
Catecholaminergic Polymorphic Ventricular Tachycardia: Clinical Characteristics, Diagnostic Evaluation and Therapeutic Strategies

 ,
,  ,
,
Abstract
1. Introduction
2. History
3. Epidemiology
4. Clinical Presentation
5. Genetics and Pathophysiology
6. Diagnosis
7. Prognosis and Risk Stratification
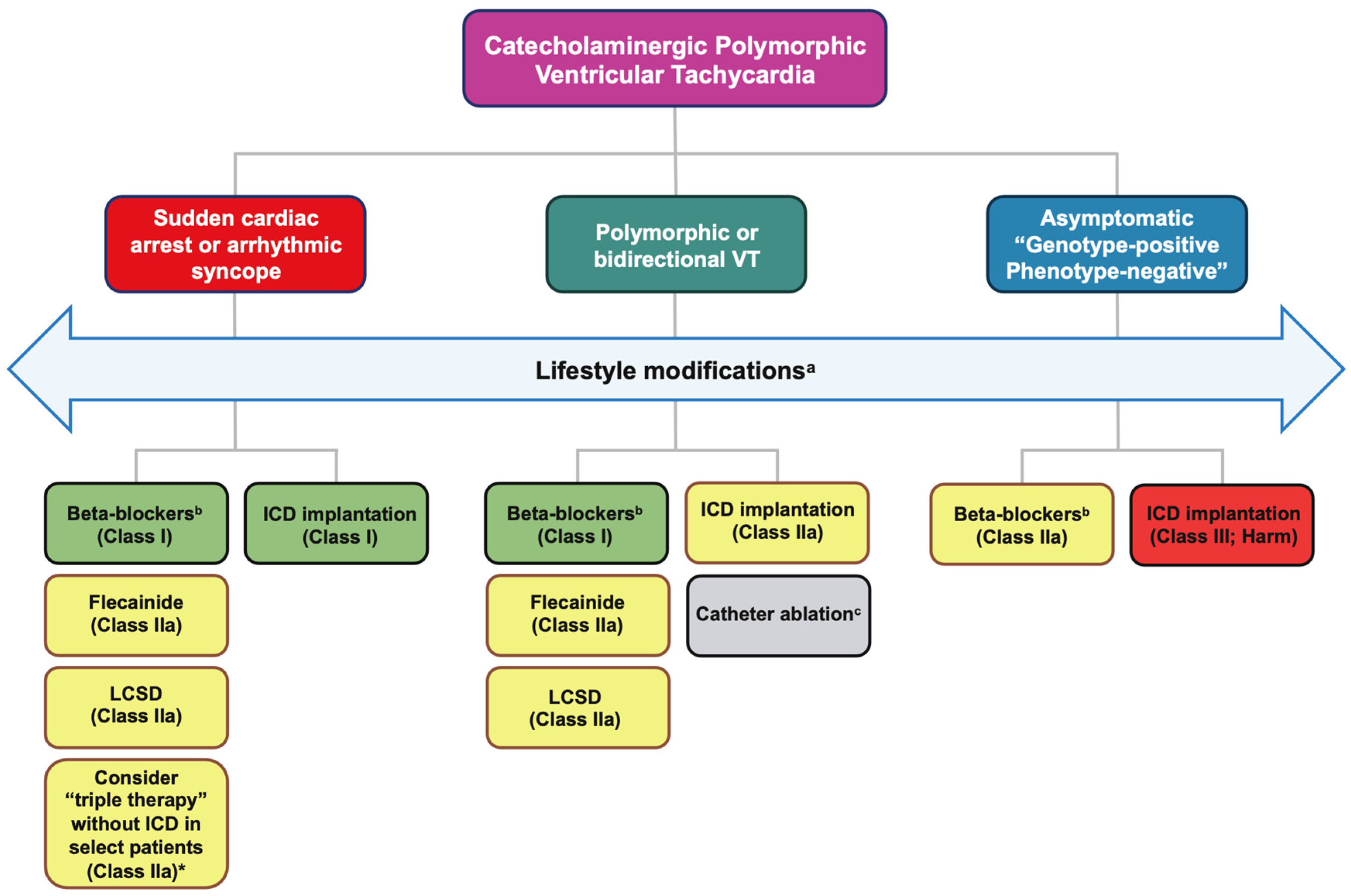
8. Therapeutic Strategies
8.1. Lifestyle Modifications
- Competitive and intensive leisure-time sports are strongly discouraged.
- Consideration of low-intensity to moderate leisure-time sports is permissible if the patient remains asymptomatic for a minimum of three months and stress tests reveal the absence of any ventricular ectopy/arrhythmia even in those with an ICD.
- Gene carriers with a pathogenic CPVT mutation lacking overt symptoms should be managed akin to patients with manifest CPVT, permitting only low-intensity sports.
- Follow-up protocols should incorporate stress tests and/or continuous electrocardiogram (ECG) monitoring (Holter) during low-intensity leisure-time sports activities to ensure control of exercise-induced ventricular arrhythmias.
- Recommendations include avoidance of stressful/emotional situations, dehydration, electrolyte disturbances or hyperthermia.
8.2. Medical Management
8.2.1. Beta-Blockers
8.2.2. Flecainide
8.2.3. Other Medications
8.3. Non-Pharmacological Therapies
8.3.1. Left Cardiac Sympathetic Denervation
8.3.2. Implantable Cardioverter-Defibrillators (ICDs)
8.3.3. Catheter Ablation
8.4. Potential Future CPVT Therapies
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACA | Aborted cardiac arrest |
| ACMG | American college of medical genetics |
| AMP | Association for molecular pathology |
| Ca2+ | Calcium |
| CASQ | Calsequestrin |
| CNV | Copy number variants |
| CPET | Cardiopulmonary exercise testing |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| CRISPR | Clustered regularly interspaced short palindromic repeat |
| CSD | Cardiac sympathetic denervation |
| CT | Computed tomography |
| EAPC | European association of preventive cardiology |
| EHRA | European heart rhythm association |
| EKG | Electrocardiogram |
| ERP | Early repolarization pattern |
| ESC | European society of cardiology |
| HCN | Hyperpolarization-activated cyclic nucleotide |
| ICD | Implantable cardioverter defibrillator |
| iPSC | Induced pluripotent stem cell |
| LBBB | Left bundle branch block |
| LCSD | Left cardiac sympathetic denervation |
| LV | Left ventricle |
| MRI | Magnetic resonance imaging |
| NSVT | Non-sustained ventricular tachycardia |
| PVC | Premature ventricular complexes |
| QTc | Corrected QT interval |
| RBBB | Right bundle branch block |
| RCSD | Right cardiac sympathetic denervation |
| RCT | Randomized controlled trials |
| RVOT | Right ventricular outflow tract |
| RWMA | Regional wall motion abnormalities |
| RyR | Ryanodine receptor |
| SCA | Sudden cardiac arrest |
| SCD | Sudden cardiac death |
| S-ICD | Subcutaneous implantable cardioverter defibrillator |
| SIDS | Sudden infant death syndrome |
| SNV | Single nucleotide variants |
| TdP | Torsades-de-Pointes |
| TECRL | Trans-2,3-Enoyl-CoA-Reductase-like |
| TRDN | Triadin |
| TTE | Transthoracic echocardiogram |
| VAT | Ventilatory anaerobic threshold |
| VF | Ventricular fibrillation |
| VT | Ventricular tachycardia |
| VUS | Variants of uncertain significance |
| WT | Wild-type |
References
- Berg, K.J. Multifocal ventricular extrasystoles with Adams-Stokes syndrome in siblings. Am. Heart J. 1960, 60, 965–970. [Google Scholar] [CrossRef]
- Reid, D.S.; Tynan, M.; Braidwood, L.; Fitzgerald, G.R. Bidirectional tachycardia in a child. A study using His bundle electrography. Br. Heart J. 1975, 37, 339–344. [Google Scholar] [CrossRef]
- Coumel, P.; Fidelle, J.; Lucet, V.; Attuel, P.; Bouvrain, Y. Catecholamine-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: Report of four cases. Br. Heart J. 1978, 40, 28–37. [Google Scholar]
- Swan, H.; Piippo, K.; Viitasalo, M.; Heikkila, P.; Paavonen, T.; Kainulainen, K.; Kere, J.; Keto, P.; Kontula, K.; Toivonen, L. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J. Am. Coll. Cardiol. 1999, 34, 2035–2042. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef]
- Obeyesekere, M.N.; Antzelevitch, C.; Krahn, A.D. Management of ventricular arrhythmias in suspected channelopathies. Circ. Arrhythm. Electrophysiol. 2015, 8, 221–231. [Google Scholar] [CrossRef]
- Mariani, M.V.; Pierucci, N.; Fanisio, F.; Laviola, D.; Silvetti, G.; Piro, A.; La Fazia, V.M.; Chimenti, C.; Rebecchi, M.; Drago, F.; et al. Inherited Arrhythmias in the Pediatric Population: An Updated Overview. Medicina 2024, 60, 94. [Google Scholar] [CrossRef]
- Choi, G.; Kopplin, L.J.; Tester, D.J.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation 2004, 110, 2119–2124. [Google Scholar] [CrossRef]
- Tester, D.J.; Spoon, D.B.; Valdivia, H.H.; Makielski, J.C.; Ackerman, M.J. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: A molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin. Proc. 2004, 79, 1380–1384. [Google Scholar] [CrossRef]
- Napolitano, C.; Mazzanti, A.; Bloise, R.; Priori, S.G. Catecholaminergic Polymorphic Ventricular Tachycardia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- van der Werf, C.; Nederend, I.; Hofman, N.; van Geloven, N.; Ebink, C.; Frohn-Mulder, I.M.; Alings, A.M.; Bosker, H.A.; Bracke, F.A.; van den Heuvel, F.; et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: Disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ. Arrhythm. Electrophysiol. 2012, 5, 748–756. [Google Scholar] [CrossRef]
- Lieve, K.V.; van der Werf, C.; Wilde, A.A. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. J. 2016, 80, 1285–1291. [Google Scholar] [CrossRef]
- Napolitano, C.; Bloise, R.; Monteforte, N.; Priori, S.G. Sudden cardiac death and genetic ion channelopathies: Long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation 2012, 125, 2027–2034. [Google Scholar] [CrossRef]
- Shauer, A.; Shor, O.; Wei, J.; Elitzur, Y.; Kucherenko, N.; Wang, R.; Chen, S.R.W.; Einav, Y.; Luria, D. Novel RyR2 Mutation (G3118R) Is Associated with Autosomal Recessive Ventricular Fibrillation and Sudden Death: Clinical, Functional, and Computational Analysis. J. Am. Heart Assoc. 2021, 10, e017128. [Google Scholar] [CrossRef]
- Wleklinski, M.J.; Kannankeril, P.J.; Knollmann, B.C. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J. Physiol. 2020, 598, 2817–2834. [Google Scholar] [CrossRef]
- Cerrone, M.; Napolitano, C.; Priori, S.G. Catecholaminergic polymorphic ventricular tachycardia: A paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm. 2009, 6, 1652–1659. [Google Scholar] [CrossRef]
- Liu, N.; Colombi, B.; Memmi, M.; Zissimopoulos, S.; Rizzi, N.; Negri, S.; Imbriani, M.; Napolitano, C.; Lai, F.A.; Priori, S.G. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: Insights from a RyR2 R4496C knock-in mouse model. Circ. Res. 2006, 99, 292–298. [Google Scholar] [CrossRef]
- Hayashi, M.; Denjoy, I.; Extramiana, F.; Maltret, A.; Buisson, N.R.; Lupoglazoff, J.M.; Klug, D.; Hayashi, M.; Takatsuki, S.; Villain, E.; et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 2009, 119, 2426–2434. [Google Scholar] [CrossRef]
- De Ferrari, G.M.; Dusi, V.; Spazzolini, C.; Bos, J.M.; Abrams, D.J.; Berul, C.I.; Crotti, L.; Davis, A.M.; Eldar, M.; Kharlap, M.; et al. Clinical Management of Catecholaminergic Polymorphic Ventricular Tachycardia: The Role of Left Cardiac Sympathetic Denervation. Circulation 2015, 131, 2185–2193. [Google Scholar] [CrossRef]
- Priori, S.G.; Chen, S.R. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Yamamoto, T.; Kobayashi, S.; Tamitani, M.; Hamada, Y.; Fukui, G.; Xu, X.; Nishimura, S.; Kato, T.; Uchinoumi, H.; et al. Ryanodine receptor-bound calmodulin is essential to protect against catecholaminergic polymorphic ventricular tachycardia. JCI Insight 2019, 4, e126112. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yano, M.; Uchinoumi, H.; Hino, A.; Suetomi, T.; Ono, M.; Tateishi, H.; Oda, T.; Okuda, S.; Doi, M.; et al. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem. Biophys. Res. Commun. 2010, 394, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Klipp, R.C.; Li, N.; Wang, Q.; Word, T.A.; Sibrian-Vazquez, M.; Strongin, R.M.; Wehrens, X.H.T.; Abramson, J.J. EL20, a potent antiarrhythmic compound, selectively inhibits calmodulin-deficient ryanodine receptor type 2. Heart Rhythm. 2018, 15, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Domingo, A.; Bhuiyan, Z.A.; Tester, D.J.; Hofman, N.; Bikker, H.; van Tintelen, J.P.; Mannens, M.M.; Wilde, A.A.; Ackerman, M.J. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: A comprehensive open reading frame mutational analysis. J. Am. Coll. Cardiol. 2009, 54, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Shimamoto, K.; Ohno, S.; Kato, K.; Takayama, K.; Sonoda, K.; Fukuyama, M.; Makiyama, T.; Okamura, S.; Asakura, K.; Imanishi, N.; et al. Impact of cascade screening for catecholaminergic polymorphic ventricular tachycardia type 1. Heart 2022, 108, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Mazzanti, A.; Santiago, D.J.; Kukavica, D.; Trancuccio, A.; Kovacic, J.C. Precision Medicine in Catecholaminergic Polymorphic Ventricular Tachycardia: JACC Focus Seminar 5/5. J. Am. Coll. Cardiol. 2021, 77, 2592–2612. [Google Scholar] [CrossRef]
- Walsh, R.; Adler, A.; Amin, A.S.; Abiusi, E.; Care, M.; Bikker, H.; Amenta, S.; Feilotter, H.; Nannenberg, E.A.; Mazzarotto, F.; et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur. Heart J. 2022, 43, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Titus, E.W.; Lieve, K.V.; Roston, T.M.; Mazzanti, A.; Deiter, F.H.; Denjoy, I.; Ingles, J.; Till, J.; Robyns, T.; et al. An International Multicenter Evaluation of Inheritance Patterns, Arrhythmic Risks, and Underlying Mechanisms of CASQ2-Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2020, 142, 932–947. [Google Scholar] [CrossRef]
- Askarinejad, A.; Esmaeili, S.; Dalili, M.; Biglari, A.; Kohansal, E.; Maleki, M.; Kalayinia, S. Catecholaminergic polymorphic ventricular tachycardia (and seizure) caused by a novel homozygous likely pathogenic variant in CASQ2 gene. Gene 2024, 895, 148012. [Google Scholar] [CrossRef]
- Coll, M.; Perez-Serra, A.; Mates, J.; Del Olmo, B.; Puigmule, M.; Fernandez-Falgueras, A.; Iglesias, A.; Pico, F.; Lopez, L.; Brugada, R.; et al. Incomplete Penetrance and Variable Expressivity: Hallmarks in Channelopathies Associated with Sudden Cardiac Death. Biology 2017, 7, 3. [Google Scholar] [CrossRef]
- Postma, A.V.; Denjoy, I.; Kamblock, J.; Alders, M.; Lupoglazoff, J.M.; Vaksmann, G.; Dubosq-Bidot, L.; Sebillon, P.; Mannens, M.M.; Guicheney, P.; et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 2005, 42, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.; Mademont-Soler, I.; Fernandez-Falgueras, A.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Garcia-Alvarez, A.; Jorda, P.; Toro, R.; Coll, M.; et al. Sudden Cardiac Death and Copy Number Variants: What Do We Know after 10 Years of Genetic Analysis? Forensic Sci. Int. Genet. 2020, 47, 102281. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Marjamaa, A.; Laitinen-Forsblom, P.; Lahtinen, A.M.; Viitasalo, M.; Toivonen, L.; Kontula, K.; Swan, H. Search for cardiac calcium cycling gene mutations in familial ventricular arrhythmias resembling catecholaminergic polymorphic ventricular tachycardia. BMC Med. Genet. 2009, 10, 12. [Google Scholar] [CrossRef]
- Ohno, S.; Omura, M.; Kawamura, M.; Kimura, H.; Itoh, H.; Makiyama, T.; Ushinohama, H.; Makita, N.; Horie, M. Exon 3 deletion of RYR2 encoding cardiac ryanodine receptor is associated with left ventricular non-compaction. EP Europace 2014, 16, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Czosek, R.J.; Hinton, R.B.; Miller, E.M. Exon 3 deletion of ryanodine receptor causes left ventricular noncompaction, worsening catecholaminergic polymorphic ventricular tachycardia, and sudden cardiac arrest. Am. J. Med. Genet. A 2015, 167, 2197–2200. [Google Scholar] [CrossRef]
- Mates, J.; Mademont-Soler, I.; Del Olmo, B.; Ferrer-Costa, C.; Coll, M.; Perez-Serra, A.; Pico, F.; Allegue, C.; Fernandez-Falgueras, A.; Alvarez, P.; et al. Role of copy number variants in sudden cardiac death and related diseases: Genetic analysis and translation into clinical practice. Eur. J. Hum. Genet. 2018, 26, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Wataru, S. Syncope in patients with inherited arrhythmias. J. Arrhythm. 2017, 33, 572–578. [Google Scholar] [CrossRef]
- Koene, R.J.; Adkisson, W.O.; Benditt, D.G. Syncope and the risk of sudden cardiac death: Evaluation, management, and prevention. J. Arrhythm. 2017, 33, 533–544. [Google Scholar] [CrossRef]
- Richter, S.; Gebauer, R.; Hindricks, G.; Brugada, P. A classic electrocardiographic manifestation of catecholaminergic polymorphic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2012, 23, 560. [Google Scholar] [CrossRef]
- Blich, M.; Marai, I.; Suleiman, M.; Lorber, A.; Gepstein, L.; Boulous, M.; Khoury, A. Electrocardiographic comparison of ventricular premature complexes during exercise test in patients with CPVT and healthy subjects. Pacing Clin. Electrophysiol. 2015, 38, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, N.; Harada, K.; Nagashima, M.; Yasuda, T.; Nakamura, Y.; Aragaki, Y.; Saito, A.; Kurosaki, K.; Jouo, K.; Koujiro, M.; et al. Catecholaminergic polymorphic ventricular tachycardia: Electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 2003, 89, 66–70. [Google Scholar] [CrossRef]
- Obeyesekere, M.N.; Klein, G.J.; Modi, S.; Leong-Sit, P.; Gula, L.J.; Yee, R.; Skanes, A.C.; Krahn, A.D. How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythm. Electrophysiol. 2011, 4, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Sy, R.W.; Gollob, M.H.; Klein, G.J.; Yee, R.; Skanes, A.C.; Gula, L.J.; Leong-Sit, P.; Gow, R.M.; Green, M.S.; Birnie, D.H.; et al. Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2011, 8, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Roston, T.M.; Kallas, D.; Davies, B.; Franciosi, S.; De Souza, A.M.; Laksman, Z.W.; Sanatani, S.; Krahn, A.D. Burst Exercise Testing Can Unmask Arrhythmias in Patients with Incompletely Penetrant Catecholaminergic Polymorphic Ventricular Tachycardia. JACC Clin. Electrophysiol. 2021, 7, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Aronoff, E.B.; Baskar, S.; Czosek, R.J.; Mays, W.A.; Spar, D.S.; Knilans, T.K.; Powell, A.W. The Relationship Between Ventilatory Anaerobic Threshold and Arrhythmia in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. JACC Clin. Electrophysiol. 2024, 10, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.V.; Dusi, V.; van der Werf, C.; Bos, J.M.; Lane, C.M.; Stokke, M.K.; Roston, T.M.; Djupsjobacka, A.; Wada, Y.; Denjoy, I.; et al. Heart Rate Recovery After Exercise Is Associated with Arrhythmic Events in Patients With Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythm. Electrophysiol. 2020, 13, e007471. [Google Scholar] [CrossRef]
- van der Werf, C.; Kannankeril, P.J.; Sacher, F.; Krahn, A.D.; Viskin, S.; Leenhardt, A.; Shimizu, W.; Sumitomo, N.; Fish, F.A.; Bhuiyan, Z.A.; et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 2011, 57, 2244–2254. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Leren, I.S.; Berge, K.E.; Bathen, J.; Loennechen, J.P.; Anfinsen, O.G.; Fruh, A.; Edvardsen, T.; Kongsgard, E.; Leren, T.P.; et al. High prevalence of exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia mutation-positive family members diagnosed by cascade genetic screening. EP Europace 2010, 12, 417–423. [Google Scholar] [CrossRef]
- Marjamaa, A.; Hiippala, A.; Arrhenius, B.; Lahtinen, A.M.; Kontula, K.; Toivonen, L.; Happonen, J.M.; Swan, H. Intravenous epinephrine infusion test in diagnosis of catecholaminergic polymorphic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2012, 23, 194–199. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011, 8, 1308–1339. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Kullo, I.J.; Ackerman, M.J. Precision Cardiovascular Medicine: State of Genetic Testing. Mayo Clin. Proc. 2017, 92, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013, 10, e85–e108. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Lieve, K.V.V.; Rohatgi, R.K.; Koca, F.; Tester, D.J.; van der Werf, C.; Martijn Bos, J.; Wilde, A.A.M.; Ackerman, M.J. Assessment and Validation of a Phenotype-Enhanced Variant Classification Framework to Promote or Demote RYR2 Missense Variants of Uncertain Significance. Circ. Genom. Precis. Med. 2019, 12, e002510. [Google Scholar] [CrossRef] [PubMed]
- Tester, D.J.; Medeiros-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac channel molecular autopsy: Insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Roston, T.M.; Vinocur, J.M.; Maginot, K.R.; Mohammed, S.; Salerno, J.C.; Etheridge, S.P.; Cohen, M.; Hamilton, R.M.; Pflaumer, A.; Kanter, R.J.; et al. Catecholaminergic polymorphic ventricular tachycardia in children: Analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ. Arrhythm. Electrophysiol. 2015, 8, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Luo, Y.; Jiang, Y.; He, J. Advances in the Molecular Genetics of Catecholaminergic Polymorphic Ventricular Tachycardia. Front. Pharmacol. 2021, 12, 718208. [Google Scholar] [CrossRef]
- Roston, T.M.; Haji-Ghassemi, O.; LaPage, M.J.; Batra, A.S.; Bar-Cohen, Y.; Anderson, C.; Lau, Y.R.; Maginot, K.; Gebauer, R.A.; Etheridge, S.P.; et al. Catecholaminergic polymorphic ventricular tachycardia patients with multiple genetic variants in the PACES CPVT Registry. PLoS ONE 2018, 13, e0205925. [Google Scholar] [CrossRef]
- Kallas, D.; Roston, T.M.; Franciosi, S.; Brett, L.; Lieve, K.V.V.; Kwok, S.Y.; Kannankeril, P.J.; Krahn, A.D.; LaPage, M.J.; Etheridge, S.; et al. Evaluation of age at symptom onset, proband status, and sex as predictors of disease severity in pediatric catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2021, 18, 1825–1832. [Google Scholar] [CrossRef]
- Tulumen, E.; Schulze-Bahr, E.; Zumhagen, S.; Stallmeyer, B.; Seebohm, G.; Beckmann, B.M.; Kaab, S.; Rudic, B.; Liebe, V.; Wolpert, C.; et al. Early repolarization pattern: A marker of increased risk in patients with catecholaminergic polymorphic ventricular tachycardia. EP Europace 2016, 18, 1587–1592. [Google Scholar] [CrossRef]
- Hayashi, M.; Denjoy, I.; Hayashi, M.; Extramiana, F.; Maltret, A.; Roux-Buisson, N.; Lupoglazoff, J.M.; Klug, D.; Maury, P.; Messali, A.; et al. The role of stress test for predicting genetic mutations and future cardiac events in asymptomatic relatives of catecholaminergic polymorphic ventricular tachycardia probands. EP Europace 2012, 14, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Lahrouchi, N.; Tadros, R.; Crotti, L.; Mizusawa, Y.; Postema, P.G.; Beekman, L.; Walsh, R.; Hasegawa, K.; Barc, J.; Ernsting, M.; et al. Transethnic Genome-Wide Association Study Provides Insights in the Genetic Architecture and Heritability of Long QT Syndrome. Circulation 2020, 142, 324–338. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Schwartz, P.J. Implantable defibrillators in primary prevention of genetic arrhythmias. A shocking choice? Eur. Heart J. 2022, 43, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Aronow, W.S.; Dutta, T.; Frenkel, D.; Frishman, W.H. Catecholaminergic Polymorphic Ventricular Tachycardia. Cardiol. Rev. 2020, 28, 325–331. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]
- Shah, M.J.; Silka, M.J.; Silva, J.N.A.; Balaji, S.; Beach, C.M.; Benjamin, M.N.; Berul, C.I.; Cannon, B.; Cecchin, F.; Cohen, M.I.; et al. 2021 PACES expert consensus statement on the indications and management of cardiovascular implantable electronic devices in pediatric patients. Cardiol. Young 2021, 31, 1738–1769. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Zipes, D.P.; Kovacs, R.J.; Maron, B.J. Eligibility and Disqualification Recommendations for Competitive Athletes with Cardiovascular Abnormalities: Task Force 10: The Cardiac Channelopathies: A Scientific Statement from the American Heart Association and American College of Cardiology. J. Am. Coll. Cardiol. 2015, 66, 2424–2428. [Google Scholar] [CrossRef]
- Pflaumer, A.; Wilde, A.A.M.; Charafeddine, F.; Davis, A.M. 50 Years of Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)—Time to Explore the Dark Side of the Moon. Heart Lung Circ. 2020, 29, 520–528. [Google Scholar] [CrossRef]
- Ostby, S.A.; Bos, J.M.; Owen, H.J.; Wackel, P.L.; Cannon, B.C.; Ackerman, M.J. Competitive Sports Participation in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia: A Single Center'’s Early Experience. JACC Clin. Electrophysiol. 2016, 2, 253–262. [Google Scholar] [CrossRef]
- Lieve, K.V.V.; Verhagen, J.M.A.; Wei, J.; Bos, J.M.; van der Werf, C.; Roses, I.N.F.; Mancini, G.M.S.; Guo, W.; Wang, R.; van den Heuvel, F.; et al. Linking the heart and the brain: Neurodevelopmental disorders in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2019, 16, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hammond-Haley, M.; Patel, R.S.; Providencia, R.; Lambiase, P.D. Exercise restrictions for patients with inherited cardiac conditions: Current guidelines, challenges and limitations. Int. J. Cardiol. 2016, 209, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Christian, S.; Somerville, M.; Giuffre, M.; Atallah, J. Physical activity restriction for children and adolescents diagnosed with an inherited arrhythmia or cardiomyopathy and its impact on body mass index. J. Cardiovasc. Electrophysiol. 2018, 29, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- van der Werf, C.; Zwinderman, A.H.; Wilde, A.A. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: State of the art and future developments. EP Europace 2012, 14, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Leren, I.S.; Saberniak, J.; Majid, E.; Haland, T.F.; Edvardsen, T.; Haugaa, K.H. Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with beta1-selective beta-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2016, 13, 433–440. [Google Scholar] [CrossRef]
- Peltenburg, P.J.; Kallas, D.; Bos, J.M.; Lieve, K.V.V.; Franciosi, S.; Roston, T.M.; Denjoy, I.; Sorensen, K.B.; Ohno, S.; Roses-Noguer, F.; et al. An International Multicenter Cohort Study on beta-Blockers for the Treatment of Symptomatic Children with Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2022, 145, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, A.; Kukavica, D.; Trancuccio, A.; Memmi, M.; Bloise, R.; Gambelli, P.; Marino, M.; Ortiz-Genga, M.; Morini, M.; Monteforte, N.; et al. Outcomes of Patients with Catecholaminergic Polymorphic Ventricular Tachycardia Treated With beta-Blockers. JAMA Cardiol. 2022, 7, 504–512. [Google Scholar] [CrossRef]
- Inoue, Y.Y.; Aiba, T.; Kawata, H.; Sakaguchi, T.; Mitsuma, W.; Morita, H.; Noda, T.; Takaki, H.; Toyohara, K.; Kanaya, Y.; et al. Different responses to exercise between Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia. EP Europace 2018, 20, 1675–1682. [Google Scholar] [CrossRef]
- Heidbuchel, H.; Arbelo, E.; D’Ascenzi, F.; Borjesson, M.; Boveda, S.; Castelletti, S.; Miljoen, H.; Mont, L.; Niebauer, J.; Papadakis, M.; et al. Recommendations for participation in leisure-time physical activity and competitive sports of patients with arrhythmias and potentially arrhythmogenic conditions. Part 2: Ventricular arrhythmias, channelopathies, and implantable defibrillators: A position statement of the Section of Sports Cardiology and Exercise from the European Association of Preventive Cardiology (EAPC) and the European Heart Rhythm Association (EHRA), both associations of the European Society of Cardiology. EP Europace 2020, 23, 147–148. [Google Scholar] [CrossRef]
- Peltenburg, P.J.; van den Heuvel, L.M.; Kallas, D.; Bell, C.; Denjoy, I.; Behr, E.R.; Field, E.; Kammeraad, J.A.E.; Yap, S.C.; Probst, V.; et al. Insights into adherence to medication and lifestyle recommendations in an international cohort of patients with catecholaminergic polymorphic ventricular tachycardia. EP Europace 2024, 26, euae044. [Google Scholar] [CrossRef]
- Miyake, C.Y.; Asaki, S.Y.; Webster, G.; Czosek, R.J.; Atallah, J.; Avasarala, K.; Rao, S.O.; Thomas, P.E.; Kim, J.J.; Valdes, S.O.; et al. Circadian Variation of Ventricular Arrhythmias in Catecholaminergic Polymorphic Ventricular Tachycardia. JACC Clin. Electrophysiol. 2017, 3, 1308–1317. [Google Scholar] [CrossRef]
- Neves, R.A.; Bains, S.; Bos, J.M.; van der Werf, C.; Bergeman, A.; Peltenburg, P.; Sanatani, S.; Swan, H.; Probst, V.; Kannankeril, P.J.; et al. An international multicenter cohort study on beta-blocker free treatment strategies for catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2023, 20, 169–170. [Google Scholar] [CrossRef]
- Sumitomo, N.; Sakurada, H.; Taniguchi, K.; Matsumura, M.; Abe, O.; Miyashita, M.; Kanamaru, H.; Karasawa, K.; Ayusawa, M.; Fukamizu, S.; et al. Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ. J. 2007, 71, 1606–1609. [Google Scholar] [CrossRef]
- Faggioni, M.; van der Werf, C.; Knollmann, B.C. Sinus node dysfunction in catecholaminergic polymorphic ventricular tachycardia: Risk factor and potential therapeutic target? Trends Cardiovasc. Med. 2014, 24, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, A.; Denjoy, I.; Guicheney, P. Catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythm. Electrophysiol. 2012, 5, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, T.K.; Manotheepan, R.; Sadredini, M.; Leren, I.S.; Edwards, A.G.; Vincent, K.P.; Lehnart, S.E.; Sejersted, O.M.; Sjaastad, I.; Haugaa, K.H.; et al. Arrhythmia initiation in catecholaminergic polymorphic ventricular tachycardia type 1 depends on both heart rate and sympathetic stimulation. PLoS ONE 2018, 13, e0207100. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.J.; Moore, J.P.; Cerrone, M.; Priori, S.G.; Kertesz, N.J.; Ro, P.S.; Batra, A.S.; Kaufman, E.S.; Fairbrother, D.L.; Saarel, E.V.; et al. Efficacy of Flecainide in the Treatment of Catecholaminergic Polymorphic Ventricular Tachycardia: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 759–766. [Google Scholar] [CrossRef]
- Watanabe, H.; Chopra, N.; Laver, D.; Hwang, H.S.; Davies, S.S.; Roach, D.E.; Duff, H.J.; Roden, D.M.; Wilde, A.A.; Knollmann, B.C. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 2009, 15, 380–383. [Google Scholar] [CrossRef]
- Kryshtal, D.O.; Blackwell, D.J.; Egly, C.L.; Smith, A.N.; Batiste, S.M.; Johnston, J.N.; Laver, D.R.; Knollmann, B.C. RYR2 Channel Inhibition Is the Principal Mechanism of Flecainide Action in CPVT. Circ. Res. 2021, 128, 321–331. [Google Scholar] [CrossRef]
- Liu, N.; Denegri, M.; Ruan, Y.; Avelino-Cruz, J.E.; Perissi, A.; Negri, S.; Napolitano, C.; Coetzee, W.A.; Boyden, P.A.; Priori, S.G. Short communication: Flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ. Res. 2011, 109, 291–295. [Google Scholar] [CrossRef]
- Bannister, M.L.; Thomas, N.L.; Sikkel, M.B.; Mukherjee, S.; Maxwell, C.; MacLeod, K.T.; George, C.H.; Williams, A.J. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ. Res. 2015, 116, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Bergeman, A.T.; Lieve, K.V.V.; Kallas, D.; Bos, J.M.; Rosés I Noguer, F.; Denjoy, I.; Zorio, E.; Kammeraad, J.A.E.; Peltenburg, P.J.; Tobert, K.; et al. Flecainide Is Associated with a Lower Incidence of Arrhythmic Events in a Large Cohort of Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation 2023, 148, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Padfield, G.J.; AlAhmari, L.; Lieve, K.V.; AlAhmari, T.; Roston, T.M.; Wilde, A.A.; Krahn, A.D.; Sanatani, S. Flecainide monotherapy is an option for selected patients with catecholaminergic polymorphic ventricular tachycardia intolerant of beta-blockade. Heart Rhythm. 2016, 13, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Hasdemir, C.; Laver, D.; Mehra, D.; Turhan, K.; Faggioni, M.; Yin, H.; Knollmann, B.C. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythm. Electrophysiol. 2011, 4, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Marx, A.; Lange, B.; Nalenz, C.; Hoffmann, B.; Rostock, T.; Konrad, T. A 35-year effective treatment of catecholaminergic polymorphic ventricular tachycardia with propafenone. Hear. Case Rep. 2019, 5, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Katz, G.; Khoury, A.; Kurtzwald, E.; Hochhauser, E.; Porat, E.; Shainberg, A.; Seidman, J.G.; Seidman, C.E.; Lorber, A.; Eldar, M.; et al. Optimizing catecholaminergic polymorphic ventricular tachycardia therapy in calsequestrin-mutant mice. Heart Rhythm. 2010, 7, 1676–1682. [Google Scholar] [CrossRef] [PubMed]
- Rosso, R.; Kalman, J.M.; Rogowski, O.; Diamant, S.; Birger, A.; Biner, S.; Belhassen, B.; Viskin, S. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007, 4, 1149–1154. [Google Scholar] [CrossRef]
- Swan, H.; Laitinen, P.; Kontula, K.; Toivonen, L. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J. Cardiovasc. Electrophysiol. 2005, 16, 162–166. [Google Scholar] [CrossRef]
- Vaksmann, G.; Klug, D. Efficacy of ivabradine to control ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Pacing Clin. Electrophysiol. 2018, 41, 1378–1380. [Google Scholar] [CrossRef]
- Bueno-Levy, H.; Weisbrod, D.; Yadin, D.; Haron-Khun, S.; Peretz, A.; Hochhauser, E.; Arad, M.; Attali, B. The Hyperpolarization-Activated Cyclic-Nucleotide-Gated Channel Blocker Ivabradine Does Not Prevent Arrhythmias in Catecholaminergic Polymorphic Ventricular Tachycardia. Front. Pharmacol. 2019, 10, 1566. [Google Scholar] [CrossRef]
- Jung, C.B.; Moretti, A.; Mederos y Schnitzler, M.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Yano, M.; Uchinoumi, H.; Suetomi, T.; Susa, T.; Ono, M.; Xu, X.; Tateishi, H.; Oda, T.; Okuda, S.; et al. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ. J. 2010, 74, 2579–2584. [Google Scholar] [CrossRef]
- Penttinen, K.; Swan, H.; Vanninen, S.; Paavola, J.; Lahtinen, A.M.; Kontula, K.; Aalto-Setala, K. Antiarrhythmic Effects of Dantrolene in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia and Replication of the Responses Using iPSC Models. PLoS ONE 2015, 10, e0125366. [Google Scholar] [CrossRef]
- Wilde, A.A.; Bhuiyan, Z.A.; Crotti, L.; Facchini, M.; De Ferrari, G.M.; Paul, T.; Ferrandi, C.; Koolbergen, D.R.; Odero, A.; Schwartz, P.J. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N. Engl. J. Med. 2008, 358, 2024–2029. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.A.; Bos, J.M.; Johnson, J.N.; Owen, H.J.; Deschamps, C.; Moir, C.; Ackerman, M.J. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2012, 5, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Ackerman, M.J. Cardiac sympathetic denervation in the prevention of genetically mediated life-threatening ventricular arrhythmias. Eur. Heart J. 2022, 43, 2096–2102. [Google Scholar] [CrossRef]
- Collura, C.A.; Johnson, J.N.; Moir, C.; Ackerman, M.J. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009, 6, 752–759. [Google Scholar] [CrossRef]
- Dusi, V.; Gornbein, J.; Do, D.H.; Sorg, J.M.; Khakpour, H.; Krokhaleva, Y.; Ajijola, O.A.; Macias, C.; Bradfield, J.S.; Buch, E.; et al. Arrhythmic Risk Profile and Outcomes of Patients Undergoing Cardiac Sympathetic Denervation for Recurrent Monomorphic Ventricular Tachycardia After Ablation. J. Am. Heart Assoc. 2021, 10, e018371. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Snebold, N.G.; Brown, A.M. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am. J. Cardiol. 1976, 37, 1034–1040. [Google Scholar] [CrossRef]
- Viskin, S. Long QT syndromes and torsade de pointes. Lancet 1999, 354, 1625–1633. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Verrier, R.L.; Lown, B. Effect of stellectomy and vagotomy on ventricular refractoriness in dogs. Circ. Res. 1977, 40, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Moe, G.K. Nonuniform Recovery of Excitability in Ventricular Muscle. Circ. Res. 1964, 14, 44–60. [Google Scholar] [CrossRef]
- Cerati, D.; Schwartz, P.J. Single cardiac vagal fiber activity, acute myocardial ischemia, and risk for sudden death. Circ. Res. 1991, 69, 1389–1401. [Google Scholar] [CrossRef]
- Vanoli, E.; De Ferrari, G.M.; Stramba-Badiale, M.; Hull, S.S., Jr.; Foreman, R.D.; Schwartz, P.J. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ. Res. 1991, 68, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Guler, T.E.; Ozcan, K.S.; Bozyel, S.; Yalin, K. Renal sympathetic denervation assisted treatment of electrical storm due to polymorphic ventricular tachycardia in a patient with cathecolaminergic polymorphic ventricular tachycardia. Turk. Kardiyol. Dern. Ars. 2017, 45, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Guler, E. Percutaneous renal sympathetic denervation in catecholaminergic polymorphic ventricular tachycardia. J. Arrhythmia 2017, 33, 245. [Google Scholar] [CrossRef] [PubMed]
- Odero, A.; Bozzani, A.; De Ferrari, G.M.; Schwartz, P.J. Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias: The surgical supraclavicular approach to cervicothoracic sympathectomy. Heart Rhythm. 2010, 7, 1161–1165. [Google Scholar] [CrossRef]
- Waddell-Smith, K.E.; Ertresvaag, K.N.; Li, J.; Chaudhuri, K.; Crawford, J.R.; Hamill, J.K.; Haydock, D.; Skinner, J.R.; Cardiac Inherited Disease Group New, Z. Physical and Psychological Consequences of Left Cardiac Sympathetic Denervation in Long-QT Syndrome and Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythm. Electrophysiol. 2015, 8, 1151–1158. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2018, 72, 1677–1749. [Google Scholar] [CrossRef]
- Olde Nordkamp, L.R.; Postema, P.G.; Knops, R.E.; van Dijk, N.; Limpens, J.; Wilde, A.A.; de Groot, J.R. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: A systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016, 13, 443–454. [Google Scholar] [CrossRef]
- Pizzale, S.; Gollob, M.H.; Gow, R.; Birnie, D.H. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J. Cardiovasc. Electrophysiol. 2008, 19, 1319–1321. [Google Scholar] [CrossRef] [PubMed]
- Palanca, V.; Quesada, A.; Trigo, A.; Jimenez, J. [Arrhythmic storm induced by AICD discharge in a patient with catecholaminergic polymorphic ventricular tachycardia]. Rev. Esp. Cardiol. 2006, 59, 1079–1080. [Google Scholar] [CrossRef]
- Adler, A.; Sadek, M.M.; Chan, A.Y.; Dell, E.; Rutberg, J.; Davis, D.; Green, M.S.; Spears, D.A.; Gollob, M.H. Patient Outcomes from a Specialized Inherited Arrhythmia Clinic. Circ. Arrhythm. Electrophysiol. 2016, 9, e003440. [Google Scholar] [CrossRef]
- Miyake, C.Y.; Webster, G.; Czosek, R.J.; Kantoch, M.J.; Dubin, A.M.; Avasarala, K.; Atallah, J. Efficacy of implantable cardioverter defibrillators in young patients with catecholaminergic polymorphic ventricular tachycardia: Success depends on substrate. Circ. Arrhythm. Electrophysiol. 2013, 6, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Roses-Noguer, F.; Jarman, J.W.; Clague, J.R.; Till, J. Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2014, 11, 58–66. [Google Scholar] [CrossRef]
- Roston, T.M.; Jones, K.; Hawkins, N.M.; Bos, J.M.; Schwartz, P.J.; Perry, F.; Ackerman, M.J.; Laksman, Z.W.M.; Kaul, P.; Lieve, K.V.V.; et al. Implantable cardioverter-defibrillator use in catecholaminergic polymorphic ventricular tachycardia: A systematic review. Heart Rhythm. 2018, 15, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- van der Werf, C.; Lieve, K.V.; Bos, J.M.; Lane, C.M.; Denjoy, I.; Roses-Noguer, F.; Aiba, T.; Wada, Y.; Ingles, J.; Leren, I.S.; et al. Implantable cardioverter-defibrillators in previously undiagnosed patients with catecholaminergic polymorphic ventricular tachycardia resuscitated from sudden cardiac arrest. Eur. Heart J. 2019, 40, 2953–2961. [Google Scholar] [CrossRef]
- Bergeman, A.T.; Wilde, A.A.M.; van der Werf, C. Catecholaminergic Polymorphic Ventricular Tachycardia: A Review of Therapeutic Strategies. Card. Electrophysiol. Clin. 2023, 15, 293–305. [Google Scholar] [CrossRef]
- Louise, R.A.; Olde Nordkamp, A.A.M.W. Implantable Cardioverter-Defibrillators in Inherited Arrhythmia Syndromes. In Clinical Cardiac Pacing, Defibrillation and Resynchronization Therapy, 5th ed.; Ellenbogen, K.A., Wilkoff, B.L., Neal Kay, G., Lau, C., Auricchio, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 566–578. [Google Scholar]
- Medeiros, P.; Santos, M.; Arantes, C.; Pereira, V.H.; Rocha, S. Implantable cardioverter-defibrillator in patients with inherited arrhythmia syndromes: A systematic review. Heart Lung 2023, 60, 1–7. [Google Scholar] [CrossRef]
- Conte, G.; Kawabata, M.; de Asmundis, C.; Taravelli, E.; Petracca, F.; Ruggiero, D.; Caputo, M.L.; Regoli, F.; Chierchia, G.B.; Chiodini, A.; et al. High rate of subcutaneous implantable cardioverter-defibrillator sensing screening failure in patients with Brugada syndrome: A comparison with other inherited primary arrhythmia syndromes. EP Europace 2018, 20, 1188–1193. [Google Scholar] [CrossRef]
- Basu-Ray, I.; Liu, J.; Jia, X.; Gold, M.; Ellenbogen, K.; DiNicolantonio, J.; Komocsi, A.; Vorobcsuk, A.; Kim, J.; Afshar, H.; et al. Subcutaneous Versus Transvenous Implantable Defibrillator Therapy: A Meta-Analysis of Case-Control Studies. JACC Clin. Electrophysiol. 2017, 3, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Eckert, H.; El-Battrawy, I.; Veith, M.; Roterberg, G.; Kowitz, J.; Lang, S.; Zhou, X.; Akin, I.; Mugge, A.; Aweimer, A. Pooled Analysis of Complications with Transvenous ICD Compared to Subcutaneous ICD in Patients with Catecholaminergic Polymorphic Ventricular Arrhythmia. J. Pers. Med. 2022, 12, 536. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.R.; Lambiase, P.D.; El-Chami, M.F.; Knops, R.E.; Aasbo, J.D.; Bongiorni, M.G.; Russo, A.M.; Deharo, J.C.; Burke, M.C.; Dinerman, J.; et al. Primary Results from the Understanding Outcomes With the S-ICD in Primary Prevention Patients with Low Ejection Fraction (UNTOUCHED) Trial. Circulation 2021, 143, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Lambiase, P.D.; Eckardt, L.; Theuns, D.A.; Betts, T.R.; Kyriacou, A.L.; Duffy, E.; Knops, R. Evaluation of subcutaneous implantable cardioverter-defibrillator performance in patients with ion channelopathies from the EFFORTLESS cohort and comparison with a meta-analysis of transvenous ICD outcomes. Heart Rhythm O2 2020, 1, 326–335. [Google Scholar] [CrossRef]
- Szumowski, L.; Walczak, F.; Przybylski, A.; Maryniak, A.; Szufladowicz, E.; Derejko, P.; Bieganowska, K.; Bodalski, R.; Orczykowski, M.; Sterlinski, M.; et al. [Ablation of a catecholaminergic polymorphic VT and VF originating from Purkinje fibers—A case report]. Kardiol. Pol. 2007, 65, 319–326. [Google Scholar] [PubMed]
- Kaneshiro, T.; Naruse, Y.; Nogami, A.; Tada, H.; Yoshida, K.; Sekiguchi, Y.; Murakoshi, N.; Kato, Y.; Horigome, H.; Kawamura, M.; et al. Successful catheter ablation of bidirectional ventricular premature contractions triggering ventricular fibrillation in catecholaminergic polymorphic ventricular tachycardia with RyR2 mutation. Circ. Arrhythm. Electrophysiol. 2012, 5, e14–e17. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Goya, M.; Ohno, S.; Horie, M.; Doi, S.; Isobe, M.; Hirao, K. Elimination of Ventricular Arrhythmia in Catecholaminergic Polymorphic Ventricular Tachycardia by Targeting “Catecholamine-Sensitive Area”: A Dominant-Subordinate Relationship between Origin Sites of Bidirectional Ventricular Premature Contractions. Pacing Clin. Electrophysiol. 2017, 40, 600–604. [Google Scholar] [CrossRef]
- Kaneshiro, T.; Nogami, A.; Kato, Y.; Kuroki, K.; Komatsu, Y.; Tada, H.; Sekiguchi, Y.; Horigome, H.; Aonuma, K. Effects of Catheter Ablation Targeting the Trigger Beats in Inherited Catecholaminergic Polymorphic Ventricular Tachycardia. JACC Clin. Electrophysiol. 2017, 3, 1062–1063. [Google Scholar] [CrossRef]
- Shen, L.; Liu, S.; Hu, F.; Zhang, Z.; Li, J.; Lai, Z.; Zheng, L.; Yao, Y. Electrophysiological Characteristics and Ablation Outcomes in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. J. Am. Heart Assoc. 2023, 12, e031768. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Prondzynski, M.; Carrier, L.; Pu, W.T. Gene therapy for inherited arrhythmias. Cardiovasc. Res. 2020, 116, 1635–1650. [Google Scholar] [CrossRef]
- Bongianino, R.; Priori, S.G. Gene therapy to treat cardiac arrhythmias. Nat. Rev. Cardiol. 2015, 12, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Denegri, M.; Avelino-Cruz, J.E.; Boncompagni, S.; De Simone, S.A.; Auricchio, A.; Villani, L.; Volpe, P.; Protasi, F.; Napolitano, C.; Priori, S.G. Viral gene transfer rescues arrhythmogenic phenotype and ultrastructural abnormalities in adult calsequestrin-null mice with inherited arrhythmias. Circ. Res. 2012, 110, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Kurtzwald-Josefson, E.; Yadin, D.; Harun-Khun, S.; Waldman, M.; Aravot, D.; Shainberg, A.; Eldar, M.; Hochhauser, E.; Arad, M. Viral delivered gene therapy to treat catecholaminergic polymorphic ventricular tachycardia (CPVT2) in mouse models. Heart Rhythm. 2017, 14, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Walton, S.D.; Ho, H.T.; Belevych, A.E.; Tikunova, S.B.; Bonilla, I.; Shettigar, V.; Knollmann, B.C.; Priori, S.G.; Volpe, P.; et al. Gene Transfer of Engineered Calmodulin Alleviates Ventricular Arrhythmias in a Calsequestrin-Associated Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. J. Am. Heart Assoc. 2018, 7, e008155. [Google Scholar] [CrossRef] [PubMed]
- Bongianino, R.; Denegri, M.; Mazzanti, A.; Lodola, F.; Vollero, A.; Boncompagni, S.; Fasciano, S.; Rizzo, G.; Mangione, D.; Barbaro, S.; et al. Allele-Specific Silencing of Mutant mRNA Rescues Ultrastructural and Arrhythmic Phenotype in Mice Carriers of the R4496C Mutation in the Ryanodine Receptor Gene (RYR2). Circ. Res. 2017, 121, 525–536. [Google Scholar] [CrossRef]
- Porteus, M.H. A New Class of Medicines through DNA Editing. N. Engl. J. Med. 2019, 380, 947–959. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Pan, X.; Philippen, L.; Lahiri, S.K.; Lee, C.; Park, S.H.; Word, T.A.; Li, N.; Jarrett, K.E.; Gupta, R.; Reynolds, J.O.; et al. In Vivo Ryr2 Editing Corrects Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2018, 123, 953–963. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Lodola, F.; Miragoli, M.; Denegri, M.; Avelino-Cruz, J.E.; Buonocore, M.; Nakahama, H.; Portararo, P.; Bloise, R.; Napolitano, C.; et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013, 4, e843. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flugel, L.; Dorn, T.; Goedel, A.; Hohnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef]
- Sebastian, S.A.; Panthangi, V.; Sethi, Y.; Padda, I.; Khan, U.; Affas, Z.R.; Mareddy, C.; Dolack, L.; Johal, G. Precision Medicine and Cardiac Channelopathies: Human iPSCs Take the Lead. Curr. Probl. Cardiol. 2023, 48, 101990. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Type | Predominant Inheritance Pattern | Protein | Gene | Chromosome Locus | Prevalence of Probands (%) |
|---|---|---|---|---|---|
| CPVT 1 | Autosomal dominant | Ryanodine receptor 2 | RYR2 | 1q42-q43 | 60–70% |
| CPVT 2 | Autosomal recessive | Calsequestrin 2 | CASQ2 | 1p13.1 | 2–5% |
| CPVT 3 | Autosomal recessive | Trans-2,3-enoyl-CoA-reductase-like | TECRL | 7p22-p14 | Rare (<1%) |
| CPVT 4 | Autosomal dominant | Calmodulin 1 | CALM1 | 14q31-q32 | Rare (<1%) |
| CPVT 5 | Autosomal recessive | Triadin | TRDN | 6q22.31 | Rare (<1%) |
| CPVT 6 | Autosomal dominant | Calmodulin 3 | CALM3 | 19q13.22 | Rare (<1%) |
| CPVT: Expert Consensus Recommendations on Diagnosis |
|---|
| 1. CPVT is diagnosed in the presence of a structurally normal heart, normal ECG and unexplained exercise or catecholamine induced bidirectional VT or polymorphic PVCs in patients < 40 years of age |
| 2. CPVT is diagnosed in patients (index case or family member) who have a pathogenic mutation. |
| 3. CPVT is diagnosed in family members of a CPVT index case with a normal heart who manifest exercise-induced PVCs or bidirectional/polymorphic VT. |
| 4. CPVT can be diagnosed in the presence of a structurally normal heart and coronary arteries, normal ECG and unexplained exercise or catecholamine-induced bidirectional VT or polymorphic PVCs or VT in an individual > 40 years of age. |
| Clinical Criteria | Points |
|---|---|
| Symptoms | |
| Exercise/activity-associated ACA/SCA | 2 |
| Exercise/activity-associated syncope or generalized seizures | 1 |
| Exercise stress test or Holter monitoring during exertional activity (REQUIRES ≥ 1 exercise stress test/ambulatory Holter finding) *† | |
| Inducible bidirectional ventricular tachycardia at HR > 100 bpm | 4 |
| Inducible PVCs in bigeminy and bidirectional couplets at HR > 100 bpm | 2 |
| Inducible PVCs at HR > 100 bpm | 1 |
| Baseline HR QTc ‡ | |
| QTc ≤ 420 ms | 0.5 |
| 421 < QTc < 460 ms | 0 |
| QTc ≥ 460 ms | −0.5 |
| CPVT genetic test | |
| Positive for ACMG-graded pathogenic variant | 4 |
| Positive for ACMG-graded likely pathogenic variant | 2 |
| Positive for a variant of uncertain significance | 0 |
| Negative CPVT genetic test (RyR2, CASQ2, TRDN and CALM1-3) | −1 |
| Holter | |
| Ambulatory ventricular ectopy (>2% of total beats) | −1 |
| Imaging (TTE or cardiac MRI/CT) § | |
| Evidence of ischemic or structural heart disease | −2 |
| Age | |
| ≥50 y of age at time of sentinel event | −1 |
| Family history * | |
| First-degree relative with definite CPVT | 1.5 |
| Suspicious autopsy-negative SCD (exertional, near drowning, etc.) in a first- or second-degree relative ≤ 45 years. | 1 |
| Unexplained autopsy-negative SCD in a first- or second-degree relative age ≤ 45 years. | 0.5 |
| CPVT score (requires an exercise stress test/ambulatory Holter finding) | |
| 3.5–12 points: high pre-test probability of CPVT (definite/probable CPVT ≥ 90% likelihood) | |
| 2–3 points: intermediate pre-test probability of CPVT (possible CPVT, ≈50% likelihood) | |
| 0.5–1.5 points: low pre-test probability of CPVT (nondiagnostic) | |
| ≤0 points: no evidence of CPVT | |
| No score: indeterminate | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aggarwal, A.; Stolear, A.; Alam, M.M.; Vardhan, S.; Dulgher, M.; Jang, S.-J.; Zarich, S.W. Catecholaminergic Polymorphic Ventricular Tachycardia: Clinical Characteristics, Diagnostic Evaluation and Therapeutic Strategies. J. Clin. Med. 2024, 13, 1781. https://doi.org/10.3390/jcm13061781
Aggarwal A, Stolear A, Alam MM, Vardhan S, Dulgher M, Jang S-J, Zarich SW. Catecholaminergic Polymorphic Ventricular Tachycardia: Clinical Characteristics, Diagnostic Evaluation and Therapeutic Strategies. Journal of Clinical Medicine. 2024; 13(6):1781. https://doi.org/10.3390/jcm13061781
Chicago/Turabian StyleAggarwal, Abhinav, Anton Stolear, Md Mashiul Alam, Swarnima Vardhan, Maxim Dulgher, Sun-Joo Jang, and Stuart W. Zarich. 2024. "Catecholaminergic Polymorphic Ventricular Tachycardia: Clinical Characteristics, Diagnostic Evaluation and Therapeutic Strategies" Journal of Clinical Medicine 13, no. 6: 1781. https://doi.org/10.3390/jcm13061781
APA StyleAggarwal, A., Stolear, A., Alam, M. M., Vardhan, S., Dulgher, M., Jang, S.-J., & Zarich, S. W. (2024). Catecholaminergic Polymorphic Ventricular Tachycardia: Clinical Characteristics, Diagnostic Evaluation and Therapeutic Strategies. Journal of Clinical Medicine, 13(6), 1781. https://doi.org/10.3390/jcm13061781