
Concise Review: Methods and Cell Types Used to Generate Down Syndrome Induced Pluripotent Stem Cells


Abstract
:1. Introduction
2. Procedures Used for the Reprogramming of T21-iPSCs

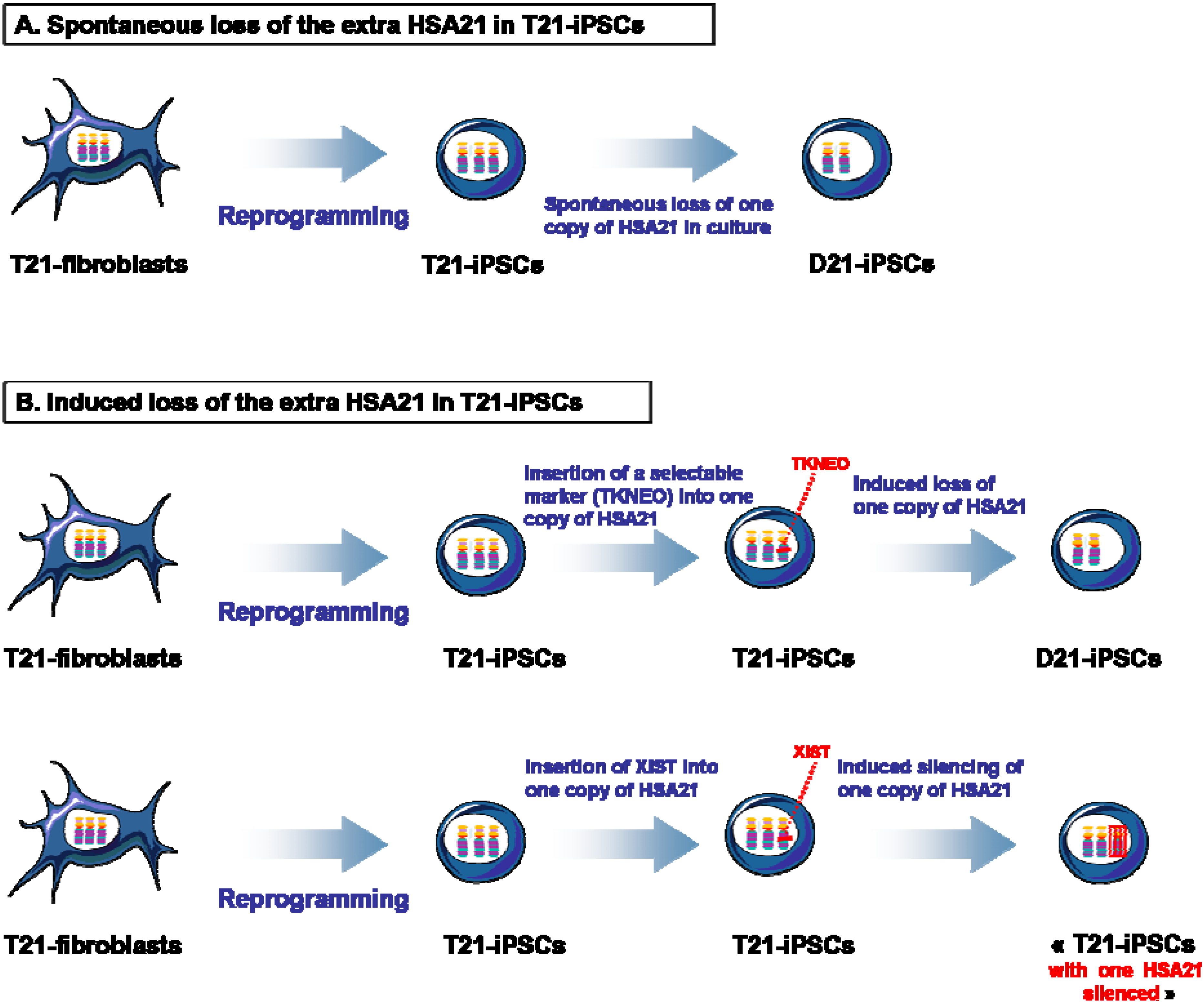
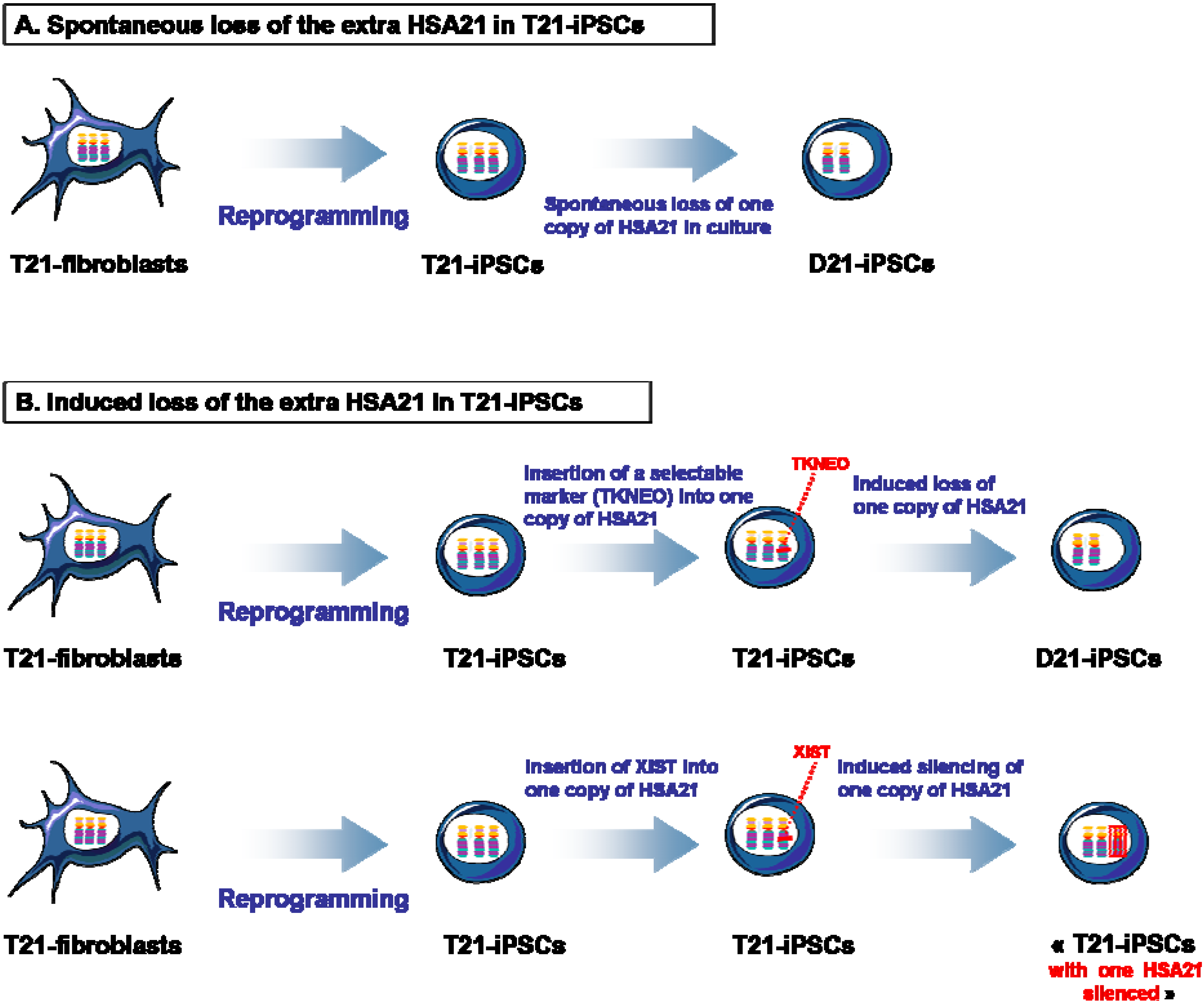{kind=link}
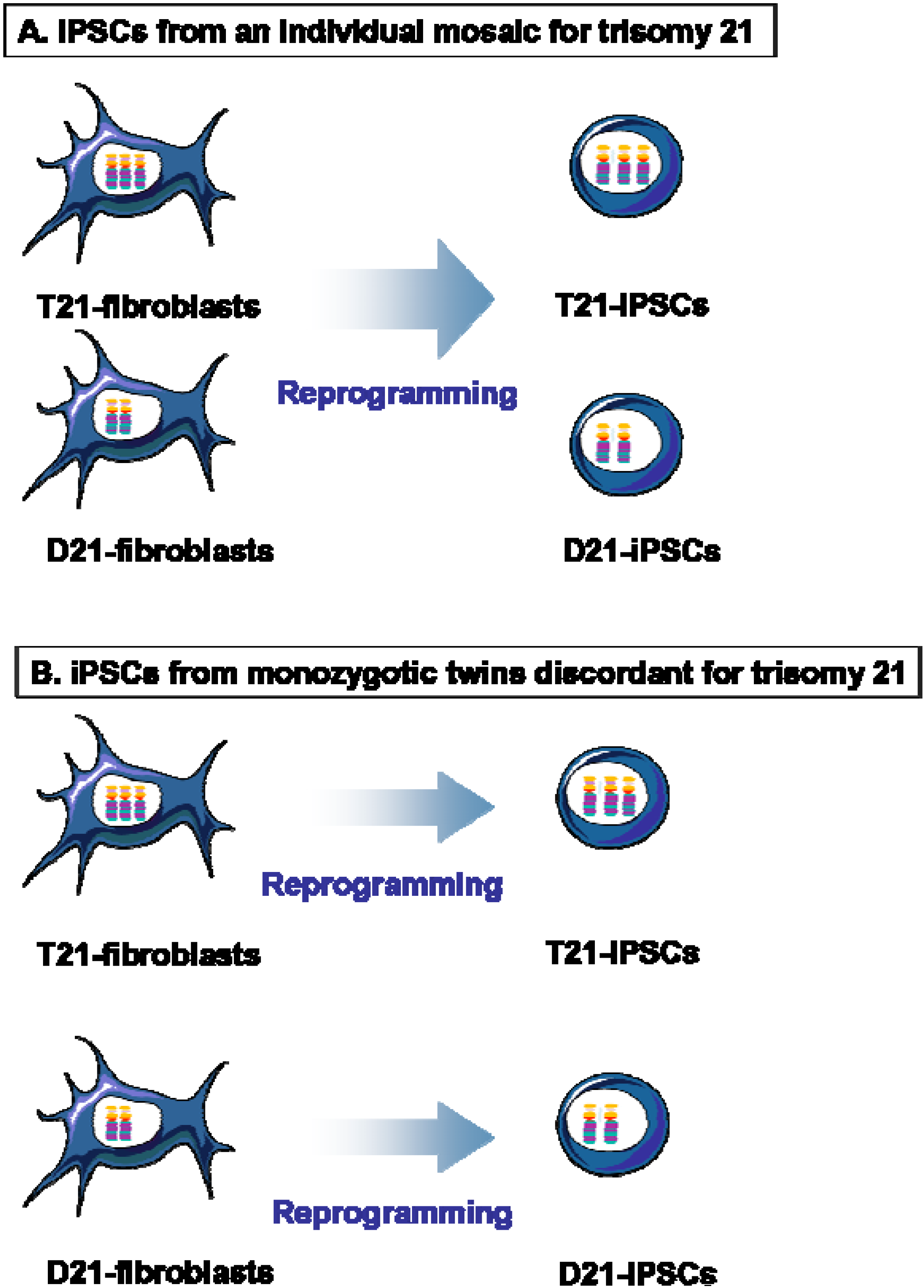{kind=link}
| Type and Age of Donor Cells | Reprogramming Method | Characteristic of the iPSCs | DS Phenotype Investigated | References |
|---|---|---|---|---|
| Fibroblasts from patients (1 year, 1 month) with unrelated controls | Retrovirus with OSKM | The first T21-iPSCs generated | [10] | |
| Fibroblasts from a DS patient (1 year) with unrelated controls | Retrovirus with OSKM | Neurons and AD associated phenotype | [10,13,14] | |
| Skin fibroblasts from DS patients (childs) with no control | Lentivirus with OSKM | T21-iPSCs with different karyotypes for DS | [15] | |
| Amniotic fluid cells (second trimester) with age match control | Lentivirus with OSKM | Reduced number of neurons | [16] | |
| Fibroblasts from DS individuals | Retrovirus with OSKM | Isogenic iPSCs | Myeloid Leukemia | [10,17] |
| Neonatal fibroblasts Fetal stromal cells Fetal mononuclear cells | Doxycycline-induced Lentivirus with OSKM, Retrovirus with OSKM | Myeloid Leukemia | [18] | |
| Fibroblasts from DS individuals | Lentivirus with OSNL | Trisomy 21 deletion through TKNEO | Proliferation and neurogenesis | [12] |
| Fibroblasts from unrelated patients and controls Fibroblasts from a mosaic DS patient. | Episomal vectors with OSK or OSNLM | Non integrating procedures Isogenic iPSCs | Neurogenesis, gliogenic shift | [19] |
| Fibroblasts from unrelated patients and controls Fibroblasts from a mosaic DS patient | Retrovirus with OSKM Sendai virus with OSKM | Isogenic iPSCs | Neuron deficit | [20] |
| Fibroblasts from a DS patient (1 year) | Retrovirus with OSKM Sendai virus with OSKM | Trisomy 21 deletion through Xist | Proliferation and neurogenesis | [10,21] |
| Fetal skin fibroblasts from monozygotic twins discordant for trisomy 21 | Lentivirus with OSKM | Monozygotic twins discordant for trisomy 21 | Neurogenesis, gliogenic shift, rescue of the phenotype | [22,23,24] |
| Fibroblasts | Retrovirus with OSKM | Non-isogenic and isogenic iPSCs | Neurogenesis, gliogenic shift | [25] |
2.1. Integrative Procedures Used for the Derivation of T21-iPSCs
2.2. Non-Integrative Procedures Used for the Derivation of T21-iPSCs
3. Age and Type of the Donor Cells Used for the Reprogramming
4. Isogenic D21-iPSCs and T21-iPSCs

5. Down Syndrome Phenotype Investigated
5.1. Brain-Related Defects
5.2. Myeloid Leukemia
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Antonarakis, S.E.; Lyle, R.; Dermitzakis, E.T.; Reymond, A.; Deutsch, S. Chromosome 21 and down syndrome: From genomics to pathophysiology. Nat. Rev. Genet. 2004, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Lange, B. The management of neoplastic disorders of haematopoeisis in children with Down’s syndrome. Br. J. Haematol. 2000, 110, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Lyle, R.; Bena, F.; Gagos, S.; Gehrig, C.; Lopez, G.; Schinzel, A.; Lespinasse, J.; Bottani, A.; Dahoun, S.; Taine, L.; et al. Genotype-phenotype correlations in Down syndrome identified by array CGH in 30 cases of partial trisomy and partial monosomy chromosome 21. Eur. J. Hum. Genet. 2008, 17, 454–466. [Google Scholar] [CrossRef]
- Korbel, J.O.; Tirosh-Wagner, T.; Urban, A.E.; Chen, X.-N.; Kasowski, M.; Dai, L.; Grubert, F.; Erdman, C.; Gao, M.C.; Lange, K.; et al. The genetic architecture of down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. USA 2009, 106, 12031–12036. [Google Scholar] [CrossRef]
- Das, I.; Reeves, R.H. The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis. Model. Mech. 2011, 4, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Hibaoui, Y.; Feki, A. Human pluripotent stem cells: Applications and challenges in neurological diseases. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q.; et al. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Li, Li B.; Chang, K.-H.; Wang, P.-R.; Hirata, R.K.; Papayannopoulou, T.; Russell, D.W. Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell 2012, 11, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kirwan, P.; Smith, J.; MacLean, G.; Orkin, S.H.; Livesey, F.J. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Vallier, L.; Touboul, T.; Brown, S.; Cho, C.; Bilican, B.; Alexander, M.; Cedervall, J.; Chandran, S.; Ährlund-Richter, L.; Weber, A.; et al. Signaling pathways controlling pluripotency and early cell fate decisions of human induced pluripotent stem cells. Stem Cells 2009, 27, 2655–2666. [Google Scholar] [CrossRef]
- Mou, X.; Wu, Y.; Cao, H.; Meng, Q.; Wang, Q.; Sun, C.; Hu, S.; Ma, Y.; Zhang, H. Generation of disease-specific induced pluripotent stem cells from patients with different karyotypes of Down syndrome. Stem Cell Res. Ther. 2012, 3. [Google Scholar] [CrossRef]
- Lu, H.-E.; Yang, Y.-C.; Chen, S.-M.; Su, H.-L.; Huang, P.-C.; Tsai, M.-S.; Wang, T.-H.; Tseng, C.-P.; Hwang, S.-M. Modeling neurogenesis impairment in Down syndrome with induced pluripotent stem cells from trisomy 21 amniotic fluid cells. Exp. Cell Res. 2013, 319, 498–505. [Google Scholar] [CrossRef] [PubMed]
- MacLean, G.A.; Menne, T.F.; Guo, G.; Sanchez, D.J.; Park, I.-H.; Daley, G.Q.; Orkin, S.H. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 17567–17572. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Byrska-Bishop, M.; Tober, J.M.; Yao, Y.; VanDorn, D.; Opalinska, J.B.; Mills, J.A.; Choi, J.K.; Speck, N.A.; Gadue, P.; et al. Trisomy 21-associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 17573–17578. [Google Scholar] [CrossRef]
- Briggs, J.A.; Sun, J.; Shepherd, J.; Ovchinnikov, D.A.; Chung, T.-L.; Nayler, S.P.; Kao, L.-P.; Morrow, C.A.; Thakar, N.Y.; Soo, S.-Y.; et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of Down syndrome etiology. Stem Cells 2013, 31, 467–478. [Google Scholar] [CrossRef]
- Weick, J.P.; Held, D.L.; Bonadurer, G.F.; Doers, M.E.; Liu, Y.; Maguire, C.; Clark, A.; Knackert, J.A.; Molinarolo, K.; Musser, M.; et al. Deficits in human trisomy 21 iPSCs and neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 9962–9967. [Google Scholar] [CrossRef]
- Jiang, J.; Jing, Y.; Cost, G.J.; Chiang, J.-C.; Kolpa, H.J.; Cotton, A.M.; Carone, D.M.; Carone, B.R.; Shivak, D.A.; Guschin, D.Y.; et al. Translating dosage compensation to trisomy 21. Nature 2013, 500, 296–300. [Google Scholar] [CrossRef]
- Letourneau, A.; Santoni, F.A.; Bonilla, X.; Sailani, M.R.; Gonzalez, D.; Kind, J.; Chevalier, C.; Thurman, R.; Sandstrom, R.S.; Hibaoui, Y.; et al. Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 2014, 508, 345–350. [Google Scholar] [CrossRef]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Sailani, M.R.; Dahoun, S.; Santoni, F.A.; Gimelli, S.; Guipponi, M.; Pelte, M.-F.; Béna, F.; et al. Modelling and rescuing neurodevelopmental defect of Down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 2014, 6, 259–277. [Google Scholar]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Santoni, F.A.; Antonarakis, S.E.; Feki, A. Data in brief: Transcriptome analysis of induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. Genomics Data 2014, 2, 226–229. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Yao, S.; Sukonnik, T.; Kean, T.; Bharadwaj, R.R.; Pasceri, P.; Ellis, J. Retrovirus silencing, variegation, extinction, and memory are controlled by a dynamic interplay of multiple epigenetic modifications. Mol. Ther. 2004, 10, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Markoulaki, S.; Hanna, J.; Saha, K.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc. Natl. Acad. Sci. USA 2009, 106, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Sommer, A.G.; Longmire, T.A.; Christodoulou, C.; Thomas, D.D.; Gostissa, M.; Alt, F.W.; Murphy, G.J.; Kotton, D.N.; Mostoslavsky, G.; et al. Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. Stem Cells 2010, 28, 64–74. [Google Scholar] [PubMed]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.-C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.-L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karyotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. High prevalence of evolutionarily conserved and species-specific genomic aberrations in mouse pluripotent stem cells. Stem Cells 2012, 30, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Martins-Taylor, K.; Nisler, B.S.; Taapken, S.M.; Compton, T.; Crandall, L.; Montgomery, K.D.; Lalande, M.; Xu, R.-H. Recurrent copy number variations in human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.E.C.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 2007, 25, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Hovatta, O.; Jaconi, M.; Töhönen, V.; Béna, F.; Gimelli, S.; Bosman, A.; Holm, F.; Wyder, S.; Zdobnov, E.M.; Irion, O.; et al. A teratocarcinoma-like human embryonic stem cell (hESC) line and four hESC lines reveal potentially oncogenic genomic changes. PLoS ONE 2010, 5, e10263. [Google Scholar] [CrossRef]
- Buzzard, J.J.; Gough, N.M.; Crook, J.M.; Colman, A. Karyotype of human ES cells during extended culture. Nat. Biotechnol. 2004, 22, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Mitalipova, M.M.; Rao, R.R.; Hoyer, D.M.; Johnson, J.A.; Meisner, L.F.; Jones, K.L.; Dalton, S.; Stice, S.L. Preserving the genetic integrity of human embryonic stem cells. Nat. Biotechnol. 2005, 23, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Olariu, V.; Harrison, N.J.; Coca, D.; Gokhale, P.J.; Baker, D.; Billings, S.; Kadirkamanathan, V.; Andrews, P.W. Modeling the evolution of culture-adapted human embryonic stem cells. Stem Cell Res. 2010, 4, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Ramirez, J.-M.; Becker, F.; Pantesco, V.; Lavabre-Bertrand, T.; Hovatta, O.; Lemaître, J.-M.; Pellestor, F.; de Vos, J. Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev. 2015, 24, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Markoulaki, S.; Hanna, J.; Beard, C.; Carey, B.W.; Cheng, A.W.; Lengner, C.J.; Dausman, J.A.; Fu, D.; Gao, Q.; Wu, S.; et al. Transgenic mice with defined combinations of drug-inducible reprogramming factors. Nat. Biotechnol. 2009, 27, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, R.; Tchieu, J.; Mason, M.J.; Yachechko, R.; Kuoy, E.; Horvath, S.; Zhou, Q.; Plath, K. Role of the murine reprogramming factors in the induction of pluripotency. Cell 2009, 136, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S.; et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.L.; Babiarz, J.E.; Venere, M.; Blelloch, R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 2009, 27, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Araki, R.; Hoki, Y.; Uda, M.; Nakamura, M.; Jincho, Y.; Tamura, C.; Sunayama, M.; Ando, S.; Sugiura, M.; Yoshida, M.A.; et al. Crucial role of c-Myc in the generation of induced pluripotent stem cells. Stem Cells 2011, 29, 1362–1370. [Google Scholar] [PubMed]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.N.; Singh, A.M.; Dalton, S. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell 2010, 7, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with Myc. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- Grad, I.; Hibaoui, Y.; Jaconi, M.; Chicha, L.; Bergström-Tengzelius, R.; Sailani, M.R.; Pelte, M.F.; Dahoun, S.; Mitsiadis, T.A.; Töhönen, V.; et al. Nanog priming before full reprogramming may generate germ cell tumours. Eur. Cells Mater. 2011, 22, 258–274. [Google Scholar]
- Hu, B.-Y.; Weick, J.P.; Yu, J.; Ma, L.-X.; Zhang, X.-Q.; Thomson, J.A.; Zhang, S.-C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed]
- Major, T.; Menon, J.; Auyeung, G.; Soldner, F.; Hockemeyer, D.; Jaenisch, R.; Tabar, V. Transgene excision has no impact on in vivo integration of human iPS derived neural precursors. PLoS ONE 2011, 6, e24687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löhle, M.; Hermann, A.; Glaß, H.; Kempe, A.; Schwarz, S.C.; Kim, J.B.; Poulet, C.; Ravens, U.; Schwarz, J.; Schöler, H.R.; et al. Differentiation efficiency of induced pluripotent stem cells depends on the number of reprogramming factors. Stem Cells 2012, 30, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, F.; Boué, S.; Belmonte, J.C.I. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011, 12, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, J.; Shi, G.; Ma, Y.; Yang, Y.; Gu, J.; Yu, H.; Jin, S.; Wei, Z.; Chen, F.; et al. Pluripotency can be rapidly and efficiently induced in human amniotic fluid-derived cells. Hum. Mol. Genet. 2009, 18, 4340–4349. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Hongguang, H.; Loh, Y.-H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Russ, H. A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitaloni, M.; Pulecio, J.; Bilic, J.; Kuebler, B.; Laricchia-Robbio, L.; Izpisua Belmonte, J.C. MicroRNAs contribute to induced pluripotent stem cell somatic donor memory. J. Biol. Chem. 2014, 289, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Georgantas, R.W.; Hildreth, R.; Morisot, S.; Alder, J.; Liu, C.-G.; Heimfeld, S.; Calin, G.A.; Croce, C.M.; Civin, C.I. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: A circuit diagram of differentiation control. Proc. Natl. Acad. Sci. USA 2007, 104, 2750–2755. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Canamero, M.; Blasco, M.A.; Serrano, M. The INK4/ARF locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Eminli, S.; Foudi, A.; Stadtfeld, M.; Maherali, N.; Ahfeldt, T.; Mostoslavsky, G.; Hock, H.; Hochedlinger, K. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat. Genet. 2009, 41, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Araúzo-Bravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; et al. Oct4-induced pluripotency in adult neural stem cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Yamanaka, S. The use of induced pluripotent stem cells in drug development. Clin. Pharmacol. Ther. 2011, 89, 655–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Lensch, M.W.; Cahan, P.; Daley, G.Q. Investigating monogenic and complex diseases with pluripotent stem cells. Nat. Rev. Genet. 2011, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Amps, K.; Andrews, P.W.; Anyfantis, G.; Armstrong, L.; Avery, S.; Baharvand, H.; Baker, J.; Baker, D.; Munoz, M.B.; Beil, S.; et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011, 29, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Devlin, L.; Morrison, P.J. Mosaic Down’s syndrome prevalence in a complete population study. Arch. Dis. Child. 2004, 89, 1177–1178. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, N. Germ-line transmission of trisomy 21: Data from 80 families suggest an implication of grandmaternal age and a high frequency of female-specific trisomy rescue. Mol. Cytogenet. 2010, 3. [Google Scholar] [CrossRef]
- Zwijnenburg, P.J.G.; Meijers-Heijboer, H.; Boomsma, D.I. Identical but not the same: The value of discordant monozygotic twins in genetic research. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1134–1149. [Google Scholar]
- Dahoun, S.; Gagos, S.; Gagnebin, M.; Gehrig, C.; Burgi, C.; Simon, F.; Vieux, C.; Extermann, P.; Lyle, R.; Morris, M.A.; et al. Monozygotic twins discordant for trisomy 21 and maternal 21q inheritance: A complex series of events. Am. J. Med. Genet. A 2008, 146A, 2086–2093. [Google Scholar] [CrossRef]
- Tunstall-Pedoe, O.; Roy, A.; Karadimitris, A.; de la Fuente, J.; Fisk, N.M.; Bennett, P.; Norton, A.; Vyas, P.; Roberts, I. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood 2008, 112, 4507–4511. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Opalinska, J.B.; Yao, Y.; Fernandes, M.A.; Kalota, A.; Brooks, J.S.J.; Choi, J.K.; Gewirtz, A.M.; Danet-Desnoyers, G.-A.; Nemiroff, R.L.; et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood 2008, 112, 4503–4506. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Bernitz, J.M.; Lee, D.-F.; Lemischka, I.R. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014, 23, 2673–2686. [Google Scholar] [CrossRef] [PubMed]
- Prandini, P.; Deutsch, S.; Lyle, R.; Gagnebin, M.; Vivier, C.D.; Delorenzi, M.; Gehrig, C.; Descombes, P.; Sherman, S.; Bricarelli, F.D.; et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am. J. Hum. Genet. 2007, 81, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Mason, E.A.; Ovchinnikov, D.A.; Wells, C.A.; Wolvetang, E.J. Concise review: New paradigms for Down syndrome research using induced pluripotent stem cells: Tackling complex human genetic disease. Stem Cells Transl. Med. 2013, 2, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Benvenisty, N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 2011, 11, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S.; Cyranoski, D. Japan stem-cell trial stirs envy. Nature 2014, 513, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Fox, I.J.; Daley, G.Q.; Goldman, S.A.; Huard, J.; Kamp, T.J.; Trucco, M. Use of differentiated pluripotent stem cells in replacement therapy for treating disease. Science 2014, 345. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hibaoui, Y.; Feki, A. Concise Review: Methods and Cell Types Used to Generate Down Syndrome Induced Pluripotent Stem Cells. J. Clin. Med. 2015, 4, 696-714. https://doi.org/10.3390/jcm4040696
Hibaoui Y, Feki A. Concise Review: Methods and Cell Types Used to Generate Down Syndrome Induced Pluripotent Stem Cells. Journal of Clinical Medicine. 2015; 4(4):696-714. https://doi.org/10.3390/jcm4040696
Chicago/Turabian StyleHibaoui, Youssef, and Anis Feki. 2015. "Concise Review: Methods and Cell Types Used to Generate Down Syndrome Induced Pluripotent Stem Cells" Journal of Clinical Medicine 4, no. 4: 696-714. https://doi.org/10.3390/jcm4040696
APA StyleHibaoui, Y., & Feki, A. (2015). Concise Review: Methods and Cell Types Used to Generate Down Syndrome Induced Pluripotent Stem Cells. Journal of Clinical Medicine, 4(4), 696-714. https://doi.org/10.3390/jcm4040696