
TGF-β1, pSmad-2/3, Smad-7, and β-Catenin Are Augmented in the Pulmonary Arteries from Patients with Idiopathic Pulmonary Fibrosis (IPF): Role in Driving Endothelial-to-Mesenchymal Transition (EndMT)
 ,
,  , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Immunohistochemical Staining for EndMT Driver Markers (TGF-β1, pSmad-2/3, Smad-7, and β-Catenin)
2.3. Measurement Strategies for Pulmonary Arteries’ Classification
2.4. Pulmonary EndMT Drivers’ (TGF-β1, pSmad-2/3, Smad-7, and β-Catenin) Total Arterial and Individual Layer Expression Measurement
2.5. Correlations between the Mesenchymal Marker Expression and Vascular Remodelling Changes
2.6. Statistical Analysis
3. Results
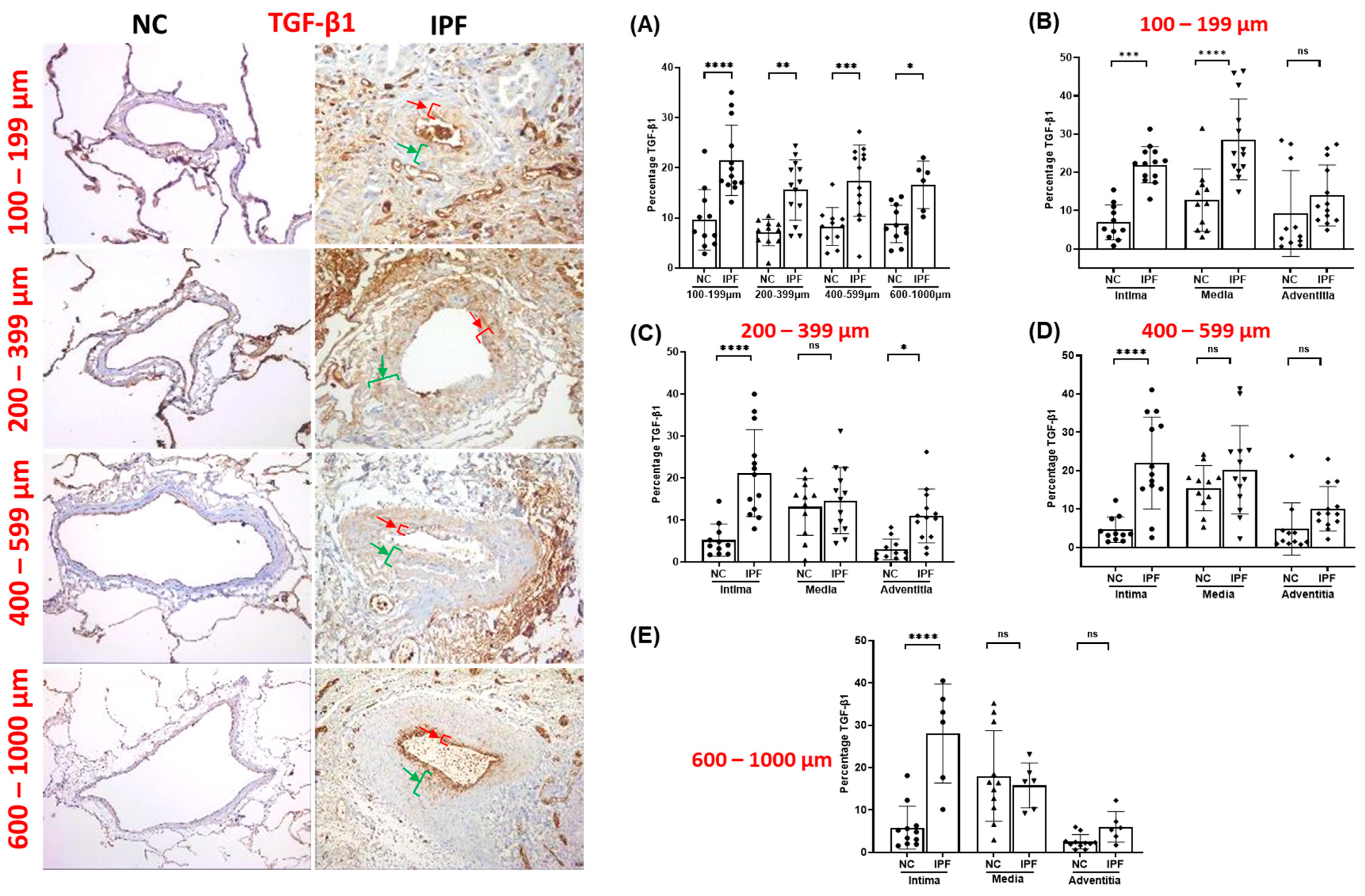
3.1. Morphological Assessment of EndMT Drivers’ Expression among Pulmonary Arteries
3.2. TGF-β1 Expression in Pulmonary Arteries
3.3. pSmad-2/3 Expression in Pulmonary Arteries
3.4. Smad-7 Expression in Pulmonary Arteries
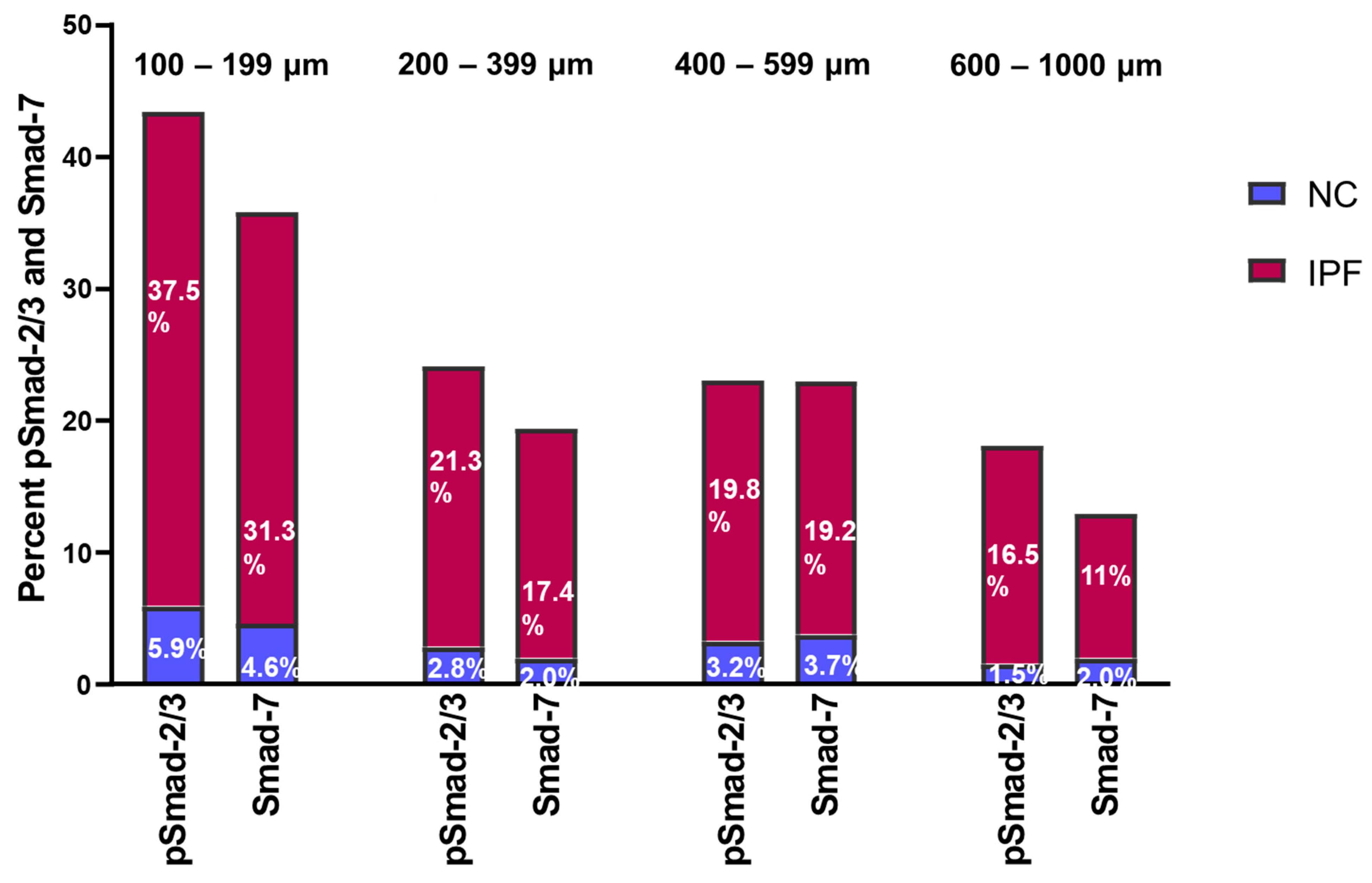
3.5. Fold Change in pSmad-2/3 and Smad-7 Expression in Pulmonary Arteries
3.6. β-Catenin Expression in Pulmonary Arteries
3.7. Correlation between EndMT Drivers’ Expression and EndMT Markers
3.8. Correlation between EndMT Drivers’ Expression and Vascular Remodelling Changes
3.9. EndMT Drivers’ Impacts on Lung Physiology
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; McDonald, J.A. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am. J. Pathol. 1991, 138, 1257–1265. [Google Scholar] [PubMed]
- Gaikwad, A.V.; Lu, W.; Dey, S.; Bhattarai, P.; Chia, C.; Larby, J.; Haug, G.; Myers, S.; Jaffar, J.; Westall, G.; et al. Vascular remodelling in idiopathic pulmonary fibrosis patients and its detrimental effect on lung physiology: Potential role of endothelial-to-mesenchymal transition. ERJ Open Res. 2022, 8, 00571-2021. [Google Scholar] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef]
- Kinoshita, T.; Goto, T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int. J. Mol. Sci. 2019, 20, 1461. [Google Scholar] [CrossRef]
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123. [Google Scholar] [CrossRef]
- Choi, S.-H.; Nam, J.-K.; Kim, B.-Y.; Jang, J.; Jin, Y.-B.; Lee, H.-J.; Park, S.; Ji, Y.H.; Cho, J.; Lee, Y.-J. HSPB1 Inhibits the Endothelial-to-Mesenchymal Transition to Suppress Pulmonary Fibrosis and Lung Tumorigenesis. Cancer Res. 2016, 76, 1019–1030. [Google Scholar] [CrossRef]
- Gaikwad, A.V.; Eapen, M.S.; McAlinden, K.D.; Chia, C.; Larby, J.; Myers, S.; Dey, S.; Haug, G.; Markos, J.; Glanville, A.R.; et al. Endothelial to mesenchymal transition (EndMT) and vascular remodeling in pulmonary hypertension and idiopathic pulmonary fibrosis. Expert. Rev. Respir. Med. 2020, 14, 1027–1043. [Google Scholar] [CrossRef]
- Beyer, C.; Schramm, A.; Akhmetshina, A.; Dees, C.; Kireva, T.; Gelse, K.; Sonnylal, S.; de Crombrugghe, B.; Taketo, M.M.; Distler, O.; et al. β-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 761–767. [Google Scholar] [CrossRef]
- Bhattarai, P.; Lu, W.; Gaikwad, A.V.; Dey, S.; Chia, C.; Larby, J.; Haug, G.; Hardikar, A.; Williams, A.; Kaur Singhera, G.; et al. Arterial remodelling in smokers and in patients with small airway disease and COPD: Implications for lung physiology and early origins of pulmonary hypertension. ERJ Open Res. 2022, 8, 00254-2022. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, P.; Lu, W.; Hardikar, A.; Dey, S.; Gaikwad, A.V.; Shahzad, A.M.; Chia, C.; Williams, A.; Singhera, G.K.; Hackett, T.L.; et al. Endothelial to mesenchymal transition is an active process in smokers and patients with early COPD contributing to pulmonary arterial pathology. ERJ Open Res. 2024, 10, 00767-02023. [Google Scholar] [CrossRef] [PubMed]
- Brake, S.J.; Lu, W.; Chia, C.; Haug, G.; Larby, J.; Hardikar, A.; Singhera, G.K.; Hackett, T.L.; Eapen, M.S.; Sohal, S.S. Transforming growth factor-beta1 and SMAD signalling pathway in the small airways of smokers and patients with COPD: Potential role in driving fibrotic type-2 epithelial mesenchymal transition. Front. Immunol. 2023, 14, 1216506. [Google Scholar] [CrossRef]
- Gaikwad, A.V.; Lu, W.; Dey, S.; Bhattarai, P.; Haug, G.; Larby, J.; Chia, C.; Jaffar, J.; Westall, G.; Singhera, G.K.; et al. Endothelial-to-mesenchymal transition: A precursor to pulmonary arterial remodelling in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2023, 9, 00487-2022. [Google Scholar] [CrossRef]
- Lu, W.; Eapen, M.S.; Hardikar, A.; Chia, C.; Robertson, I.; Singhera, G.K.; Hackett, T.L.; Sohal, S.S. Epithelial-mesenchymal transition changes in nonsmall cell lung cancer patients with early COPD. ERJ Open Res. 2023, 9, 00581-2023. [Google Scholar] [CrossRef]
- Lu, W.; Eapen, M.S.; Singhera, G.K.; Markos, J.; Haug, G.; Chia, C.; Larby, J.; Brake, S.J.; Westall, G.P.; Jaffar, J.; et al. Angiotensin-Converting Enzyme 2 (ACE2), Transmembrane Peptidase Serine 2 (TMPRSS2), and Furin Expression Increases in the Lungs of Patients with Idiopathic Pulmonary Fibrosis (IPF) and Lymphangioleiomyomatosis (LAM): Implications for SARS-CoV-2 (COVID-19) Infections. J. Clin. Med. 2022, 11, 777. [Google Scholar]
- Eapen, M.S.; Lu, W.; Gaikwad, A.V.; Bhattarai, P.; Chia, C.; Hardikar, A.; Haug, G.; Sohal, S.S. Endothelial to mesenchymal transition: A precursor to post-COVID-19 interstitial pulmonary fibrosis and vascular obliteration? Eur. Respir. J. 2020, 56, 2003167. [Google Scholar] [CrossRef]
- Cantor, J.O.; Parshley, M.S.; Mandl, I.; Turino, G.M. Elastin synthesis by endothelial cells. In Biology of Endothelial Cells; Jaffe, E.A., Ed.; Springer: Boston, MA, USA, 1984; pp. 189–193. [Google Scholar]
- Stenmark, K.R.; Frid, M.; Perros, F. Endothelial-to-Mesenchymal Transition. Circulation 2016, 133, 1734–1737. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Y. TGF-β1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; O’Connor, R.N.; Flanders, K.C.; Unruh, H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: An immunohistochemical study. Am. J. Respir. Cell Mol. Biol. 1996, 14, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Coker, R.K.; Laurent, G.J.; Shahzeidi, S.; Lympany, P.A.; du Bois, R.M.; Jeffery, P.K.; McAnulty, R.J. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am. J. Pathol. 1997, 150, 981–991. [Google Scholar] [PubMed]
- Roach, K.M.; Wulff, H.; Feghali-Bostwick, C.; Amrani, Y.; Bradding, P. Increased constitutive αSMA and Smad2/3 expression in idiopathic pulmonary fibrosis myofibroblasts is KCa3.1-dependent. Respir. Res. 2014, 15, 155. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, J.; Castro, S.V.; Jimenez, S.A. Narrative Review: Fibrotic Diseases: Cellular and Molecular Mechanisms and Novel Therapies. Ann. Intern. Med. 2010, 152, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef]
- Medici, D.; Potenta, S.; Kalluri, R. Transforming growth factor-β2 promotes Snail-mediated endothelial–mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem. J. 2011, 437, 515–520. [Google Scholar] [CrossRef]
- Guan, S.; Zhou, J. CXCR7 attenuates the TGF-β-induced endothelial-to-mesenchymal transition and pulmonary fibrosis. Mol. BioSystems 2017, 13, 2116–2124. [Google Scholar] [CrossRef]
- Katsuno, Y.; Derynck, R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The Essential Autophagy Gene ATG7 Modulates Organ Fibrosis via Regulation of Endothelial-to-Mesenchymal Transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone Jr, M.A.; Wrana, J.L.; et al. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem.-Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef]
- Ard, S.; Reed, E.B.; Smolyaninova, L.V.; Orlov, S.N.; Mutlu, G.M.; Guzy, R.D.; Dulin, N.O. Sustained Smad2 Phosphorylation Is Required for Myofibroblast Transformation in Response to TGF-β. Am. J. Respir. Cell Mol. Biol. 2019, 60, 367–369. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, X.; Hu, R.; Dai, A. TGF-β1-induced SMAD2/3/4 activation promotes RELM-β transcription to modulate the endothelium-mesenchymal transition in human endothelial cells. Int. J. Biochem. Cell Biol. 2018, 105, 52–60. [Google Scholar] [CrossRef]
- Yung, L.M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12, eaaz5660. [Google Scholar] [CrossRef]
- Oliveira, S.D.S.; Castellon, M.; Chen, J.; Bonini, M.G.; Gu, X.; Elliott, M.H.; Machado, R.F.; Minshall, R.D. Inflammation-induced caveolin-1 and BMPRII depletion promotes endothelial dysfunction and TGF-β-driven pulmonary vascular remodeling. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 312, L760–L771. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, H.; Sun, Z.; Xiang, Z.; Ge, Y.; Ni, C.; Luo, Z.; Qian, W.; Han, X. Inhibition of Wnt/β-catenin signaling promotes epithelial differentiation of mesenchymal stem cells and repairs bleomycin-induced lung injury. Am. J. Physiol.-Cell Physiol. 2014, 307, C234–C244. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef]
- Lv, H.; Nan, Z.; Jiang, P.; Wang, Z.; Song, M.; Ding, H.; Liu, D.; Zhao, G.; Zheng, Y.; Hu, Y. Vascular endothelial growth factor 165 inhibits pro-fibrotic differentiation of stromal cells via the DLL4/Notch4/smad7 pathway. Cell Death Dis. 2019, 10, 681. [Google Scholar] [CrossRef]
- Qin, H.; Wen, H.-T.; Gu, K.-J.; Hu, X.-D.; Yang, T.; Yan, X.-F.; Ye, T.-J.; Huo, J.-L.; Hu, J. Total extract of Xin Jia Xuan Bai Cheng Qi decoction inhibits pulmonary fibrosis via the TGF-β/Smad signaling pathways in vivo and in vitro. Drug Des. Dev. Ther. 2019, 13, 2873–2886. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, A.; Li, Y.; Tan, C.; La Regina, G.; Silvestri, R.; Wang, H.; Qi, W. RS4651 suppresses lung fibroblast activation via the TGF-β1/SMAD signalling pathway. Eur. J. Pharmacol. 2021, 903, 174135. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/β-Catenin Pathway Activation in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.H.; Sakmar, T.P.; Rafii, S.; Ding, B.S. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162. [Google Scholar] [CrossRef]
- Andersson-Sjöland, A.; Karlsson, J.C.; Rydell-Törmänen, K. ROS-induced endothelial stress contributes to pulmonary fibrosis through pericytes and Wnt signaling. Lab. Investig. 2016, 96, 206–217. [Google Scholar] [CrossRef]
- Laumanns, I.P.; Fink, L.; Wilhelm, J.; Wolff, J.-C.; Mitnacht-Kraus, R.; Graef-Hoechst, S.; Stein, M.M.; Bohle, R.M.; Klepetko, W.; Hoda, M.A.R.; et al. The Noncanonical WNT Pathway Is Operative in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2009, 40, 683–691. [Google Scholar] [CrossRef]
- Liu, C.-T.; Hsu, S.-C.; Hsieh, H.-L.; Chen, C.-H.; Chen, C.-Y.; Sue, Y.-M.; Chen, T.-H.; Hsu, Y.-H.; Lin, F.-Y.; Shih, C.-M.; et al. Inhibition of β-catenin signaling attenuates arteriovenous fistula thickening in mice by suppressing myofibroblasts. Mol. Med. 2022, 28, 7. [Google Scholar] [CrossRef]
- Boucherat, O.; Bonnet, S. MicroRNA signature of end-stage idiopathic pulmonary arterial hypertension: Clinical correlations and regulation of WNT signaling. J. Mol. Med. 2016, 94, 849–851. [Google Scholar] [CrossRef][Green Version]
- Tyson, J.; Bundy, K.; Roach, C.; Douglas, H.; Ventura, V.; Segars, M.F.; Schwartz, O.; Simpson, C.L. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering 2020, 7, 88. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Herazo-Maya, J.D.; Sennello, J.A.; Flozak, A.S.; Russell, S.; Mutlu, G.M.; Budinger, G.R.; DasGupta, R.; Varga, J.; Kaminski, N.; et al. Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Cui, W.-H.; Zhou, W.-C.; Li, D.-L.; Li, L.-C.; Zhao, P.; Mo, X.-T.; Zhang, Z.; Gao, J. Activation of Wnt/β-catenin signalling is required for TGF-β/Smad2/3 signalling during myofibroblast proliferation. J. Cell. Mol. Med. 2017, 21, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Kadota, T.; Fujita, Y.; Araya, J.; Watanabe, N.; Fujimoto, S.; Kawamoto, H.; Minagawa, S.; Hara, H.; Ohtsuka, T.; Yamamoto, Y.; et al. Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-β-WNT crosstalk. J. Extracell. Vesicles 2021, 10, e12124. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.S.; Sohal, S.S. WNT/beta-catenin pathway: A novel therapeutic target for attenuating airway remodelling and EMT in COPD. EBioMedicine 2020, 62, 103095. [Google Scholar] [CrossRef]
- Aghaei, M.; Dastghaib, S.; Aftabi, S.; Aghanoori, M.R.; Alizadeh, J.; Mokarram, P.; Mehrbod, P.; Ashrafizadeh, M.; Zarrabi, A.; McAlinden, K.D.; et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life 2020, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ward, C.; Eapen, M.S.; Myers, S.; Hallgren, O.; Levine, H.; Sohal, S.S. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. 2018, 247, 346–358. [Google Scholar] [CrossRef]
- Guo, M.; Breslin, J.W.; Wu, M.H.; Gottardi, C.J.; Yuan, S.Y. VE-cadherin and beta-catenin binding dynamics during histamine-induced endothelial hyperpermeability. Am. J. Physiol. Cell Physiol. 2008, 294, C977–C984. [Google Scholar] [CrossRef]
- Mahmood, M.Q.; Walters, E.H.; Shukla, S.D.; Weston, S.; Muller, H.K.; Ward, C.; Sohal, S.S. beta-catenin, Twist and Snail: Transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci. Rep. 2017, 7, 10832. [Google Scholar] [CrossRef]
- Guo, M.; Wu, M.H.; Granger, H.J.; Yuan, S.Y. Transference of recombinant VE-cadherin cytoplasmic domain alters endothelial junctional integrity and porcine microvascular permeability. J. Physiol. 2004, 554, 78–88. [Google Scholar] [CrossRef]
- Delgado-Bellido, D.; Zamudio-Martínez, E.; Fernández-Cortés, M.; Herrera-Campos, A.B.; Olmedo-Pelayo, J.; Perez, C.J.; Expósito, J.; de Álava, E.; Amaral, A.T.; Valle, F.O.; et al. VE-Cadherin modulates β-catenin/TCF-4 to enhance Vasculogenic Mimicry. Cell Death Dis. 2023, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Deckers, M.; Bertolino, P.; ten Dijke, P. TGF-β receptor function in the endothelium. Cardiovasc. Res. 2005, 65, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Colombat, M.; Mal, H.; Groussard, O.; Capron, F.; Thabut, G.; Jebrak, G.; Brugière, O.; Dauriat, G.; Castier, Y.; Lesèche, G.; et al. Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: Histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum. Pathol. 2007, 38, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Ruffenach, G.; Hong, J.; Vaillancourt, M.; Medzikovic, L.; Eghbali, M. Pulmonary hypertension secondary to pulmonary fibrosis: Clinical data, histopathology and molecular insights. Respir. Res. 2020, 21, 303. [Google Scholar] [CrossRef]
- Farkas, L.; Farkas, D.; Ask, K.; Möller, A.; Gauldie, J.; Margetts, P.; Inman, M.; Kolb, M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J. Clin. Investig. 2009, 119, 1298–1311. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Control (NC) | IPF | |
|---|---|---|
| Factor | Values * | |
| Total number (n) | 11 | 13 |
| Age | 41 ± 15.1 | 64 ± 5.06 |
| Gender (F/M) | 6/5 | 6/7 |
| Body mass index | NA | 26.69 ± 3.03 |
| Smoking status: | ||
| Current, ex-smoker, and never (n) | Non-smokers | 0/7/6 |
| Smoking pack-years | - | 20.84 ± 23.16 |
| Lung physiology | ||
| FEV1 (L) | NA | 1.70 ± 0.40 |
| FEV1 (%) predicted | NA | 60.17 ± 12.22 |
| FVC (L) | NA | 1.97 ± 0.51 |
| FVC (%) predicted | NA | 53.5 ± 12.98 |
| DLCO (mL/min/mmHg) | NA | 5.91 ± 2.92 |
| DLCO corrected (%) predicted | NA | 25.85 ± 15.30 |
| Correlation Analysis | |||||
|---|---|---|---|---|---|
| TGF-β1 vs. EndMT Markers | |||||
| (A) Total TGF-β1 vs. total vimentin | (B) Intimal TGF-β1 vs. intimal vimentin | (C) Total TGF-β1 vs. total N-cadherin | (D) Intimal TGF-β1 vs. intimal N-cadherin | (E) Intimal TGF-β1 vs. VE-cadherin junctional-positive | (F) Intimal TGF-β1 vs. VE-cadherin cytoplasm-positive |
| r′ = 0.65 p = 0.02 | r′ = 0.54 p = 0.05 | r′ = 0.69 p = 0.01 | r′ = 0.62 p = 0.03 | r′ = 0.59 p = 0.03 | r′ = 0.61 p = 0.04 |
| β-Catenin vs. EndMT markers | |||||
| (G) Total β-catenin vs. total vimentin | (H) Intimal β-catenin vs. intimal vimentin | (I) Total β-catenin vs. total N-cadherin | (J) Intimal β-catenin vs. intimal N-cadherin | (K) Intimal β-catenin vs. VE-cadherin junctional-positive | (L) Intimal β-catenin vs. VE-cadherin cytoplasm-positive |
| r′ = 0.53 p = 0.03 | r′ = 0.61 p = 0.02 | r′ = 0.58 p = 0.03 | r′ = 0.70 p = 0.001 | r′ = −0.44 p = 0.09 | r′ = 0.70 p = 0.02 |
| Correlation Analysis | |||
|---|---|---|---|
| TGF-β1 vs. Arterial thickness | TGF-β1 vs. Arterial elastin | ||
| (A) Total TGF-β1 vs. total arterial thickness | (B) Intimal TGF-β1 vs. intimal thickness | (C) Total TGF-β1 vs. total arterial elastin | (D) Intimal TGF-β1 vs. intimal elastin |
| r′ = 0.52 p = 0.04 | r′ = 0.52 p = 0.04 | r′ = 0.68 p = 0.01 | r′ = 0.79 p = 0.002 |
| β-Catenin vs. Arterial thickness | β-Catenin vs. Arterial elastin | ||
| (E) Total β-catenin vs. total arterial thickness | (F) Intimal β-catenin vs. intimal thickness | (G) Total β-catenin vs. total arterial elastin | (H) Intimal β-catenin vs. intimal elastin |
| r′ = 0.56 p = 0.05 | r′ = 0.52 p = 0.04 | r′ = 0.22 p = 0.25 | r′ = 0.03 p = 0.46 |
| TGF-β1 vs. DLCO (%) | β-Catenin vs. DLCO (%) | ||
| (I) Total TGF-β1 vs. DLCO (%) | (J) Intimal β-catenin vs. DLCO (%) | (K) Total β-catenin vs. DLCO (%) | (L) Intimal β-catenin vs. DLCO (%) |
| r′ = −0.61 p = 0.03 | r′ = −0.13 p = 0.35 | r′ = −0.16 p = 0.31 | r′ = −0.13 p = 0.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaikwad, A.V.; Eapen, M.S.; Dey, S.; Bhattarai, P.; Shahzad, A.M.; Chia, C.; Jaffar, J.; Westall, G.; Sutherland, D.; Singhera, G.K.; et al. TGF-β1, pSmad-2/3, Smad-7, and β-Catenin Are Augmented in the Pulmonary Arteries from Patients with Idiopathic Pulmonary Fibrosis (IPF): Role in Driving Endothelial-to-Mesenchymal Transition (EndMT). J. Clin. Med. 2024, 13, 1160. https://doi.org/10.3390/jcm13041160
Gaikwad AV, Eapen MS, Dey S, Bhattarai P, Shahzad AM, Chia C, Jaffar J, Westall G, Sutherland D, Singhera GK, et al. TGF-β1, pSmad-2/3, Smad-7, and β-Catenin Are Augmented in the Pulmonary Arteries from Patients with Idiopathic Pulmonary Fibrosis (IPF): Role in Driving Endothelial-to-Mesenchymal Transition (EndMT). Journal of Clinical Medicine. 2024; 13(4):1160. https://doi.org/10.3390/jcm13041160
Chicago/Turabian StyleGaikwad, Archana Vijay, Mathew Suji Eapen, Surajit Dey, Prem Bhattarai, Affan Mahmood Shahzad, Collin Chia, Jade Jaffar, Glen Westall, Darren Sutherland, Gurpreet Kaur Singhera, and et al. 2024. "TGF-β1, pSmad-2/3, Smad-7, and β-Catenin Are Augmented in the Pulmonary Arteries from Patients with Idiopathic Pulmonary Fibrosis (IPF): Role in Driving Endothelial-to-Mesenchymal Transition (EndMT)" Journal of Clinical Medicine 13, no. 4: 1160. https://doi.org/10.3390/jcm13041160
APA StyleGaikwad, A. V., Eapen, M. S., Dey, S., Bhattarai, P., Shahzad, A. M., Chia, C., Jaffar, J., Westall, G., Sutherland, D., Singhera, G. K., Hackett, T.-L., Lu, W., & Sohal, S. S. (2024). TGF-β1, pSmad-2/3, Smad-7, and β-Catenin Are Augmented in the Pulmonary Arteries from Patients with Idiopathic Pulmonary Fibrosis (IPF): Role in Driving Endothelial-to-Mesenchymal Transition (EndMT). Journal of Clinical Medicine, 13(4), 1160. https://doi.org/10.3390/jcm13041160