Adult Stem Cells and Diseases of Aging

Abstract
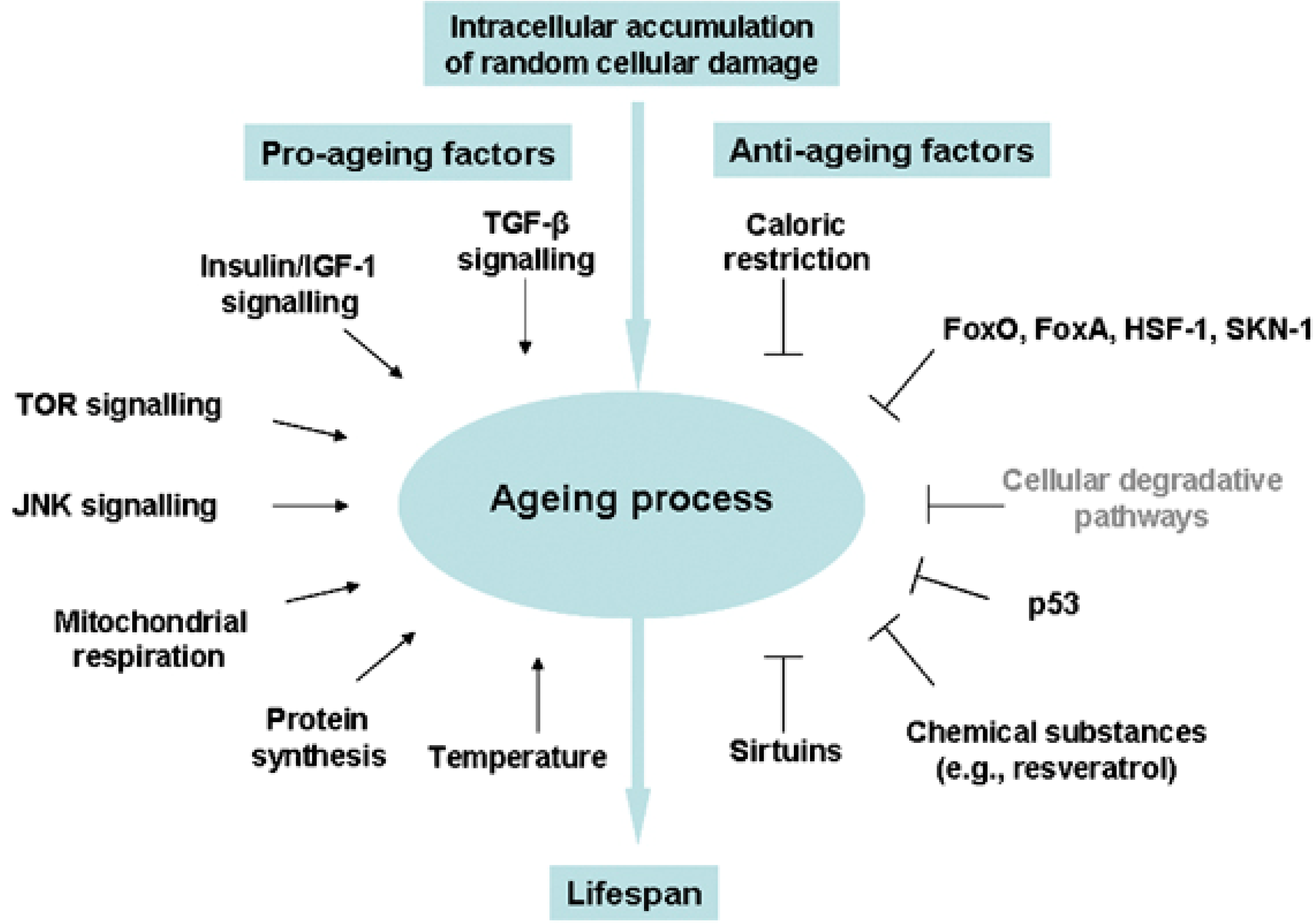
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | H. sapiens | M. musculus | D. melanogaster | C. elegans |
|---|---|---|---|---|
| Decreased cardiac function | Yes | Yes | Yes | NA |
| Apoptosis, senescence (somatic cells) | Yes | Yes | Yes | ? |
| Cancer, hyperplasia | Yes | Yes | No | No |
| Genome instability | Yes | Yes | Yes | Yes |
| Macromolecular aggregates | Yes | Yes | Yes | Yes |
| Reduced memory and learning | Yes | Yes | Yes | NA |
| Decline in growth hormone (GH), dehydroepiandrosterone (DHEA), testosterone, IGF | Yes | Yes | ? | ? |
| Increase in gonadotropins, insulin | Yes | Yes | ? | ? |
| Decreased thyroid function | Yes | Yes | NA | NA |
| Decrease in innate immunity | Yes | Yes | Yes | Yes |
| Increase in inflammation | Yes | Yes | No | No |
| Skin/Cuticle morphology changes | Yes | Yes | ? | Yes |
| Decreased mitochondrial function | Yes | Yes | Yes | Yes |
| Sarcopenia | Yes | Yes | Yes | Yes |
| Osteoporosis | Yes | Yes | NA | NA |
| Abnormal sleep/rest patterns | Yes | Yes | Yes | ? |
| Decrease in vision | Yes | Yes | ? | NA |
| Demyelination | Yes | Yes | ? | No |
| Decreased fitness | Yes | Yes | Yes | Yes |
| Arteriosclerosis | Yes | No | NA | NA |
| Changes in fat | Yes | Yes | ? | ? |
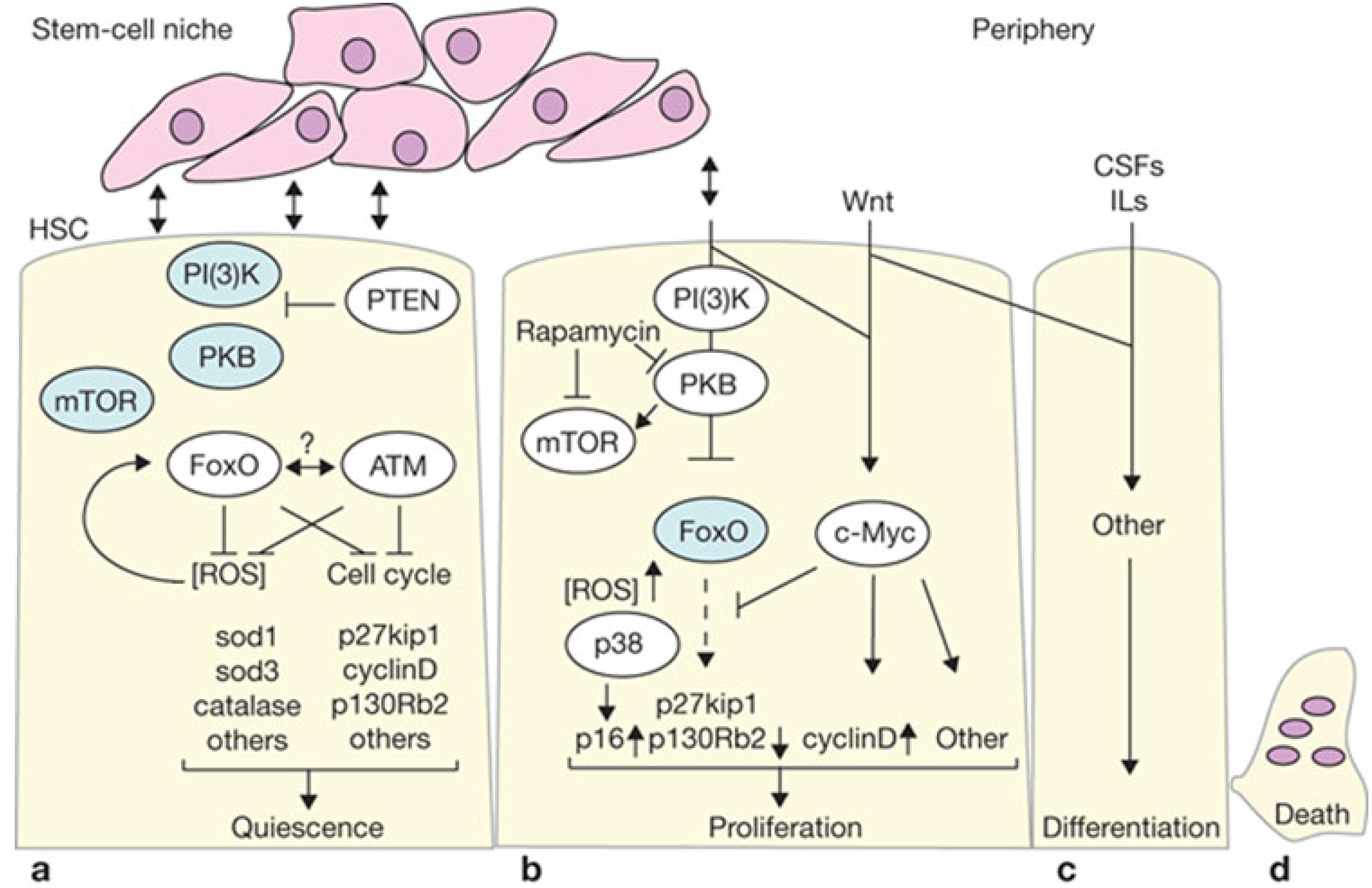
2. Adults Stem Cells and Causes of Aging
2.1. Self-Renewal and Maintenance of Stem Cell Pools
2.2. An Aging Immune Milieu
2.3. Genomic and Transcriptomic Data
2.4. Heritable Longevity
2.5. Premature Aging
2.6. Ex vivo Stem Cell Aging
3. Metabolic Stress and Adult Stem Cell Aging
3.1. Oxidative Stress
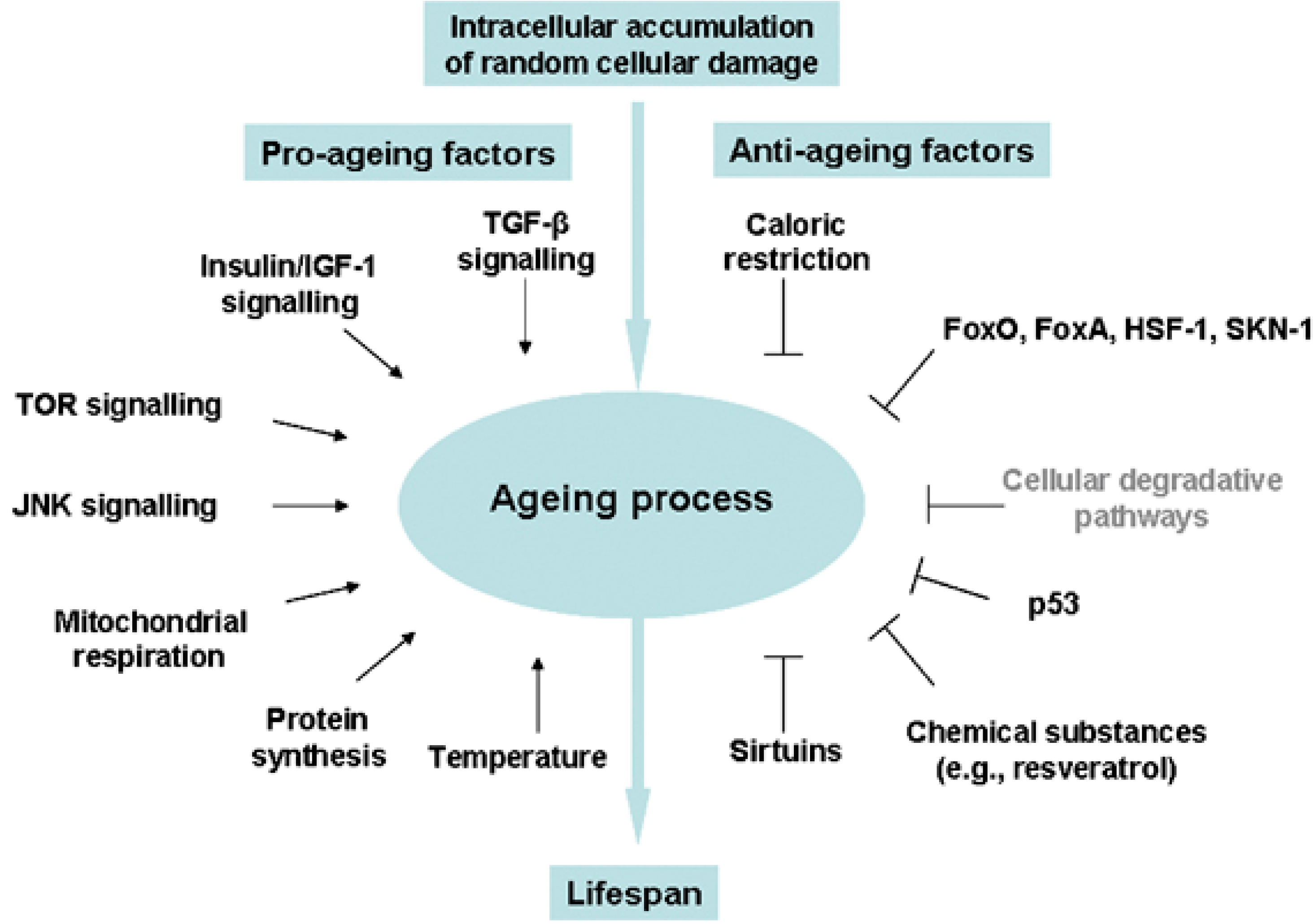
3.2. Stress Response and Homeostasis
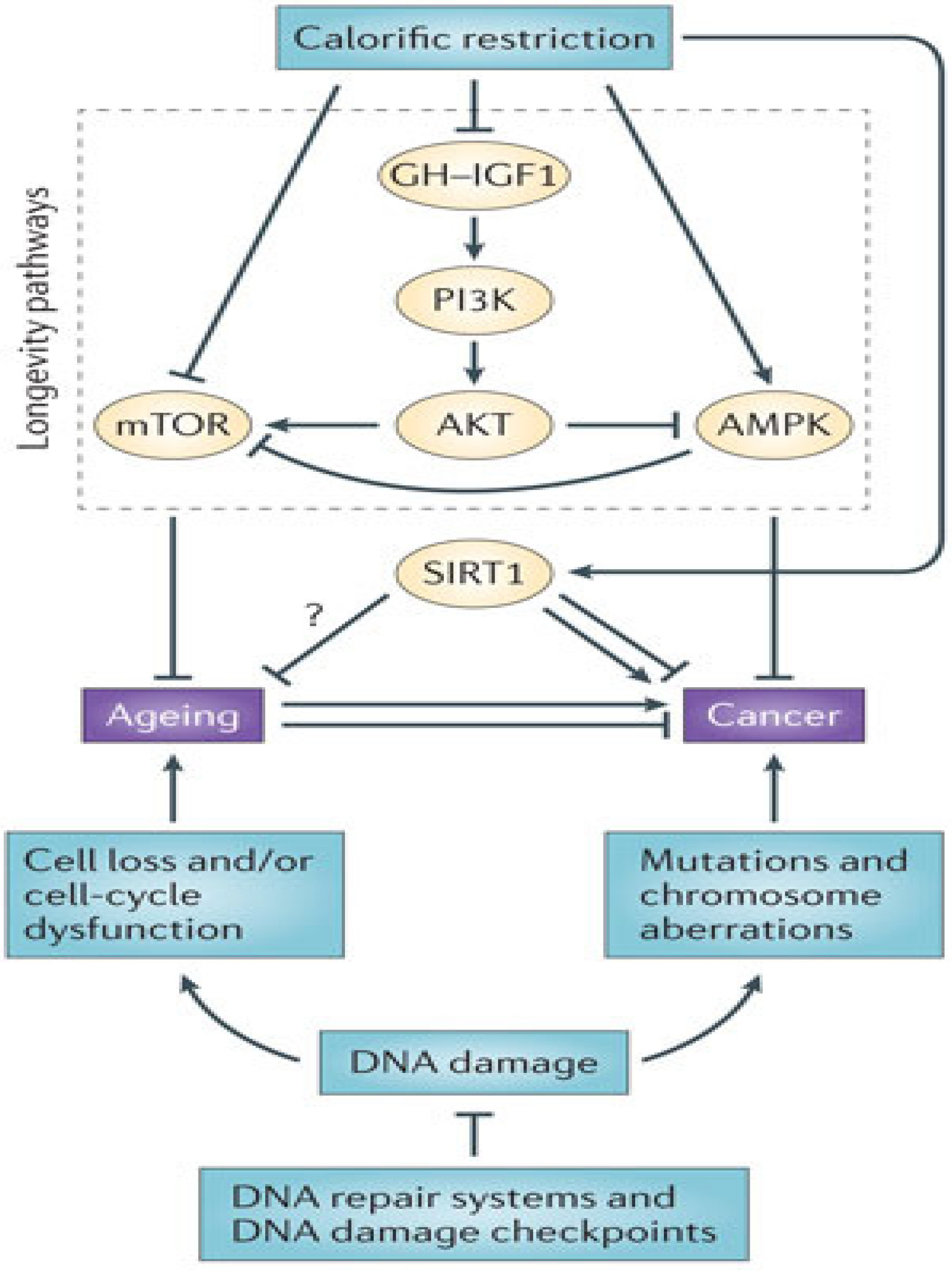
4. Adult Stem Cells: Caught in the Balance between Cancer and Metabolic Disease
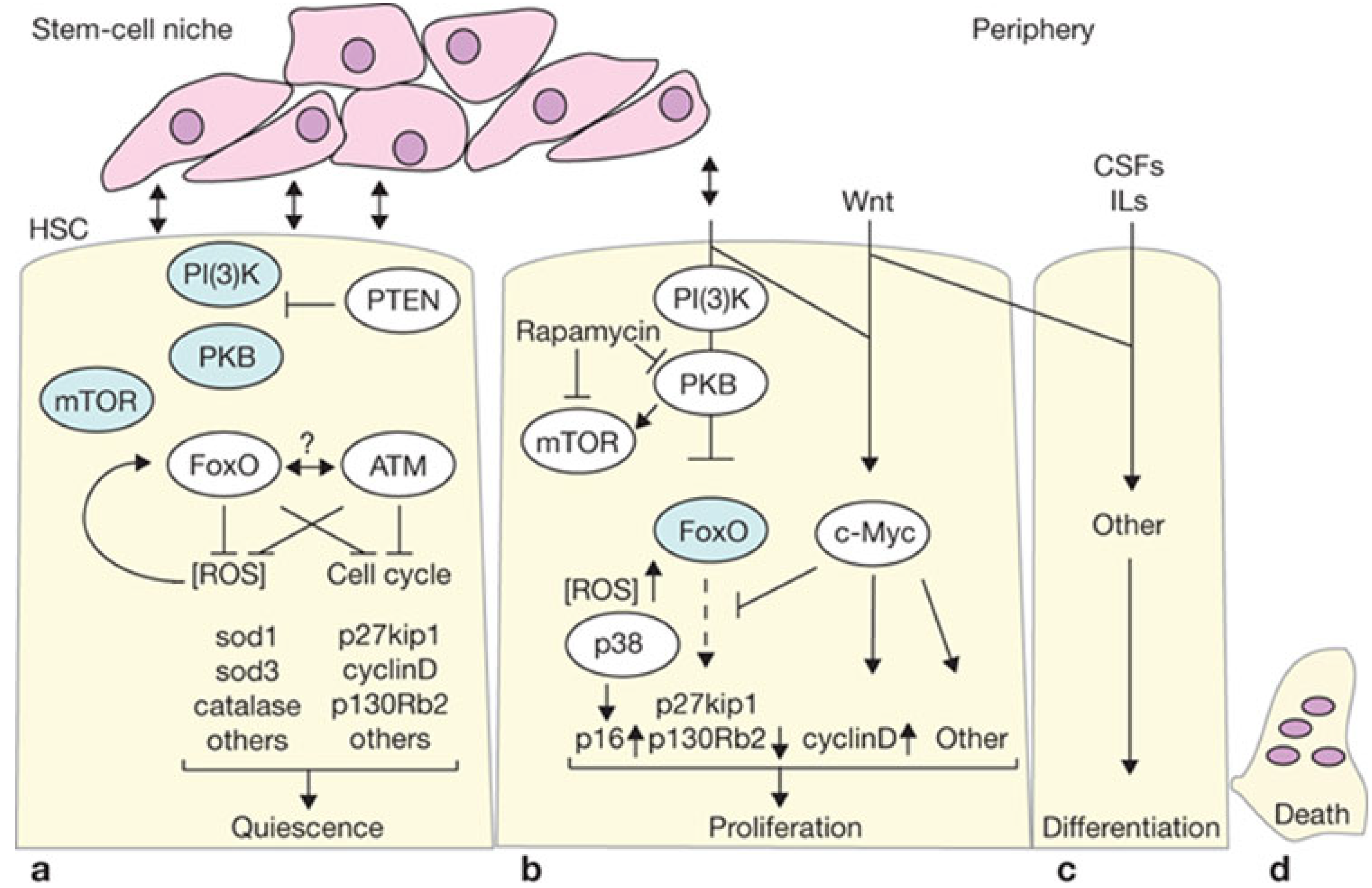
4.1. Proliferation vs. Oncogenic Resistance at the Cellular Level
4.2. Growth versus Oncogenic Resistance at the Organismal Level
4.3. Manipulation of Growth Pathways
4.4. Metabolic Dysregulation
5.Illustrations of Deranged Signaling in Aging
5.1. Canonical Wnt Signaling: A Critical Pathway in Aging Stem Cells
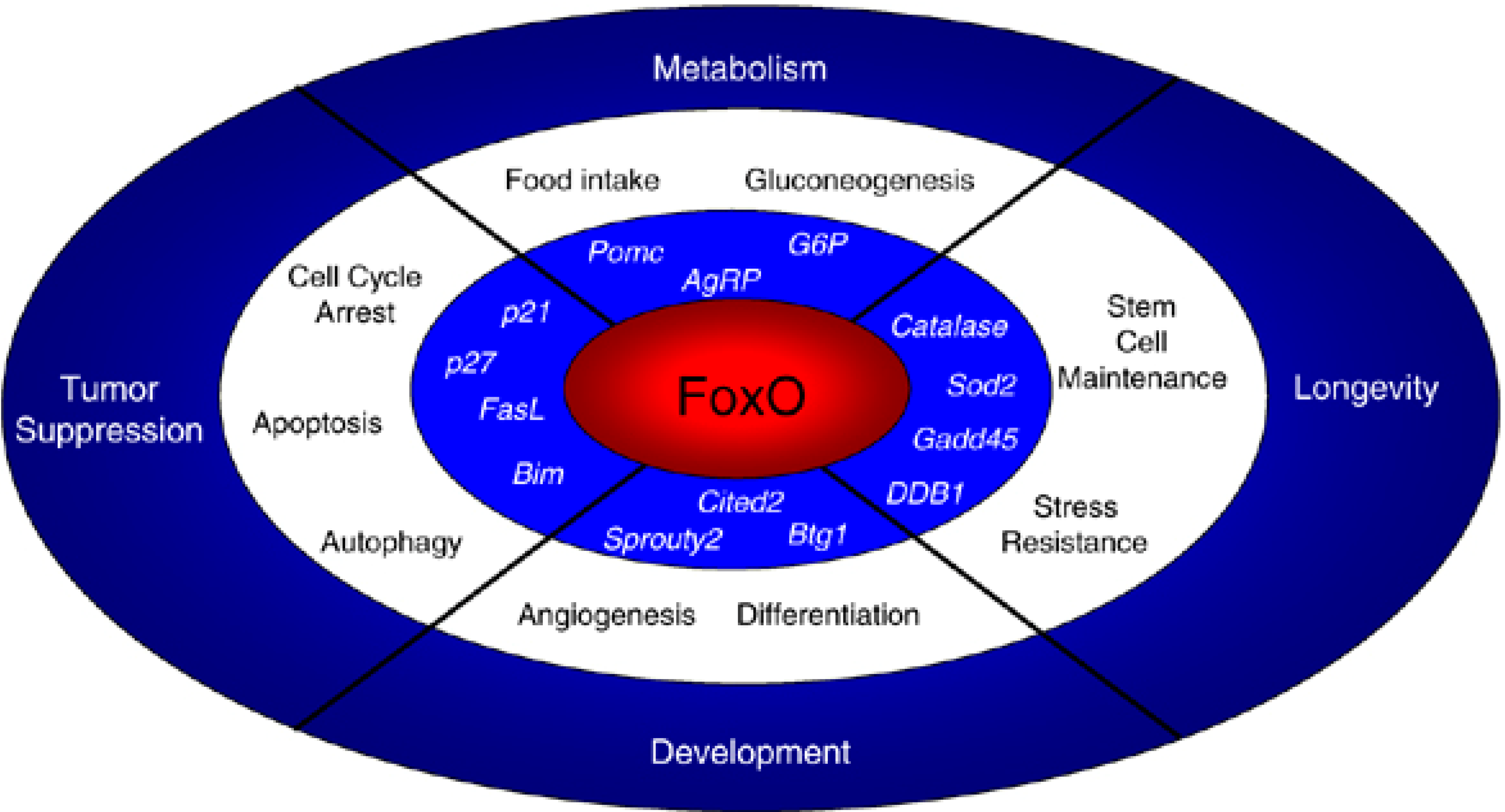
5.2. The FoxO Family: Stem Cell Stress Response and Indirect Effects
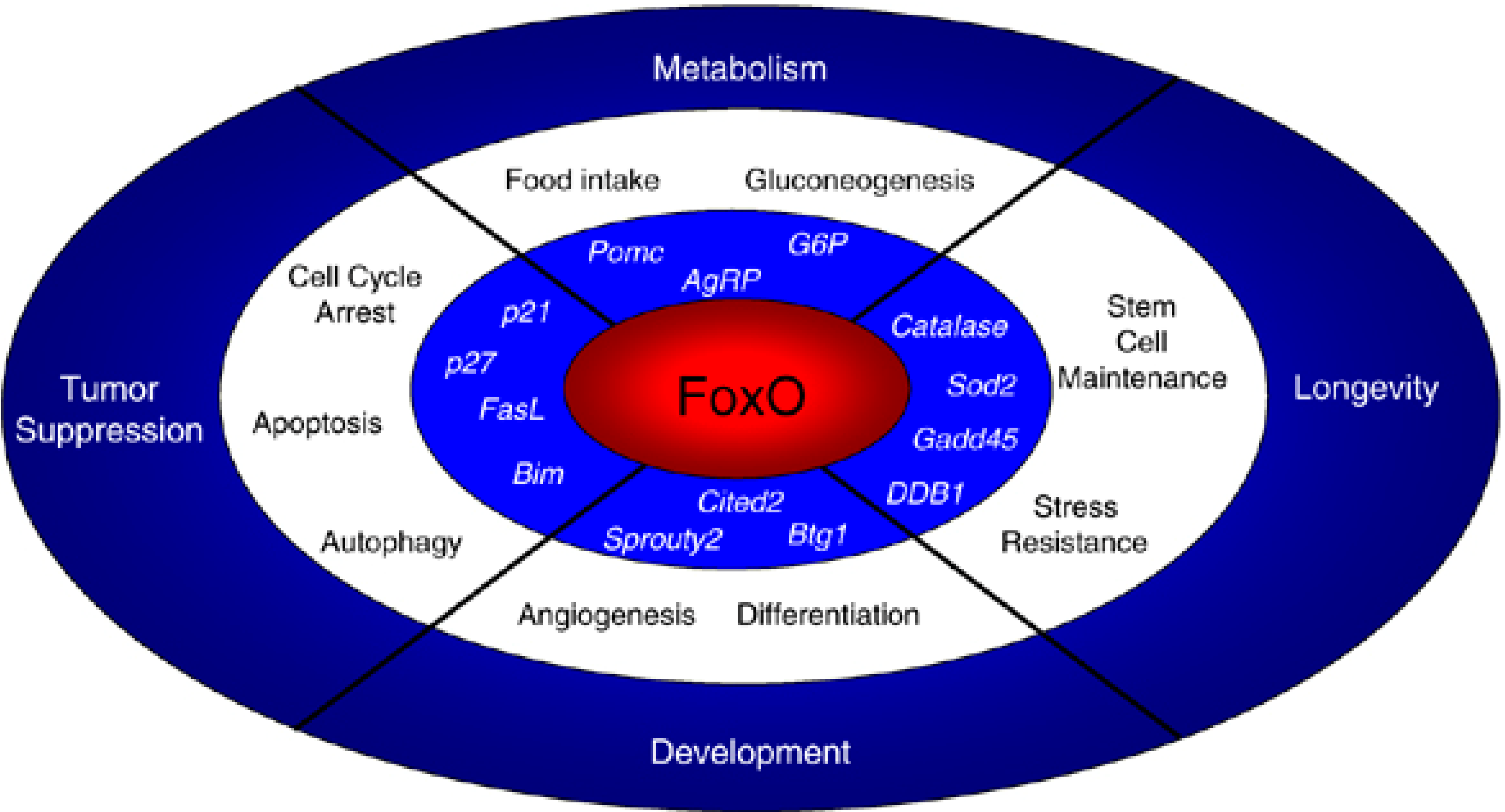
6. Mechanisms of Stem Cell Aging: Lessons from Transcriptional Reprogramming
6.1. Partial Reprogramming
6.2. Molecular Mechanisms of Reprogramming
7. Mechanisms of Stem Cell Aging: Lessons from Reprogramming Efficiency Studies
Reprogramming Efficiency and Metabolic Stress
8. Therapeutic Approaches under Investigation
8.1. Transcriptional Reprogramming
8.2. Calorie Restriction and Pharmacologic Mimicry of Calorie Restriction
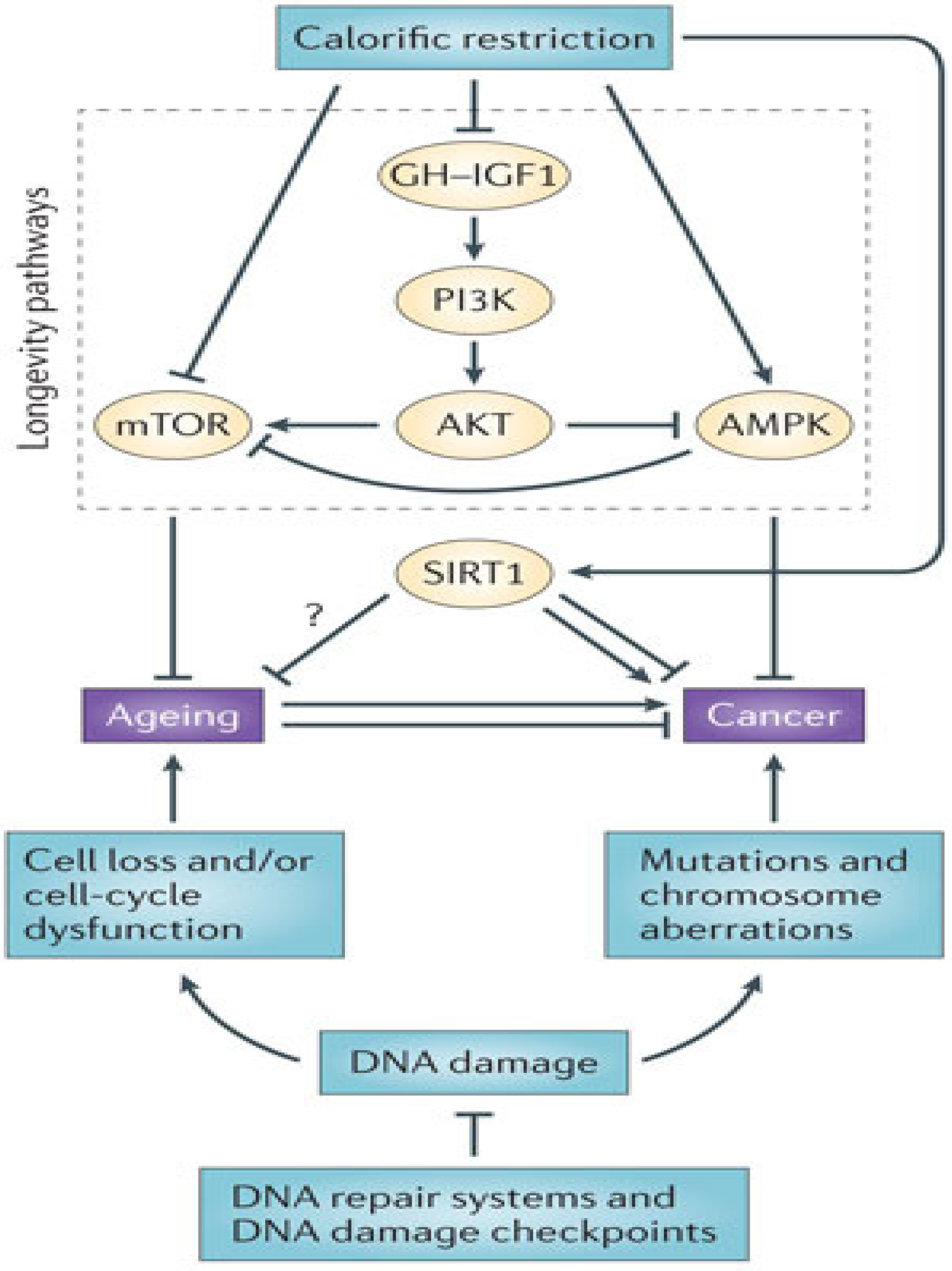
8.3. Epigenetic Modification
8.4. Strategies to Delay Senescence
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Vijg, J.; Campisi, J. Puzzles, promises and a cure for ageing. Nature 2008, 454, 1065–1071. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Hou, J.; Shang, Y.C. The “O” class: Crafting clinical care with FoxO transcription factors. Adv. Exp. Med. Biol. 2009, 665, 242–260. [Google Scholar] [CrossRef]
- Jeck, W.R.; Siebold, A.P.; Sharpless, N.E. Review: A meta-analysis of GWAS and age-associated diseases. Aging Cell 2012, 11, 727–731. [Google Scholar] [CrossRef]
- Newgard, C.B.; Sharpless, N.E. Coming of age: Molecular drivers of aging and therapeutic opportunities. J. Clin. Investig. 2013, 123, 946–950. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Shanti, R.M.; Li, W.J.; Nesti, L.J.; Wang, X.; Tuan, R.S. Adult mesenchymal stem cells: Biological properties, characteristics, and applications in maxillofacial surgery. J. Oral Maxillofac. Surg. 2007, 65, 1640–1647. [Google Scholar] [CrossRef]
- Chen, F.H.; Tuan, R.S. Mesenchymal stem cells in arthritic diseases. Arthritis Res. Ther. 2008, 10. [Google Scholar] [CrossRef]
- Janjanin, S.; Djouad, F.; Shanti, R.M.; Baksh, D.; Gollapudi, K.; Prgomet, D.; Rackwitz, L.; Joshi, A.S.; Tuan, R.S. Human palatine tonsil: A new potential tissue source of multipotent mesenchymal progenitor cells. Arthritis Res. Ther. 2008, 10. [Google Scholar] [CrossRef]
- Noth, U.; Steinert, A.F.; Tuan, R.S. Technology insight: Adult mesenchymal stem cells for osteoarthritis therapy. Nat. Clin. Pract. Rheumatol. 2008, 4, 371–380. [Google Scholar]
- Tuan, R.S. Stemming cartilage degeneration: Adult mesenchymal stem cells as a cell source for articular cartilage tissue engineering. Arthritis Rheum. 2006, 54, 3075–3078. [Google Scholar] [CrossRef]
- Noth, U.; Osyczka, A.M.; Tuli, R.; Hickok, N.J.; Danielson, K.G.; Tuan, R.S. Multilineage mesenchymal differentiation potential of human trabecular bone-derived cells. J. Orthop. Res. 2002, 20, 1060–1069. [Google Scholar] [CrossRef]
- Song, L.; Tuan, R.S. Transdifferentiation potential of human mesenchymal stem cells derived from bone marrow. FASEB J. 2004, 18, 980–982. [Google Scholar]
- Tuan, R.S.; Boland, G.; Tuli, R. Adult mesenchymal stem cells and cell-based tissue engineering. Arthritis Res. Ther. 2003, 5, 32–45. [Google Scholar] [CrossRef]
- Caterson, E.J.; Nesti, L.J.; Danielson, K.G.; Tuan, R.S. Human marrow-derived mesenchymal progenitor cells: Isolation, culture expansion, and analysis of differentiation. Mol. Biotechnol. 2002, 20, 245–256. [Google Scholar] [CrossRef]
- Caterson, E.J.; Nesti, L.J.; Albert, T.; Danielson, K.; Tuan, R. Application of mesenchymal stem cells in the regeneration of musculoskeletal tissues. MedGenMed 2001, 3, 1. [Google Scholar]
- Baksh, D.; Song, L.; Tuan, R.S. Adult mesenchymal stem cells: Characterization, differentiation, and application in cell and gene therapy. J. Cell. Mol. Med. 2004, 8, 301–316. [Google Scholar] [CrossRef]
- Jackson, W.M.; Aragon, A.B.; Djouad, F.; Song, Y.; Koehler, S.M.; Nesti, L.J.; Tuan, R.S. Mesenchymal progenitor cells derived from traumatized human muscle. J. Tissue Eng. Regen. Med. 2009, 3, 129–138. [Google Scholar] [CrossRef]
- Steigman, S.A.; Ahmed, A.; Shanti, R.M.; Tuan, R.S.; Valim, C.; Fauza, D.O. Sternal repair with bone grafts engineered from amniotic mesenchymal stem cells. J. Pediatr. Surg. 2009, 44, 1120–1126. [Google Scholar] [CrossRef]
- Charbord, P. Bone marrow mesenchymal stem cells: Historical overview and concepts. Hum. Gene Ther. 2010, 21, 1045–1056. [Google Scholar] [CrossRef]
- Chen, E.; Finkel, T. The tortoise, the hare, and the FoxO. Cell Stem Cell 2009, 5, 451–452. [Google Scholar] [CrossRef]
- Paik, J.H.; Ding, Z.; Narurkar, R.; Ramkissoon, S.; Muller, F.; Kamoun, W.S.; Chae, S.S.; Zheng, H.; Ying, H.; Mahoney, J.; et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 2009, 5, 540–553. [Google Scholar] [CrossRef]
- Wagner, W.; Bork, S.; Horn, P.; Krunic, D.; Walenda, T.; Diehlmann, A.; Benes, V.; Blake, J.; Huber, F.X.; Eckstein, V.; et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS One 2009, 4, e5846. [Google Scholar] [CrossRef]
- Mansilla, E.; Diaz Aquino, V.; Zambon, D.; Marin, G.H.; Martire, K.; Roque, G.; Ichim, T.; Riordan, N.H.; Patel, A.; Sturla, F.; et al. Could metabolic syndrome, lipodystrophy, and aging be mesenchymal stem cell exhaustion syndromes? Stem Cells Int. 2011, 2011. [Google Scholar] [CrossRef]
- Kahn, A.; Gibbons, R.; Perkins, S.; Gazit, D. Age-related bone loss. A hypothesis and initial assessment in mice. Clin. Orthop. Relat. Res. 1995, 313, 69–75. [Google Scholar]
- Lichtman, M.A.; Rowe, J.M. The relationship of patient age to the pathobiology of the clonal myeloid diseases. Sem. Oncol. 2004, 31, 185–197. [Google Scholar] [CrossRef]
- Zhou, T.; Hasty, P.; Walter, C.A.; Bishop, A.J.; Scott, L.M.; Rebel, V.I. Myelodysplastic syndrome: An inability to appropriately respond to damaged DNA? Exp. Hematol. 2013, 41, 665–674. [Google Scholar] [CrossRef]
- Noda, S.; Ichikawa, H.; Miyoshi, H. Hematopoietic stem cell aging is associated with functional decline and delayed cell cycle progression. Biochem. Biophys. Res. Commun. 2009, 383, 210–215. [Google Scholar] [CrossRef]
- Macas, J.; Nern, C.; Plate, K.H.; Momma, S. Increased generation of neuronal progenitors after ischemic injury in the aged adult human forebrain. J. Neurosci. 2006, 26, 13114–13119. [Google Scholar] [CrossRef]
- Mieno, S.; Boodhwani, M.; Clements, R.T.; Ramlawi, B.; Sodha, N.R.; Li, J.; Sellke, F.W. Aging is associated with an impaired coronary microvascular response to vascular endothelial growth factor in patients. J. Thorac. Cardiovasc. Surg. 2006, 132, 1348–1355. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Tripathi, P.; Kumar, A.; Ahmad, R.; Singh, R.K.; Balapure, A.K.; Vishwakermad, A.L. S-Phase fraction as a useful marker for prognosis and therapeutic response in patients with aplastic anemia. Hematol. Oncol. Stem Cell Ther. 2008, 1, 216–220. [Google Scholar]
- Harris, L.J.; Zhang, P.; Abdollahi, H.; Tarola, N.A.; DiMatteo, C.; McIlhenny, S.E.; Tulenko, T.N.; DiMuzio, P.J. Availability of adipose-derived stem cells in patients undergoing vascular surgical procedures. J. Surg. Res. 2010, 163, 105–112. [Google Scholar] [CrossRef]
- DiMuzio, P.; Tulenko, T. Tissue engineering applications to vascular bypass graft development: The use of adipose-derived stem cells. J. Vasc. Surg. 2007, 45, 99–103. [Google Scholar]
- Scheubel, R.J.; Kahrstedt, S.; Weber, H.; Holtz, J.; Friedrich, I.; Borgermann, J.; Silber, R.E.; Simm, A. Depression of progenitor cell function by advanced glycation endproducts (ages): Potential relevance for impaired angiogenesis in advanced age and diabetes. Exp. Gerontol. 2006, 41, 540–548. [Google Scholar] [CrossRef]
- Yamagishi, S.; Nakamura, K.; Inoue, H. Possible participation of advanced glycation end products in the pathogenesis of osteoporosis in diabetic patients. Med. Hypoth. 2005, 65, 1013–1015. [Google Scholar] [CrossRef]
- Zhang, P.; Moudgill, N.; Hager, E.; Tarola, N.; Dimatteo, C.; McIlhenny, S.; Tulenko, T.; DiMuzio, P.J. Endothelial differentiation of adipose-derived stem cells from elderly patients with cardiovascular disease. Stem Cells Dev. 2011, 20, 977–988. [Google Scholar] [CrossRef]
- Fan, M.; Chen, W.; Liu, W.; Du, G.Q.; Jiang, S.L.; Tian, W.C.; Sun, L.; Li, R.K.; Tian, H. The effect of age on the efficacy of human mesenchymal stem cell transplantation after a myocardial infarction. Rejuven. Res. 2010, 13, 429–438. [Google Scholar] [CrossRef]
- Hermann, A.; List, C.; Habisch, H.J.; Vukicevic, V.; Ehrhart-Bornstein, M.; Brenner, R.; Bernstein, P.; Fickert, S.; Storch, A. Age-dependent neuroectodermal differentiation capacity of human mesenchymal stromal cells: Limitations for autologous cell replacement strategies. Cytotherapy 2010, 12, 17–30. [Google Scholar] [CrossRef]
- Dexheimer, V.; Mueller, S.; Braatz, F.; Richter, W. Reduced reactivation from dormancy but maintained lineage choice of human mesenchymal stem cells with donor age. PLoS One 2011, 6, e22980. [Google Scholar]
- Lepperdinger, G. Inflammation and mesenchymal stem cell aging. Curr. Opin. Immunol. 2011, 23, 518–524. [Google Scholar] [CrossRef]
- Walenda, T.; Bork, S.; Horn, P.; Wein, F.; Saffrich, R.; Diehlmann, A.; Eckstein, V.; Ho, A.D.; Wagner, W. Co-Culture with mesenchymal stromal cells increases proliferation and maintenance of haematopoietic progenitor cells. J. Cell. Mol. Med. 2010, 14, 337–350. [Google Scholar] [CrossRef]
- Wagner, W.; Horn, P.; Bork, S.; Ho, A.D. Aging of hematopoietic stem cells is regulated by the stem cell niche. Exp. Gerontol. 2008, 43, 974–980. [Google Scholar] [CrossRef]
- De Gonzalo-Calvo, D.; Neitzert, K.; Fernandez, M.; Vega-Naredo, I.; Caballero, B.; Garcia-Macia, M.; Suarez, F.M.; Rodriguez-Colunga, M.J.; Solano, J.J.; Coto-Montes, A. Differential inflammatory responses in aging and disease: TNF-alpha and IL-6 as possible biomarkers. Free Radic. Biol. Med. 2010, 49, 733–737. [Google Scholar] [CrossRef]
- Osorio, F.G.; Barcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; Lopez-Otin, C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef]
- Murabito, J.M.; Yuan, R.; Lunetta, K.L. The search for longevity and healthy aging genes: Insights from epidemiological studies and samples of long-lived individuals. J. Gerontol. 2012, 67, 470–479. [Google Scholar] [CrossRef]
- Jiang, S.S.; Chen, C.H.; Tseng, K.Y.; Tsai, F.Y.; Wang, M.J.; Chang, I.S.; Lin, J.L.; Lin, S. Gene expression profiling suggests a pathological role of human bone marrow-derived mesenchymal stem cells in aging-related skeletal diseases. Aging 2011, 3, 672–684. [Google Scholar]
- Benisch, P.; Schilling, T.; Klein-Hitpass, L.; Frey, S.P.; Seefried, L.; Raaijmakers, N.; Krug, M.; Regensburger, M.; Zeck, S.; Schinke, T.; et al. The transcriptional profile of mesenchymal stem cell populations in primary osteoporosis is distinct and shows overexpression of osteogenic inhibitors. PLoS One 2012, 7, e45142. [Google Scholar]
- Swindell, W.R.; Johnston, A.; Sun, L.; Xing, X.; Fisher, G.J.; Bulyk, M.L.; Elder, J.T.; Gudjonsson, J.E. Meta-profiles of gene expression during aging: Limited similarities between mouse and human and an unexpectedly decreased inflammatory signature. PLoS One 2012, 7, e33204. [Google Scholar] [CrossRef]
- Halaschek-Wiener, J.; Amirabbasi-Beik, M.; Monfared, N.; Pieczyk, M.; Sailer, C.; Kollar, A.; Thomas, R.; Agalaridis, G.; Yamada, S.; Oliveira, L.; et al. Genetic variation in healthy oldest-old. PLoS One 2009, 4, e6641. [Google Scholar] [CrossRef]
- Deelen, J.; Beekman, M.; Uh, H.W.; Helmer, Q.; Kuningas, M.; Christiansen, L.; Kremer, D.; van der Breggen, R.; Suchiman, H.E.; Lakenberg, N.; et al. Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell 2011, 10, 686–698. [Google Scholar] [CrossRef]
- Pawlikowska, L.; Hu, D.; Huntsman, S.; Sung, A.; Chu, C.; Chen, J.; Joyner, A.H.; Schork, N.J.; Hsueh, W.C.; Reiner, A.P.; et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell 2009, 8, 460–472. [Google Scholar] [CrossRef]
- Prokocimer, M.; Barkan, R.; Gruenbaum, Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell 2013, 12, 533–543. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Y.; Jiang, H.; Nie, D. Promotion of tumor development in prostate cancer by progerin. Cancer Cell Int. 2010, 10, 1–10. [Google Scholar] [CrossRef]
- Ding, S.L.; Shen, C.Y. Model of human aging: Recent findings on Werner’s and Hutchinson-Gilford progeria syndromes. Clin. Interv. Aging 2008, 3, 431–444. [Google Scholar]
- Kudlow, B.A.; Stanfel, M.N.; Burtner, C.R.; Johnston, E.D.; Kennedy, B.K. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol. Biol. Cell 2008, 19, 5238–5248. [Google Scholar] [CrossRef]
- Benson, E.K.; Lee, S.W.; Aaronson, S.A. Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J. Cell Sci. 2010, 123, 2605–2612. [Google Scholar] [CrossRef]
- Cao, K.; Blair, C.D.; Faddah, D.A.; Kieckhaefer, J.E.; Olive, M.; Erdos, M.R.; Nabel, E.G.; Collins, F.S. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J. Clin. Invest. 2011, 121, 2833–2844. [Google Scholar] [CrossRef]
- Nissan, X.; Blondel, S.; Peschanski, M. In vitro pathological modelling using patient-specific induced pluripotent stem cells: The case of progeria. Biochem. Soc. Trans. 2011, 39, 1775–1779. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef]
- Osorio, F.G.; Navarro, C.L.; Cadinanos, J.; Lopez-Mejia, I.C.; Quiros, P.M.; Bartoli, C.; Rivera, J.; Tazi, J.; Guzman, G.; Varela, I.; et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Leung, G.K.; Schmidt, W.K.; Bergo, M.O.; Gavino, B.; Wong, D.H.; Tam, A.; Ashby, M.N.; Michaelis, S.; Young, S.G. Biochemical studies of Zmpste24-deficient mice. J. Biol. Chem. 2001, 276, 29051–29058. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A progeroid syndrome in mice is caused by defects in a-type lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef]
- Sagelius, H.; Rosengardten, Y.; Schmidt, E.; Sonnabend, C.; Rozell, B.; Eriksson, M. Reversible phenotype in a mouse model of Hutchinson-Gilford progeria syndrome. J. Med. Genet. 2008, 45, 794–801. [Google Scholar] [CrossRef]
- Pendas, A.M.; Zhou, Z.; Cadinanos, J.; Freije, J.M.; Wang, J.; Hultenby, K.; Astudillo, A.; Wernerson, A.; Rodriguez, F.; Tryggvason, K.; et al. Defective prelamin a processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002, 31, 94–99. [Google Scholar]
- Best, B.P. Nuclear DNA damage as a direct cause of aging. Rejuven. Res. 2009, 12, 199–208. [Google Scholar] [CrossRef]
- Pekovic, V.; Hutchison, C.J. Adult stem cell maintenance and tissue regeneration in the ageing context: The role for A-type lamins as intrinsic modulators of ageing in adult stem cells and their niches. J. Anat. 2008, 213, 5–25. [Google Scholar] [CrossRef]
- McClintock, D.; Ratner, D.; Lokuge, M.; Owens, D.M.; Gordon, L.B.; Collins, F.S.; Djabali, K. The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One 2007, 2, e1269. [Google Scholar]
- Wenzel, V.; Roedl, D.; Gabriel, D.; Gordon, L.B.; Herlyn, M.; Schneider, R.; Ring, J.; Djabali, K. Naive adult stem cells from patients with Hutchinson-Gilford progeria syndrome express low levels of progerin in vivo. Biol. Open 2012, 1, 516–526. [Google Scholar] [CrossRef]
- Pekovic, V.; Gibbs-Seymour, I.; Markiewicz, E.; Alzoghaibi, F.; Benham, A.M.; Edwards, R.; Wenhert, M.; von Zglinicki, T.; Hutchison, C.J. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 2011, 10, 1067–1079. [Google Scholar] [CrossRef]
- Halaschek-Wiener, J.; Brooks-Wilson, A. Progeria of stem cells: Stem cell exhaustion in Hutchinson-Gilford progeria syndrome. J. Gerontol. 2007, 62, 3–8. [Google Scholar] [CrossRef]
- Campisi, J. From cells to organisms: Can we learn about aging from cells in culture? Exp. Gerontol. 2001, 36, 607–618. [Google Scholar] [CrossRef]
- Trudeau, M.A.; Wong, J.M. Genetic variations in telomere maintenance, with implications on tissue renewal capacity and chronic disease pathologies. Curr. Pharm. Pers. Med. 2010, 8, 7–24. [Google Scholar]
- Armanios, M. Telomeres and age-related disease: How telomere biology informs clinical paradigms. J. Clin. Invest. 2013, 123, 996–1002. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Bonab, M.M.; Alimoghaddam, K.; Talebian, F.; Ghaffari, S.H.; Ghavamzadeh, A.; Nikbin, B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol. 2006, 7. [Google Scholar] [CrossRef]
- Li, Z.; Liu, C.; Xie, Z.; Song, P.; Zhao, R.C.; Guo, L.; Liu, Z.; Wu, Y. Epigenetic dysregulation in mesenchymal stem cell aging and spontaneous differentiation. PLoS One 2011, 6, e20526. [Google Scholar]
- Kasper, G.; Mao, L.; Geissler, S.; Draycheva, A.; Trippens, J.; Kuhnisch, J.; Tschirschmann, M.; Kaspar, K.; Perka, C.; Duda, G.N.; et al. Insights into mesenchymal stem cell aging: Involvement of antioxidant defense and actin cytoskeleton. Stem Cells 2009, 27, 1288–1297. [Google Scholar] [CrossRef]
- De Barros, S.; Dehez, S.; Arnaud, E.; Barreau, C.; Cazavet, A.; Perez, G.; Galinier, A.; Casteilla, L.; Planat-Benard, V. Aging-related decrease of human ASC angiogenic potential is reversed by hypoxia preconditioning through ROS production. Mol. Ther. 2013, 21, 399–408. [Google Scholar] [CrossRef]
- Sethe, S.; Scutt, A.; Stolzing, A. Aging of mesenchymal stem cells. Aging Res. Rev. 2006, 5, 91–116. [Google Scholar] [CrossRef]
- Stolzing, A.; Jones, E.; McGonagle, D.; Scutt, A. Age-related changes in human bone marrow-derived mesenchymal stem cells: Consequences for cell therapies. Mech. Aging Dev. 2008, 129, 163–173. [Google Scholar] [CrossRef]
- Miettinen, J.A.; Salonen, R.J.; Ylitalo, K.; Niemela, M.; Kervinen, K.; Saily, M.; Koistinen, P.; Savolainen, E.R.; Makikallio, T.H.; Huikuri, H.V.; et al. The effect of bone marrow microenvironment on the functional properties of the therapeutic bone marrow-derived cells in patients with acute myocardial infarction. J. Transl. Med. 2012, 10, 1–11. [Google Scholar]
- Haines, D.D.; Juhasz, B.; Tosaki, A. Management of multicellular senescence and oxidative stress. J. Cell. Mol. Med. 2013, 17, 936–957. [Google Scholar] [CrossRef]
- Rafalski, V.A.; Brunet, A. Energy metabolism in adult neural stem cell fate. Prog. Neurobiol. 2011, 93, 182–203. [Google Scholar] [CrossRef]
- Oellerich, M.F.; Potente, M. FOXOs and sirtuins in vascular growth, maintenance, and aging. Circ. Res. 2012, 110, 1238–1251. [Google Scholar] [CrossRef]
- Rafalski, V.A.; Mancini, E.; Brunet, A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 2012, 125, 5597–5608. [Google Scholar] [CrossRef]
- Vellai, T. Autophagy genes and ageing. Cell Death Differ. 2009, 16, 94–102. [Google Scholar] [CrossRef]
- Harries, L.W.; Fellows, A.D.; Pilling, L.C.; Hernandez, D.; Singleton, A.; Bandinelli, S.; Guralnik, J.; Powell, J.; Ferrucci, L.; Melzer, D. Advancing age is associated with gene expression changes resembling mTOR inhibition: Evidence from two human populations. Mech. Ageing Dev. 2012, 133, 556–562. [Google Scholar] [CrossRef]
- Pan, H.; Cai, N.; Li, M.; Liu, G.H.; Izpisua Belmonte, J.C. Autophagic control of cell “stemness”. EMBO Mol. Med. 2013, 5, 327–331. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Hall, J.A.; Dominy, J.E.; Lee, Y.; Puigserver, P. The sirtuin family’s role in aging and age-associated pathologies. J. Clin. Invest. 2013, 123, 973–979. [Google Scholar] [CrossRef]
- Saunders, L.R.; Sharma, A.D.; Tawney, J.; Nakagawa, M.; Okita, K.; Yamanaka, S.; Willenbring, H.; Verdin, E. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging 2010, 2, 415–431. [Google Scholar]
- Matsui, K.; Ezoe, S.; Oritani, K.; Shibata, M.; Tokunaga, M.; Fujita, N.; Tanimura, A.; Sudo, T.; Tanaka, H.; McBurney, M.W.; et al. NAD-Dependent histone deacetylase, SIRT1, plays essential roles in the maintenance of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 2012, 418, 811–817. [Google Scholar] [CrossRef]
- Tseng, P.C.; Hou, S.M.; Chen, R.J.; Peng, H.W.; Hsieh, C.F.; Kuo, M.L.; Yen, M.L. Resveratrol promotes osteogenesis of human mesenchymal stem cells by upregulating RUNX2 gene expression via the SIRT1/FOXO3A axis. J. Bone Miner. Res. 2011, 26, 2552–2563. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, F.; Ge, X.; Yan, T.; Chen, X.; Shi, X.; Zhai, Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007, 6, 307–319. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Ito, K.; Bernardi, R.; Pandolfi, P.P. A novel signaling network as a critical rheostat for the biology and maintenance of the normal stem cell and the cancer-initiating cell. Curr. Opin. Genet. Dev. 2009, 19, 51–59. [Google Scholar] [CrossRef]
- Coffer, P.J.; Burgering, B.M. Stressed marrow: FoxOs stem tumour growth. Nat. Cell Biol. 2007, 9, 251–253. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; Menendez, J.A. Pharmacological mimicking of caloric restriction elicits epigenetic reprogramming of differentiated cells to stem-like self-renewal states. Rejuven. Res. 2010, 13, 519–526. [Google Scholar] [CrossRef]
- Lu, Y. Physical Interation of Parathyroid Hormone-Related Protein with the Epigenetic Regulator Bmi1; McGill University: Montreal, QC, Canada, 2011. [Google Scholar]
- Zhang, H.W.; Ding, J.; Jin, J.L.; Guo, J.; Liu, J.N.; Karaplis, A.; Goltzman, D.; Miao, D. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J. Bone Miner. Res. 2010, 25, 640–652. [Google Scholar] [CrossRef]
- Han, J.D. An aging program at the systems level? Birth Defects Res. C: Embryo Today 2012, 96, 206–211. [Google Scholar] [CrossRef]
- Pardal, R.; Molofsky, A.V.; He, S.; Morrison, S.J. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 177–185. [Google Scholar] [CrossRef]
- Oh, Y.S.; Kim, D.G.; Kim, G.; Choi, E.C.; Kennedy, B.K.; Suh, Y.; Park, B.J.; Kim, S. Downregulation of lamin A by tumor suppressor AIMP3/p18 leads to a progeroid phenotype in mice. Aging Cell 2010, 9, 810–822. [Google Scholar] [CrossRef]
- Lepperdinger, G.; Brunauer, R.; Gassner, R.; Jamnig, A.; Kloss, F.; Laschober, G.T. Changes of the functional capacity of mesenchymal stem cells due to aging or age-associated disease-implications for clinical applications and donor recruitment. Transfus. Med. Hemother. 2008, 35, 299–305. [Google Scholar] [CrossRef]
- Magee, J.A.; Ikenoue, T.; Nakada, D.; Lee, J.Y.; Guan, K.L.; Morrison, S.J. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 2012, 11, 415–428. [Google Scholar] [CrossRef]
- Gems, D.; Partridge, L. Genetics of longevity in model organisms: Debates and paradigm shifts. Annu. Rev. Physiol. 2013, 75, 621–644. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell senescence: Hypertrophic arrest beyond the restriction point. J. Cell. Physiol. 2006, 209, 592–597. [Google Scholar] [CrossRef]
- Marino, G.; Ugalde, A.P.; Fernandez, A.F.; Osorio, F.G.; Fueyo, A.; Freije, J.M.; Lopez-Otin, C. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc. Natl. Acad. Sci. USA 2010, 107, 16268–16273. [Google Scholar]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef]
- Garinis, G.A.; Uittenboogaard, L.M.; Stachelscheid, H.; Fousteri, M.; van Ijcken, W.; Breit, T.M.; van Steeg, H.; Mullenders, L.H.; van der Horst, G.T.; Bruning, J.C.; et al. Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nat. Cell Biol. 2009, 11, 604–615. [Google Scholar] [CrossRef]
- Cohen, D.H.; LeRoith, D. Obesity, type 2 diabetes, and cancer: The insulin and IGF connection. Endocr.-Relat. Cancer 2012, 19, 27–45. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010, 9, 1057–1064. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Diabetes, cancer, and metformin: Connections of metabolism and cell proliferation. Ann. N. Y. Acad. Sci. 2011, 1243, 54–68. [Google Scholar]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Herst, P.M.; Berridge, M.V. Cell hierarchy, metabolic flexibility and systems approaches to cancer treatment. Curr. Pharm. Biotechnol. 2013, 14, 289–299. [Google Scholar] [CrossRef]
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): From cancer stem cells to aging-associated fibrosis. Cell Cycle 2010, 9, 4461–4468. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Vellon, L.; Quiros, P.M.; Cufi, S.; Ruiz de Galarreta, E.; Oliveras-Ferraros, C.; Martin, A.G.; Martin-Castillo, B.; Lopez-Otin, C.; Menendez, J.A. Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells. Cell Cycle 2012, 11, 974–989. [Google Scholar] [CrossRef]
- Menendez, J.A.; Vazquez-Martin, A. Rejuvenating regeneration: Metformin activates endogenous adult stem cells. Cell Cycle 2012, 11, 3521–3522. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. PTEN dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012, 15, 713–724. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Morrison, S.J. The PI-3kinase pathway in hematopoietic stem cells and leukemia-initiating cells: A mechanistic difference between normal and cancer stem cells. Blood Cells Mol. Dis. 2008, 41, 73–76. [Google Scholar] [CrossRef]
- Cheng, T.; Rodrigues, N.; Shen, H.; Yang, Y.; Dombkowski, D.; Sykes, M.; Scadden, D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 2000, 287, 1804–1808. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Cufi, S.; Lopez-Bonet, E.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Menendez, J.A. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef]
- Gao, Y.; Xue, J.; Li, X.; Jia, Y.; Hu, J. Metformin regulates osteoblast and adipocyte differentiation of rat mesenchymal stem cells. J. Pharm. Pharmacol. 2008, 60, 1695–1700. [Google Scholar] [CrossRef]
- Viccica, G.; Francucci, C.M.; Marcocci, C. The role of PPARγ for the osteoblastic differentiation. J. Endocrinol. Investig. 2010, 33, 9–12. [Google Scholar]
- Wu, W.; Ye, Z.; Zhou, Y.; Tan, W.S. AICAR, a small chemical molecule, primes osteogenic differentiation of adult mesenchymal stem cells. Int. J. Artif. Organs 2011, 34, 1128–1136. [Google Scholar] [CrossRef]
- Kasai, T.; Bandow, K.; Suzuki, H.; Chiba, N.; Kakimoto, K.; Ohnishi, T.; Kawamoto, S.; Nagaoka, E.; Matsuguchi, T. Osteoblast differentiation is functionally associated with decreased AMP kinase activity. J. Cell. Physiol. 2009, 221, 740–749. [Google Scholar] [CrossRef]
- Fadini, G.P.; Ceolotto, G.; Pagnin, E.; de Kreutzenberg, S.; Avogaro, A. At the crossroads of longevity and metabolism: The metabolic syndrome and lifespan determinant pathways. Aging Cell 2011, 10, 10–17. [Google Scholar] [CrossRef]
- Morley, J.E. Diabetes and aging: Epidemiologic overview. Clin. Geriatr. Med. 2008, 24, 395–405. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar]
- Matsuzawa, Y.; Funahashi, T.; Kihara, S.; Shimomura, I. Adiponectin and metabolic syndrome. Arterioscl. Thromb. Vasc. Biol. 2004, 24, 29–33. [Google Scholar] [CrossRef]
- Ferder, L.; Inserra, F.; Martinez-Maldonado, M. Inflammation and the metabolic syndrome: Role of angiotensin II and oxidative stress. Curr. Hypertens. Rep. 2006, 8, 191–198. [Google Scholar] [CrossRef]
- Kim, D.H.; Puri, N.; Sodhi, K.; Falck, J.R.; Abraham, N.G.; Shapiro, J.; Schwartzman, M.L. Cyclooxygenase-2 dependent metabolism of 20-HETE increases adiposity and adipocyte enlargement in mesenchymal stem cell-derived adipocytes. J. Lipid Res. 2013, 54, 786–793. [Google Scholar] [CrossRef]
- Cao, J.J. Effects of obesity on bone metabolism. J. Orthop. Surg. Res. 2011, 6, 1–7. [Google Scholar] [CrossRef]
- Lecka-Czernik, B.; Rosen, C.J.; Kawai, M. Skeletal aging and the adipocyte program: New insights from an “old” molecule. Cell Cycle 2010, 9, 3648–3654. [Google Scholar] [CrossRef]
- Jin, C.; Li, J.; Green, C.D.; Yu, X.; Tang, X.; Han, D.; Xian, B.; Wang, D.; Huang, X.; Cao, X.; et al. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 2011, 14, 161–172. [Google Scholar] [CrossRef]
- Campbell, P.T.; Newton, C.C.; Patel, A.V.; Jacobs, E.J.; Gapstur, S.M. Diabetes and cause-specific mortality in a prospective cohort of one million US adults. Diabetes Care 2012, 35, 1835–1844. [Google Scholar] [CrossRef]
- Johnson, J.A.; Carstensen, B.; Witte, D.; Bowker, S.L.; Lipscombe, L.; Renehan, A.G. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Rasi, C.; Razzaghian, H.R.; Pakalapati, G.; Waite, L.; Thilbeault, K.S.; Ronowicz, A.; Wineinger, N.E.; Tiwari, H.K.; Boomsma, D.; et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Hum. Genet. 2012, 90, 217–228. [Google Scholar] [CrossRef]
- Bonnefond, A.; Skrobek, B.; Lobbens, S.; Eury, E.; Thuillier, D.; Cauchi, S.; Lantieri, O.; Balkau, B.; Riboli, E.; Marre, M.; et al. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat. Genet. 2013, 45, 1040–1043. [Google Scholar] [CrossRef]
- Castillo, J.J.; Mull, N.; Reagan, J.L.; Nemr, S.; Mitri, J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: A meta-analysis of observational studies. Blood 2012, 119, 4845–4850. [Google Scholar] [CrossRef]
- Almeida, M.; Han, L.; Martin-Millan, M.; O’Brien, C.A.; Manolagas, S.C. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J. Biol. Chem. 2007, 282, 27298–27305. [Google Scholar] [CrossRef]
- Manolagas, S.C.; Almeida, M. Gone with the Wnts: Beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol. Endocrinol. 2007, 21, 2605–2614. [Google Scholar] [CrossRef]
- Castilho, R.M.; Squarize, C.H.; Chodosh, L.A.; Williams, B.O.; Gutkind, J.S. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 2009, 5, 279–289. [Google Scholar] [CrossRef]
- Wood, K.C.; Sabatini, D.M. Growth signaling at the nexus of stem cell life and death. Cell Stem Cell 2009, 5, 232–234. [Google Scholar] [CrossRef]
- White, B.D.; Nguyen, N.K.; Moon, R.T. Wnt signaling: It gets more humorous with age. Curr. Biol. 2007, 17, 923–925. [Google Scholar] [CrossRef]
- Katoh, M. Wnt signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef]
- Katoh, M. Networking of WNT, FGF, Notch, BMP, and hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007, 3, 30–38. [Google Scholar] [CrossRef]
- Giarre, M.; Semenov, M.V.; Brown, A.M. Wnt signaling stabilizes the dual-function protein beta-catenin in diverse cell types. Ann. N. Y. Acad. Sci. 1998, 857, 43–55. [Google Scholar] [CrossRef]
- Shimizu, T.; Kagawa, T.; Inoue, T.; Nonaka, A.; Takada, S.; Aburatani, H.; Taga, T. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol. Cell. Biol. 2008, 28, 7427–7441. [Google Scholar] [CrossRef]
- Baksh, D.; Boland, G.M.; Tuan, R.S. Cross-talk between Wnt signaling pathways in human mesenchymal stem cells leads to functional antagonism during osteogenic differentiation. J. Cell. Biochem. 2007, 101, 1109–1124. [Google Scholar] [CrossRef]
- Baksh, D.; Tuan, R.S. Canonical and non-canonical Wnts differentially affect the development potential of primary isolate of human bone marrow mesenchymal stem cells. J. Cell. Physiol. 2007, 212, 817–826. [Google Scholar] [CrossRef]
- Boland, G.M.; Perkins, G.; Hall, D.J.; Tuan, R.S. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J. Cell. Biochem. 2004, 93, 1210–1230. [Google Scholar] [CrossRef]
- Chen, C.C.; Gau, J.P.; You, J.Y.; Lee, K.D.; Yu, Y.B.; Lu, C.H.; Lin, J.T.; Lan, C.; Lo, W.H.; Liu, J.M.; et al. Prognostic significance of beta-catenin and topoisomerase IIalpha in de novo acute myeloid leukemia. Am. J. Hematol. 2009, 84, 87–92. [Google Scholar] [CrossRef]
- Meshorer, E.; Gruenbaum, Y. Gone with the Wnt/Notch: Stem cells in laminopathies, progeria, and aging. J. Cell Biol. 2008, 181, 9–13. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 2008, 10, 452–459. [Google Scholar] [CrossRef]
- Espada, J.; Varela, I.; Flores, I.; Ugalde, A.P.; Cadinanos, J.; Pendas, A.M.; Stewart, C.L.; Tryggvason, K.; Blasco, M.A.; Freije, J.M.; et al. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J. Cell Biol. 2008, 181, 27–35. [Google Scholar] [CrossRef]
- Hernandez, L.; Roux, K.J.; Wong, E.S.; Mounkes, L.C.; Mutalif, R.; Navasankari, R.; Rai, B.; Cool, S.; Jeong, J.W.; Wang, H.; et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev. Cell 2010, 19, 413–425. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef]
- Kirstetter, P.; Anderson, K.; Porse, B.T.; Jacobsen, S.E.; Nerlov, C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat. Immunol. 2006, 7, 1048–1056. [Google Scholar] [CrossRef]
- Scheller, M.; Huelsken, J.; Rosenbauer, F.; Taketo, M.M.; Birchmeier, W.; Tenen, D.G.; Leutz, A. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat. Immunol. 2006, 7, 1037–1047. [Google Scholar] [CrossRef]
- Schmidt, E.; Nilsson, O.; Koskela, A.; Tuukkanen, J.; Ohlsson, C.; Rozell, B.; Eriksson, M. Expression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J. Biol. Chem. 2012, 287, 33512–33522. [Google Scholar]
- Xiao, N.M.; Zhang, Y.M.; Zheng, Q.; Gu, J. Klotho is a serum factor related to human aging. Chin. Med. J. 2004, 117, 742–747. [Google Scholar]
- Salih, D.A.; Brunet, A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 2008, 20, 126–136. [Google Scholar] [CrossRef]
- Daitoku, H.; Fukamizu, A. FOXO transcription factors in the regulatory networks of longevity. J. Biochem. 2007, 141, 769–774. [Google Scholar] [CrossRef]
- Rollo, C.D. Aging and the mammalian regulatory triumvirate. Aging Dis. 2010, 1, 105–138. [Google Scholar]
- Brosens, J.J.; Wilson, M.S.; Lam, E.W. FOXO transcription factors: From cell fate decisions to regulation of human female reproduction. Adv. Exp. Med. Biol. 2009, 665, 227–241. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef]
- Essers, M.A.; de Vries-Smits, L.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.; Korswagen, H.C. Functional interaction between beta-catenin and FoxO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]
- Birkenkamp, K.U.; Coffer, P.J. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem. Soc. Trans. 2003, 31, 292–297. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Kobayashi, Y.; Chen, C.; Motoyama, N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid. Redox Signal. 2005, 7, 752–760. [Google Scholar] [CrossRef]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef]
- He, P.; Shen, Y. Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer’s disease. J. Neurosci. 2009, 29, 6545–6557. [Google Scholar] [CrossRef]
- Hoogeboom, D.; Burgering, B.M. Should I stay or should I go: Beta-Catenin decides under stress. Biochim. Biophys. Acta 2009, 1796, 63–74. [Google Scholar]
- Rached, M.T.; Kode, A.; Xu, L.; Yoshikawa, Y.; Paik, J.H.; Depinho, R.A.; Kousteni, S. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010, 11, 147–160. [Google Scholar] [CrossRef]
- Rached, M.T.; Kode, A.; Silva, B.C.; Jung, D.Y.; Gray, S.; Ong, H.; Paik, J.H.; DePinho, R.A.; Kim, J.K.; Karsenty, G.; et al. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J. Clin. Invest. 2010, 120, 357–368. [Google Scholar] [CrossRef]
- Dejean, A.S.; Hedrick, S.M.; Kerdiles, Y.M. Highly specialized role of Forkhead box O transcription factors in the immune system. Antioxid. Redox Signal. 2011, 14, 663–674. [Google Scholar] [CrossRef]
- Nelson, T.J.; Behfar, A.; Yamada, S.; Martinez-Fernandez, A.; Terzic, A. Stem cell platforms for regenerative medicine. Clin. Transl. Sci. 2009, 2, 222–227. [Google Scholar] [CrossRef]
- Yamanaka, S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 2007, 1, 39–49. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induced pluripotent stem cells in medicine and biology. Development 2013, 140, 2457–2461. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Yu, J.; Thomson, J.A. Pluripotent stem cell lines. Genes Dev. 2008, 22, 1987–1997. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef]
- Shi, Y.; Desponts, C.; Do, J.T.; Hahm, H.S.; Scholer, H.R.; Ding, S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell 2008, 3, 568–574. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Diederichs, S.; Shine, K.M.; Tuan, R.S. The promise and challenges of stem cell-based therapies for skeletal diseases: Stem cell applications in skeletal medicine: Potential, cell sources and characteristics, and challenges of clinical translation. BioEssays 2013, 35, 220–230. [Google Scholar] [CrossRef]
- Yamanaka, S. Elite and stochastic models for induced pluripotent stem cell generation. Nature 2009, 460, 49–52. [Google Scholar] [CrossRef]
- Morris, S.A.; Daley, G.Q. A blueprint for engineering cell fate: Current technologies to reprogram cell identity. Cell Res. 2013, 23, 33–48. [Google Scholar] [CrossRef]
- Hotta, A.; Ellis, J. Retroviral vector silencing during iPS cell induction: An epigenetic beacon that signals distinct pluripotent states. J. Cell. Biochem. 2008, 105, 940–948. [Google Scholar] [CrossRef]
- Wong, C.J.; Casper, R.F.; Rogers, I.M. Epigenetic changes to human umbilical cord blood cells cultured with three proteins indicate partial reprogramming to a pluripotent state. Exp. Cell Res. 2010, 316, 927–939. [Google Scholar] [CrossRef]
- Ruhnke, M.; Ungefroren, H.; Nussler, A.; Martin, F.; Brulport, M.; Schormann, W.; Hengstler, J.G.; Klapper, W.; Ulrichs, K.; Hutchinson, J.A.; et al. Differentiation of in vitro-modified human peripheral blood monocytes into hepatocyte-like and pancreatic islet-like cells. Gastroenterology 2005, 128, 1774–1786. [Google Scholar] [CrossRef]
- Chang, M.Y.; Kim, D.; Kim, C.H.; Kang, H.C.; Yang, E.; Moon, J.I.; Ko, S.; Park, J.; Park, K.S.; Lee, K.A.; et al. Direct reprogramming of rat neural precursor cells and fibroblasts into pluripotent stem cells. PLoS One 2010, 5, e9838. [Google Scholar] [CrossRef]
- Metzler, K.R. Directing smooth muscle cell fate: A partial reprogramming approach to engineer vessels. Circ. Res. 2013, 112, 1402–1404. [Google Scholar] [CrossRef]
- Karamariti, E.; Margariti, A.; Winkler, B.; Wang, X.; Hong, X.; Baban, D.; Ragoussis, J.; Huang, Y.; Han, J.D.; Wong, M.M.; et al. Smooth muscle cells differentiated from reprogrammed embryonic lung fibroblasts through DKK3 signaling are potent for tissue engineering of vascular grafts. Circ. Res. 2013, 112, 1433–1443. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef]
- Inagawa, K.; Miyamoto, K.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Umei, T.; Wada, R.; Katsumata, Y.; Kaneda, R.; Nakade, K.; et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 1147–1156. [Google Scholar] [CrossRef]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.D.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Kim, J.; Efe, J.A.; Zhu, S.; Talantova, M.; Yuan, X.; Wang, S.; Lipton, S.A.; Zhang, K.; Ding, S. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 7838–7843. [Google Scholar]
- Efe, J.A.; Hilcove, S.; Kim, J.; Zhou, H.; Ouyang, K.; Wang, G.; Chen, J.; Ding, S. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat. Cell Biol. 2011, 13, 215–222. [Google Scholar] [CrossRef]
- Wagner, W.; Bork, S.; Lepperdinger, G.; Joussen, S.; Ma, N.; Strunk, D.; Koch, C. How to track cellular aging of mesenchymal stromal cells? Aging 2010, 2, 224–230. [Google Scholar]
- Wagner, W.; Ho, A.D.; Zenke, M. Different facets of aging in human mesenchymal stem cells. Tissue Eng. Part B: Rev. 2010, 16, 445–453. [Google Scholar] [CrossRef]
- Singh, S.; Dhaliwal, N.; Crawford, R.; Xiao, Y. Cellular senescence and longevity of osteophyte-derived mesenchymal stem cells compared to patient-matched bone marrow stromal cells. J. Cell. Biochem. 2009, 108, 839–850. [Google Scholar] [CrossRef]
- Ksiazek, K. A comprehensive review on mesenchymal stem cell growth and senescence. Rejuven. Res. 2009, 12, 105–116. [Google Scholar] [CrossRef]
- Melone, M.A.; Giuliano, M.; Squillaro, T.; Alessio, N.; Casale, F.; Mattioli, E.; Cipollaro, M.; Giordano, A.; Galderisi, U. Genes involved in regulation of stem cell properties: Studies on their expression in a small cohort of neuroblastoma patients. Cancer Biol. Ther. 2009, 8, 1300–1306. [Google Scholar] [CrossRef]
- Galderisi, U.; Helmbold, H.; Squillaro, T.; Alessio, N.; Komm, N.; Khadang, B.; Cipollaro, M.; Bohn, W.; Giordano, A. In vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev. 2009, 18, 1033–1042. [Google Scholar] [CrossRef]
- Chew, J.L.; Loh, Y.H.; Zhang, W.; Chen, X.; Tam, W.L.; Yeap, L.S.; Li, P.; Ang, Y.S.; Lim, B.; Robson, P.; et al. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol. Cell. Biol. 2005, 25, 6031–6046. [Google Scholar] [CrossRef]
- Rodda, D.J.; Chew, J.L.; Lim, L.H.; Loh, Y.H.; Wang, B.; Ng, H.H.; Robson, P. Transcriptional regulation of Nanog by Oct4 and Sox2. J. Biol. Chem. 2005, 280, 24731–24737. [Google Scholar]
- Johnson, R.; Teh, C.H.; Kunarso, G.; Wong, K.Y.; Srinivasan, G.; Cooper, M.L.; Volta, M.; Chan, S.S.; Lipovich, L.; Pollard, S.M.; et al. Rest regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol. 2008, 6, e256. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, Y.S.; Loh, Y.H.; Cai, J.; Tong, G.Q.; Lim, C.A.; Robson, P.; Zhong, S.; Ng, H.H. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 2008, 10, 353–360. [Google Scholar] [CrossRef]
- Pan, G.; Li, J.; Zhou, Y.; Zheng, H.; Pei, D. A negative feedback loop of transcription factors that controls stem cell pluripotency and self-renewal. FASEB J. 2006, 20, 1730–1732. [Google Scholar] [CrossRef]
- Chambers, I.; Tomlinson, S.R. The transcriptional foundation of pluripotency. Development 2009, 136, 2311–2322. [Google Scholar] [CrossRef]
- Som, A.; Harder, C.; Greber, B.; Siatkowski, M.; Paudel, Y.; Warsow, G.; Cap, C.; Scholer, H.; Fuellen, G. The PluriNetWork: An electronic representation of the network underlying pluripotency in mouse, and its applications. PLoS One 2010, 5, e15165. [Google Scholar] [CrossRef]
- Dowell, K.G.; Simons, A.K.; Bai, H.; Kell, B.; Wang, Z.Z.; Yun, K.; Hibbs, M.A. Novel insights into embryonic stem cell self-renewal revealed through comparative human and mouse systems biology networks. Stem Cells 2013. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef]
- Kashyap, V.; Rezende, N.C.; Scotland, K.B.; Shaffer, S.M.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. Regulation of stem cell pluripotency and differentiation involves a mutual regulatory circuit of the Nanog, Oct4, and Sox2 pluripotency transcription factors with polycomb repressive complexes and stem cell microRNAs. Stem Cells Dev. 2009, 18, 1093–1108. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Lunyak, V.V. Epigenetics: Judge, jury and executioner of stem cell fate. Epigenetics 2012, 7, 823–840. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Brunet, A. Aging and reprogramming: A two-way street. Curr. Opin. Cell Biol. 2012, 24, 744–756. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Norddahl, G.L.; Sten, G.; Ugale, A.; Frisk, M.A.; Mattsson, R.; Deierborg, T.; Sigvardsson, M.; Bryder, D. An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood 2013, 121, 4257–4264. [Google Scholar] [CrossRef]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Vaziri, H.; Chapman, K.B.; Guigova, A.; Teichroeb, J.; Lacher, M.D.; Sternberg, H.; Singec, I.; Briggs, L.; Wheeler, J.; Sampathkumar, J.; et al. Spontaneous reversal of the developmental aging of normal human cells following transcriptional reprogramming. Regen. Med. 2010, 5, 345–363. [Google Scholar] [CrossRef]
- Wen, Y.; Wani, P.; Zhou, L.; Baer, T.; Phadnis, S.M.; Reijo Pera, R.A.; Chen, B. Reprogramming of fibroblasts from older women with pelvic floor disorders alters cellular behavior associated with donor age. Stem Cells Transl. Med. 2013, 2, 118–128. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Corominas-Faja, B.; Cufi, S.; Vellon, L.; Oliveras-Ferraros, C.; Menendez, O.J.; Joven, J.; Lupu, R.; Menendez, J.A. The mitochondrial H(+)-ATP synthase and the lipogenic switch: New core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle 2013, 12, 207–218. [Google Scholar] [CrossRef]
- Lee, E.K.; Jeong, J.U.; Chang, J.W.; Yang, W.S.; Kim, S.B.; Park, S.K.; Park, J.S.; Lee, S.K. Activation of AMP-activated protein kinase inhibits albumin-induced endoplasmic reticulum stress and apoptosis through inhibition of reactive oxygen species. Nephron. Exp. Nephrol. 2012, 121, 38–48. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef]
- Chen, T.; Shen, L.; Yu, J.; Wan, H.; Guo, A.; Chen, J.; Long, Y.; Zhao, J.; Pei, G. Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 2011, 10, 908–911. [Google Scholar] [CrossRef]
- Shi, Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat. Rev. Genet. 2007, 8, 829–833. [Google Scholar] [CrossRef]
- Menendez, J.A.; Vellon, L.; Oliveras-Ferraros, C.; Cufi, S.; Vazquez-Martin, A. Mtor-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: A roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011, 10, 3658–3677. [Google Scholar] [CrossRef]
- Shen, Y.A.; Lin, C.H.; Chi, W.H.; Wang, C.Y.; Hsieh, Y.T.; Wei, Y.H.; Chen, Y.J. Resveratrol impedes the stemness, epithelial-mesenchymal transition, and metabolic reprogramming of cancer stem cells in nasopharyngeal carcinoma through p53 activation. Evid.-Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Liu, G.X. Anti-aging effect of transplantation of mouse fetus-derived mesenchymal stem cells. Acta Physiol. Sin. 2010, 62, 79–85. [Google Scholar]
- Asahara, T.; Kalka, C.; Isner, J.M. Stem cell therapy and gene transfer for regeneration. Gene Ther. 2000, 7, 451–457. [Google Scholar] [CrossRef]
- Schiavetta, A.; Maione, C.; Botti, C.; Marino, G.; Lillo, S.; Garrone, A.; Lanza, L.; Pagliari, S.; Silvestroni, A.; Signoriello, G.; et al. A phase II trial of autologous transplantation of bone marrow stem cells for critical limb ischemia: Results of the Naples and Pietra Ligure evaluation of stem cells study. Stem Cells Transl. Med. 2012, 1, 572–578. [Google Scholar] [CrossRef]
- Liew, A.; O’Brien, T. Therapeutic potential for mesenchymal stem cell transplantation in critical limb ischemia. Stem Cell Res. Ther. 2012, 3, 1–14. [Google Scholar] [CrossRef]
- Deng, W.; Bivalacqua, T.J.; Hellstrom, W.J.; Kadowitz, P.J. Gene and stem cell therapy for erectile dysfunction. Int. J. Impot. Res. 2005, 17, 57–63. [Google Scholar] [CrossRef]
- Qiu, X.; Sun, C.; Yu, W.; Lin, H.; Sun, Z.; Chen, Y.; Wang, R.; Dai, Y. Combined strategy of mesenchymal stem cell injection with vascular endothelial growth factor gene therapy for the treatment of diabetes-associated erectile dysfunction. J. Androl. 2012, 33, 37–44. [Google Scholar] [CrossRef]
- McGuckin, C.P.; Jurga, M.; Miller, A.M.; Sarnowska, A.; Wiedner, M.; Boyle, N.T.; Lynch, M.A.; Jablonska, A.; Drela, K.; Lukomska, B.; et al. Ischemic brain injury: A consortium analysis of key factors involved in mesenchymal stem cell-mediated inflammatory reduction. Arch. Biochem. Biophys. 2013, 534, 88–97. [Google Scholar] [CrossRef]
- Hu, X.; Yu, S.P.; Fraser, J.L.; Lu, Z.; Ogle, M.E.; Wang, J.A.; Wei, L. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J. Thorac. Cardiovasc. Surg. 2008, 135, 799–808. [Google Scholar]
- Rosova, I.; Dao, M.; Capoccia, B.; Link, D.; Nolta, J.A. Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells 2008, 26, 2173–2182. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, P.J.; Tian, T.; Jin, C.; Li, Y.; Feng, M.; Liu, X.Y.; Jie, L.; Tao, L.D. Role of vascular endothelial growth factor in protection of intrahepatic cholangiocytes mediated by hypoxic preconditioning after liver transplantation in rats. Transplant. Proc. 2010, 42, 2457–2462. [Google Scholar] [CrossRef]
- Yagi, H.; Tan, J.; Tuan, R.S. Polyphenols suppress hydrogen peroxide-induced oxidative stress in human bone-marrow derived mesenchymal stem cells. J. Cell. Biochem. 2013, 114, 1163–1173. [Google Scholar] [CrossRef]
- Kang, K.; Sun, L.; Xiao, Y.; Li, S.H.; Wu, J.; Guo, J.; Jiang, S.L.; Yang, L.; Yau, T.M.; Weisel, R.D.; et al. Aged human cells rejuvenated by cytokine enhancement of biomaterials for surgical ventricular restoration. J. Am. Coll. Cardiol. 2012, 60, 2237–2249. [Google Scholar] [CrossRef]
- Turgeman, G.; Zilberman, Y.; Zhou, S.; Kelly, P.; Moutsatsos, I.K.; Kharode, Y.P.; Borella, L.E.; Bex, F.J.; Komm, B.S.; Bodine, P.V.; et al. Systemically administered rhBMP-2 promotes MSC activity and reverses bone and cartilage loss in osteopenic mice. J. Cell. Biochem. 2002, 86, 461–474. [Google Scholar] [CrossRef]
- Madonna, R.; Taylor, D.A.; Geng, Y.J.; de Caterina, R.; Shelat, H.; Perin, E.C.; Willerson, J.T. Transplantation of mesenchymal cells rejuvenated by the overexpression of telomerase and myocardin promotes revascularization and tissue repair in a murine model of hindlimb ischemia. Circ. Res. 2013, 113, 902–914. [Google Scholar] [CrossRef]
- Yao, J.; Jiang, S.L.; Liu, W.; Liu, C.; Chen, W.; Sun, L.; Liu, K.Y.; Jia, Z.B.; Li, R.K.; Tian, H. Tissue inhibitor of matrix metalloproteinase-3 or vascular endothelial growth factor transfection of aged human mesenchymal stem cells enhances cell therapy after myocardial infarction. Rejuven. Res. 2012, 15, 495–506. [Google Scholar] [CrossRef]
- Tapia, N.; Han, D.W.; Scholer, H.R. Restoring stem cell function in aged tissues by direct reprogramming? Cell Stem Cell 2012, 10, 653–656. [Google Scholar] [CrossRef]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Glorioso, J.C.; Robbins, P.D. Dedifferentiation rescues senescence of progeria cells but only while pluripotent. Stem Cell Res. Ther. 2011, 2, 1–4. [Google Scholar] [CrossRef]
- Milone, G.; Mercurio, S.; Strano, A.; Leotta, S.; Pinto, V.; Battiato, K.; Coppoletta, S.; Murgano, P.; Farsaci, B.; Privitera, A.; et al. Adverse events after infusions of cryopreserved hematopoietic stem cells depend on non-mononuclear cells in the infused suspension and patient age. Cytotherapy 2007, 9, 348–355. [Google Scholar] [CrossRef]
- Alousi, A.M.; Le-Rademacher, J.; Saliba, R.M.; Appelbaum, F.R.; Artz, A.; Benjamin, J.; Devine, S.M.; Kan, F.; Laughlin, M.J.; Lazarus, H.M.; et al. Who is the better donor for older hematopoietic transplant recipients: An older-aged sibling or a young, matched unrelated volunteer? Blood 2013, 121, 2567–2573. [Google Scholar] [CrossRef]
- Han, J.; Mistriotis, P.; Lei, P.; Wang, D.; Liu, S.; Andreadis, S.T. Nanog reverses the effects of organismal aging on mesenchymal stem cell proliferation and myogenic differentiation potential. Stem Cells 2012, 30, 2746–2759. [Google Scholar] [CrossRef]
- Curtis, R.; Geesaman, B.J.; DiStefano, P.S. Ageing and metabolism: Drug discovery opportunities. Nat. Rev. Drug Discov. 2005, 4, 569–580. [Google Scholar] [CrossRef]
- Geiger, L.E.; Wsd, D.J.; Lewis, C.B.; Liu, K.C.; Newsholme, S.J. Rat carcinogenicity study with gw501516, a ppar delta agonist. Toxicologist 2009, 108, 185. [Google Scholar]
- Newsholme, S.J.; Dunsford, W.S.; Brodie, T.; Brennan, C.; Brown, M.; Geiger, L.E. Mouse carcinogenicity study with gw501516, a ppar delta agonist. Toxicologist 2009, 108, 185. [Google Scholar]
- De Magalhaes, J.P. How ageing processes influence cancer. Nat. Rev. Cancer 2013, 13, 357–365. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS One 2010, 5, e8758. [Google Scholar] [CrossRef]
- Kirk, H.; Cefalu, W.T.; Ribnicky, D.; Liu, Z.; Eilertsen, K.J. Botanicals as epigenetic modulators for mechanisms contributing to development of metabolic syndrome. Metabolism 2008, 57, 16–23. [Google Scholar]
- Selcuklu, S.D.; Spillane, C. Translational epigenetics: Clinical approaches to epigenome therapeutics for cancer. Epigenetics 2008, 3, 107–112. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Z.; Zhang, L.; Ghosh, S.; Zheng, H.; Zhou, Z. Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Krishnan, V.; Chow, M.Z.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Invest. 2013, 123, 966–972. [Google Scholar] [CrossRef]
- Bilsland, A.E.; Revie, J.; Keith, W. MicroRNA and senescence: The senectome, integration and distributed control. Crit. Rev. Oncog. 2013, 18, 373–390. [Google Scholar] [CrossRef]
- Bitar, M.S.; Abdel-Halim, S.M.; Al-Mulla, F. Caveolin-1/PTRF upregulation in diabetic fibroblasts and wounded tissues: Implication for understanding the underlying mechanisms of non-healing diabetic ulcers. Am. J. Physiol. Endocrinol. Metab. 2013, 305, 951–963. [Google Scholar]
- Hayashi, T.; Iguchi, A. Possibility of the regression of atherosclerosis through the prevention of endothelial senescence by the regulation of nitric oxide and free radical scavengers. Geriatr. Gerontol. Int. 2010, 10, 115–130. [Google Scholar]
- Effros, R.B. Telomerase induction in T cells: A cure for aging and disease? Exp. Gerontol. 2007, 42, 416–420. [Google Scholar] [CrossRef]
- Sharp, Z.D.; Curiel, T.J.; Livi, C.B. Chronic mechanistic target of rapamycin inhibition: Preventing cancer to delay aging, or vice versa? Cancer Aging 2013, 38, 1–16. [Google Scholar] [CrossRef]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Ibrahim, M.X.; Sayin, V.I.; Akula, M.K.; Liu, M.; Fong, L.G.; Young, S.G.; Bergo, M.O. Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 2013, 340, 1330–1333. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Invest. 2013, 123, 980–989. [Google Scholar] [CrossRef]
- Yuen, D.A.; Zhang, Y.; Thai, K.; Spring, C.; Chan, L.; Guo, X.; Advani, A.; Sivak, J.M.; Gilbert, R.E. Angiogenic dysfunction in bone marrow-derived early outgrowth cells from diabetic animals is attenuated by SIRT1 activation. Stem Cells Transl. Med. 2012, 1, 921–926. [Google Scholar] [CrossRef]
- Matsushita, K.; Wu, Y.; Okamoto, Y.; Pratt, R.E.; Dzau, V.J. Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension 2006, 48, 1095–1102. [Google Scholar] [CrossRef]
- Kim, D.H.; Vanella, L.; Inoue, K.; Burgess, A.; Gotlinger, K.; Manthati, V.L.; Koduru, S.R.; Zeldin, D.C.; Falck, J.R.; Schwartzman, M.L.; et al. Epoxyeicosatrienoic acid agonist regulates human mesenchymal stem cell-derived adipocytes through activation of HO-1-pAKT signaling and a decrease in PPARγ. Stem Cells Dev. 2010, 19, 1863–1873. [Google Scholar] [CrossRef]
- Sodhi, K.; Inoue, K.; Gotlinger, K.H.; Canestraro, M.; Vanella, L.; Kim, D.H.; Manthati, V.L.; Koduru, S.R.; Falck, J.R.; Schwartzman, M.L.; et al. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J. Pharmacol. Exp. Ther. 2009, 331, 906–916. [Google Scholar] [CrossRef]
- Gimble, J.M.; Floyd, Z.E.; Bunnell, B.A. The 4th dimension and adult stem cells: Can timing be everything? J. Cell. Biochem. 2009, 107, 569–578. [Google Scholar] [CrossRef]
- Yagita, K.; Horie, K.; Koinuma, S.; Nakamura, W.; Yamanaka, I.; Urasaki, A.; Shigeyoshi, Y.; Kawakami, K.; Shimada, S.; Takeda, J.; et al. Development of the circadian oscillator during differentiation of mouse embryonic stem cells in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 3846–3851. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Ahmad, N. SIRT1 controls circadian clock circuitry and promotes cell survival: A connection with age-related neoplasms. FASEB J. 2009, 23, 2803–2809. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef]
- Tevy, M.F.; Giebultowicz, J.; Pincus, Z.; Mazzoccoli, G.; Vinciguerra, M. Aging signaling pathways and circadian clock-dependent metabolic derangements. Trends Endocrinol. Metab. 2013, 24, 229–237. [Google Scholar] [CrossRef]
- Chen, C.C.; Chuong, C.M. Multi-layered environmental regulation on the homeostasis of stem cells: The saga of hair growth and alopecia. J. Dermatol. Sci. 2012, 66, 3–11. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boyette, L.B.; Tuan, R.S. Adult Stem Cells and Diseases of Aging. J. Clin. Med. 2014, 3, 88-134. https://doi.org/10.3390/jcm3010088
Boyette LB, Tuan RS. Adult Stem Cells and Diseases of Aging. Journal of Clinical Medicine. 2014; 3(1):88-134. https://doi.org/10.3390/jcm3010088
Chicago/Turabian StyleBoyette, Lisa B., and Rocky S. Tuan. 2014. "Adult Stem Cells and Diseases of Aging" Journal of Clinical Medicine 3, no. 1: 88-134. https://doi.org/10.3390/jcm3010088
APA StyleBoyette, L. B., & Tuan, R. S. (2014). Adult Stem Cells and Diseases of Aging. Journal of Clinical Medicine, 3(1), 88-134. https://doi.org/10.3390/jcm3010088