A.; Rosivall, L.; Mucsi, I.; Kapus, A. Central role for Rho in TGF-β1-induced α-smooth muscle actin expression during epithelial-mesenchymal transition. Am. J. Physiol. Ren. Physiol. 2003, 284, F911–F924. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.M.; Ng, Y.Y.; Hill, P.A.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Transforming growth factor-β regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999, 56, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Hosper, N.A.; van den Berg, P.P.; de Rond, S.; Popa, E.R.; Wilmer, M.J.; Masereeuw, R.; Bank, R.A. Epithelial-to-mesenchymal transition in fibrosis: Collagen type I expression is highly upregulated after EMT, but does not contribute to collagen deposition. Exp. Cell Res. 2013, 319, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Danoff, T.M.; Kalluri, R.; Neilson, E.G. Early role of Fsp1 in epithelial-mesenchymal transformation. Am. J. Physiol. 1997, 273, F563–F574. [Google Scholar] [PubMed]
- Kong, P.; Christia, P.; Saxena, A.; Su, Y.; Frangogiannis, N.G. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1363–H1372. [Google Scholar] [CrossRef] [PubMed]
- Osterreicher, C.H.; Penz-Osterreicher, M.; Grivennikov, S.I.; Guma, M.; Koltsova, E.K.; Datz, C.; Sasik, R.; Hardiman, G.; Karin, M.; Brenner, D.A. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. USA 2011, 108, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Le Hir, M.; Hegyi, I.; Cueni-Loffing, D.; Loffing, J.; Kaissling, B. Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem. Cell Biol. 2005, 123, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Plieth, D.; Venkov, C.D.; Xu, C.; Neilson, E.G. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005, 67, 2488–2493. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Koesters, R.; Kaissling, B.; Lehir, M.; Picard, N.; Theilig, F.; Gebhardt, R.; Glick, A.B.; Hahnel, B.; Hosser, H.; Grone, H.J.; et al. Tubular overexpression of transforming growth factor-β1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am. J. Pathol. 2010, 177, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Nakasatomi, M.; Maeshima, A.; Mishima, K.; Ikeuchi, H.; Sakairi, T.; Kaneko, Y.; Hiromura, K.; Nojima, Y. Novel approach for the detection of tubular cell migration into the interstitium during renal fibrosis in rats. Fibrogenesis Tissue Repair 2015, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Kaissling, B.; le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Investig. 2011, 121, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; de Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Anglani, F.; Ceol, M.; Mezzabotta, F.; Torregrossa, R.; Tiralongo, E.; Tosetto, E.; Del Prete, D.; D’Angelo, A. The renal stem cell system in kidney repair and regeneration. Front. Biosci. A J. Virtual Libr. 2008, 13, 6395–6405. [Google Scholar] [CrossRef]
- Jiang, Y.S.; Jiang, T.; Huang, B.; Chen, P.S.; Ouyang, J. Epithelial-mesenchymal transition of renal tubules: Divergent processes of repairing in acute or chronic injury? Med. Hypotheses 2013, 81, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Swetha, G.; Chandra, V.; Phadnis, S.; Bhonde, R. Glomerular parietal epithelial cells of adult murine kidney undergo EMT to generate cells with traits of renal progenitors. J. Cell. Mol. Med. 2011, 15, 396–413. [Google Scholar] [PubMed]
- Huang, B.; Pi, L.; Chen, C.; Yuan, F.; Zhou, Q.; Teng, J.; Jiang, T. WT1 and Pax2 re-expression is required for epithelial-mesenchymal transition in 5/6 nephrectomized rats and cultured kidney tubular epithelial cells. Cells Tissues Organs 2012, 195, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, T.; Ouyang, J.; Zhou, Q.; Liang, Y.; Cui, Y.; Chen, P.; Huang, B. Cell atavistic transition: Paired box 2 re-expression occurs in mature tubular epithelial cells during acute kidney injury and is regulated by Angiotensin II. PLoS ONE 2014, 9, e93563. [Google Scholar] [CrossRef] [PubMed]
- Hendry, C.E.; Vanslambrouck, J.M.; Ineson, J.; Suhaimi, N.; Takasato, M.; Rae, F.; Little, M.H. Direct transcriptional reprogramming of adult cells to embryonic nephron progenitors. J. Am. Soc. Nephrol. JASN 2013, 24, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Leroy, P.; Mostov, K.E. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol. Biol. Cell 2007, 18, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, L.; Fagoonee, S.; Ranghino, A.; Bruno, S.; Camussi, G.; Tolosano, E.; Silengo, L.; Altruda, F. Renal cells from spermatogonial germline stem cells protect against kidney injury. J. Am. Soc. Nephrol. JASN 2014, 25, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schondorf, D.C.; Wagner, L.; Glatza, M.; Hoing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.J.; Dariolli, R.; Jorge, F.M.; Monteiro, M.R.; Maximino, J.R.; Martins, R.S.; Strauss, B.E.; Krieger, J.E.; Callegaro, D.; Chadi, G. Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 2015, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Saha, K.; Pando, B.; van Zon, J.; Lengner, C.J.; Creyghton, M.P.; van Oudenaarden, A.; Jaenisch, R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 2009, 462, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Hu, K. All roads lead to induced pluripotent stem cells: The technologies of iPSC generation. Stem Cells Dev. 2014, 23, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Ruetz, T.; Kaji, K. Routes to induced pluripotent stem cells. Curr. Opin. Genet. Dev. 2014, 28, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Revilla, A.; Gonzalez, C.; Iriondo, A.; Fernandez, B.; Prieto, C.; Marin, C.; Liste, I. Current advances in the generation of human iPS cells: Implications in cell-based regenerative medicine. J. Tissue Eng. Regen. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Csobonyeiova, M.; Polak, S.; Koller, J.; Danisovic, L. Induced pluripotent stem cells and their implication for regenerative medicine. Cell Tissue Bank. 2015, 16, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Rao, M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [PubMed]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Card, D.A.; Hebbar, P.B.; Li, L.; Trotter, K.W.; Komatsu, Y.; Mishina, Y.; Archer, T.K. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 2008, 28, 6426–6438. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wilson, K.D.; Ghosh, Z.; Han, L.; Wang, Y.; Lan, F.; Ransohoff, K.J.; Burridge, P.; Wu, J.C. MicroRNA-302 increases reprogramming efficiency via repression of NR2F2. Stem Cells 2013, 31, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lipchina, I.; Studer, L.; Betel, D. The expanding role of miR-302-367 in pluripotency and reprogramming. Cell Cycle 2012, 11, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Faherty, N.; Curran, S.P.; O’Donovan, H.; Martin, F.; Godson, C.; Brazil, D.P.; Crean, J.K. CCN2/CTGF increases expression of miR-302 microRNAs, which target the TGFβ type II receptor with implications for nephropathic cell phenotypes. J. Cell Sci. 2012, 125, 5621–5629. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, L.; Andrews, D.; Godson, C.; Crean, J. Targeting the Polycomb Repressor Complex Chromatin Remodeling Machinery for Therapeutic Benefit in Diabetic Nephropathy; American Society of Nephrology, ASN, Kidney Week: San Diego, CA, USA, 2015. [Google Scholar]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the microRNA-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriction in differentiating ESCs. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Unternaehrer, J.J.; Zhao, R.; Kim, K.; Cesana, M.; Powers, J.T.; Ratanasirintrawoot, S.; Onder, T.; Shibue, T.; Weinberg, R.A.; Daley, G.Q. The epithelial-mesenchymal transition factor SNAIL paradoxically enhances reprogramming. Stem Cell Rep. 2014, 3, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Melton, C.; Judson, R.L.; Blelloch, R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature 2010, 463, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Li, M.A.; He, L. microRNAs as novel regulators of stem cell pluripotency and somatic cell reprogramming. Bioessays 2012, 34, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Ahfeldt, T.; Rigamonti, A.; Utikal, J.; Cowan, C.; Hochedlinger, K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 2008, 3, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014, 2014, 768391. [Google Scholar] [CrossRef] [PubMed]
- Gingold, J.A.; Fidalgo, M.; Guallar, D.; Lau, Z.; Sun, Z.; Zhou, H.; Faiola, F.; Huang, X.; Lee, D.F.; Waghray, A.; et al. A genome-wide RNAi screen identifies opposing functions of Snai1 and Snai2 on the Nanog dependency in reprogramming. Mol. Cell 2014, 56, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Lalonde, M.E.; Cote, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; LeRoy, G.; Drury, W.J., 3rd; Zee, B.M.; Son, J.; Beck, D.B.; Young, N.L.; Garcia, B.A.; Reinberg, D. Asymmetrically modified nucleosomes. Cell 2012, 151, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Van Kruijsbergen, I.; Hontelez, S.; Veenstra, G.J. Recruiting polycomb to chromatin. Int. J. Biochem. Cell Biol. 2015, 67, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Moore, H.M.; Li, X.; Toy, K.A.; Huang, W.; Sabel, M.S.; Kidwell, K.M.; Kleer, C.G. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 3098–3103. [Google Scholar] [CrossRef] [PubMed]
- Volkel, P.; Dupret, B.; le Bourhis, X.; Angrand, P.O. Diverse involvement of EZH2 in cancer epigenetics. Am. J. Transl. Res. 2015, 7, 175–193. [Google Scholar] [PubMed]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu, Z.; Zhong, L.; Wang, H.; Jiang, S.; Long, Q.; Xu, J.; Guo, J. Enhancer of zeste homolog 2 (EZH2) promotes tumour cell migration and invasion via epigenetic repression of E-cadherin in renal cell carcinoma. BJU Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, X.; Chen, Z.; Huang, H.; Jin, Y.; Kolokythas, A.; Wang, A.; Dai, Y.; Wong, D.T.; Zhou, X. Polycomb group protein EZH2-mediated E-cadherin repression promotes metastasis of oral tongue squamous cell carcinoma. Mol. Carcinog. 2013, 52, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Yu, J.; Dhanasekaran, S.M.; Kim, J.H.; Mani, R.S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Yu, J.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Pasini, D.; Diaz, V.M.; Franci, C.; Gutierrez, A.; Dave, N.; Escriva, M.; Hernandez-Munoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Tien, C.L.; Jones, A.; Wang, H.; Gerigk, M.; Nozell, S.; Chang, C. Snail2/Slug cooperates with Polycomb repressive complex 2 (PRC2) to regulate neural crest development. Development 2015, 142, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Oktyabri, D.; Tange, S.; Terashima, M.; Ishimura, A.; Suzuki, T. EED regulates epithelial-mesenchymal transition of cancer cells induced by TGF-β. Biochem. Biophys. Res. Commun. 2014, 453, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Taylor, R.J.; La Torre, A.; Wilken, M.S.; Cox, K.E.; Reh, T.A.; Vetter, M.L. Ezh2 maintains retinal progenitor proliferation, transcriptional integrity, and the timing of late differentiation. Dev. Biol. 2015, 403, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Metsuyanim, S.; Pode-Shakked, N.; Schmidt-Ott, K.M.; Keshet, G.; Rechavi, G.; Blumental, D.; Dekel, B. Accumulation of malignant renal stem cells is associated with epigenetic changes in normal renal progenitor genes. Stem Cells 2008, 26, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, N.; Wang, F.; Saifudeen, Z.; el-Dahr, S.S. In situ histone landscape of nephrogenesis. Epigenetics 2014, 9, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Buzhor, E.; Omer, D.; Harari-Steinberg, O.; Dotan, Z.; Vax, E.; Pri-Chen, S.; Metsuyanim, S.; Pleniceanu, O.; Goldstein, R.S.; Dekel, B. Reactivation of NCAM1 defines a subpopulation of human adult kidney epithelial cells with clonogenic and stem/progenitor properties. Am. J. Pathol. 2013, 183, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.A.; Dhele, N.; Cheemadan, S.; Ketkar, A.; Jayandharan, G.R.; Palakodeti, D.; Rampalli, S. Ezh2 mediated H3K27me3 activity facilitates somatic transition during human pluripotent reprogramming. Sci. Rep. 2015, 5, 8229. [Google Scholar] [CrossRef] [PubMed]
- Akpa, M.M.; Iglesias, D.M.; Chu, L.L.; Cybulsky, M.; Bravi, C.; Goodyer, P.R. Wilms tumor suppressor, WT1, suppresses epigenetic silencing of the β-catenin gene. J. Biol. Chem. 2015, 290, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Tange, S.; Oktyabri, D.; Terashima, M.; Ishimura, A.; Suzuki, T. JARID2 is involved in transforming growth factor-β-induced epithelial-mesenchymal transition of lung and colon cancer cell lines. PLoS ONE 2014, 9, e115684. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.S.; Qu, Y.; Cheng, Y.; Li, W.C.; Rotter, V.; Oyan, A.M.; Kalland, K.H. Global profiling of histone and DNA methylation reveals epigenetic-based regulation of gene expression during epithelial to mesenchymal transition in prostate cells. BMC Genom. 2010, 11, 669. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Jiang, Y.P.; Chen, W.; Li, K.D.; Liu, X.; Gao, S.Y.; Feng, H.; Wang, S.S.; Jiang, J.; Ma, X.R.; et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget 2015, 6, 6797–6810. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor β-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Miyazono, K. Roles of TGF-β family signaling in stem cell renewal and differentiation. Cell Res. 2009, 19, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Huang, J.S. TGF-β control of cell proliferation. J. Cell. Biochem. 2005, 96, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Hill, C.S. TGF-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Loven, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Ruetz, T.; Pfisterer, U.; DiStefano, B.; Johnsson, A.; Choen, E.; Linnarsson, S.; Graf, T.; Parmar, M.; Kaji, K. Identification of a potent signaling pathway that orchestrates both reprogramming and transdifferentiation. In Proceedings of the International Society for Stem Cell Research, Annual Meeting 2015, Stockholm, Sweden, 26 June 2015.
- Andrews, D.; Oliviero, G.; De Chiara, L.; Cagney, G.; Crean, J.; University College Dublin, Dublin, Ireland. Unpublished work. 2015.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Chiara, L.; Crean, J. Emerging Transcriptional Mechanisms in the Regulation of Epithelial to Mesenchymal Transition and Cellular Plasticity in the Kidney. J. Clin. Med. 2016, 5, 6. https://doi.org/10.3390/jcm5010006
De Chiara L, Crean J. Emerging Transcriptional Mechanisms in the Regulation of Epithelial to Mesenchymal Transition and Cellular Plasticity in the Kidney. Journal of Clinical Medicine. 2016; 5(1):6. https://doi.org/10.3390/jcm5010006
Chicago/Turabian StyleDe Chiara, Letizia, and John Crean. 2016. "Emerging Transcriptional Mechanisms in the Regulation of Epithelial to Mesenchymal Transition and Cellular Plasticity in the Kidney" Journal of Clinical Medicine 5, no. 1: 6. https://doi.org/10.3390/jcm5010006
APA StyleDe Chiara, L., & Crean, J. (2016). Emerging Transcriptional Mechanisms in the Regulation of Epithelial to Mesenchymal Transition and Cellular Plasticity in the Kidney. Journal of Clinical Medicine, 5(1), 6. https://doi.org/10.3390/jcm5010006