
Progress in Brain Magnetic Resonance Imaging of Individuals with Prader–Willi Syndrome
Abstract
1. Introduction
2. Molecular Genetics
3. Pathophysiological Mechanisms
4. Clinical Manifestations
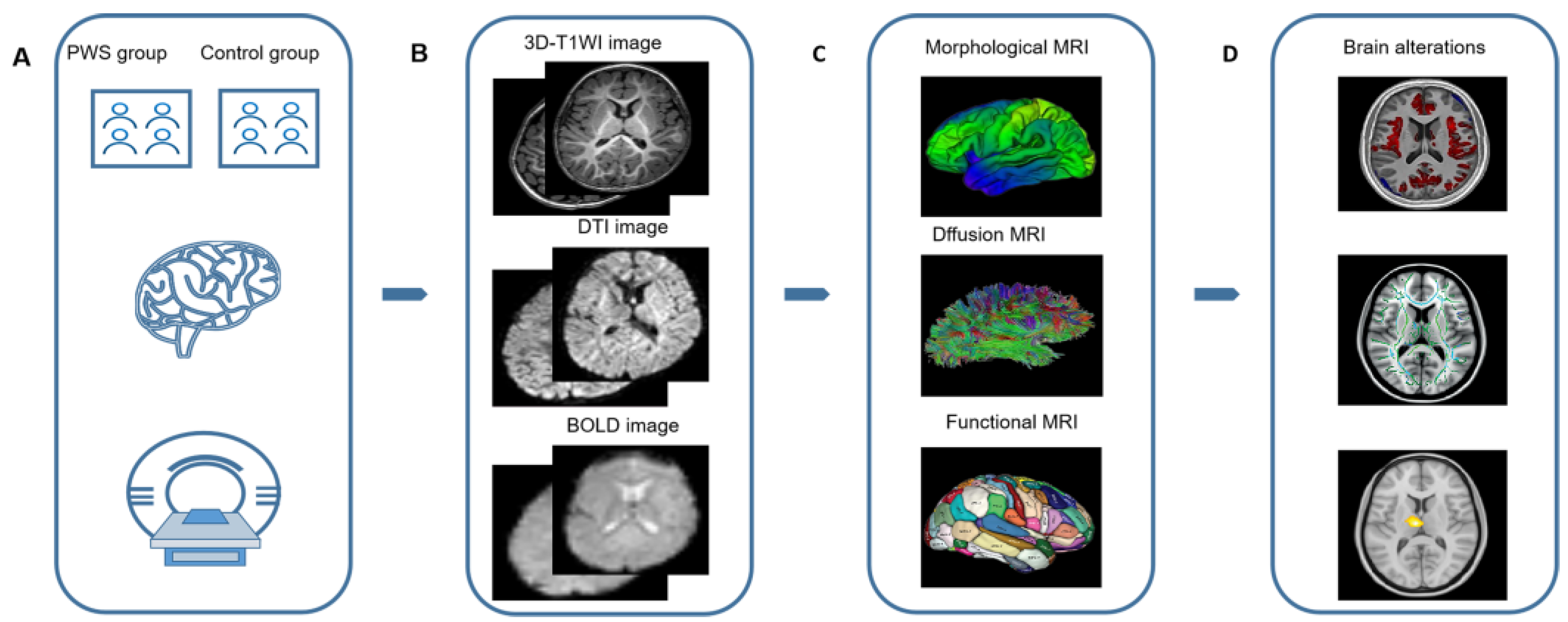
5. MRI
5.1. PWS-Related Brain Regions
5.2. PWS-Related MRI Methods
5.2.1. Conventional Structural MRI
5.2.2. Morphological MRI (mMRI)
5.2.3. Diffusion MRI (dMRI)
5.2.4. Functional MRI (fMRI)
Overeating
Other Behaviors
6. Challenges and Future Prospects
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prader, A. Ein syndrom von adipositas, kleinwuchs, kryptorchismus und oligophrenie nach myatonieartigem zustand im neugeborenenalter. Schweiz. Med. Wochenschr. 1956, 86, 1260–1261. [Google Scholar]
- Lionti, T.; Reid, S.M.; White, S.M.; Rowell, M.M. A population-based profile of 160 Australians with Prader-Willi syndrome: Trends in diagnosis, birth prevalence and birth characteristics. Am. J. Med. Genet. A 2015, 167, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Bar, C.; Diene, G.; Molinas, C.; Bieth, E.; Casper, C.; Tauber, M. Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet J. Rare Dis. 2017, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Kimonis, V.; Dykens, E.; Gold, J.A.; Miller, J.; Tamura, R.; Driscoll, D.J. Prader-Willi syndrome and early-onset morbid obesity NIH rare disease consortium: A review of natural history study. Am. J. Med. Genet. A 2018, 176, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Whittington, J.E.; Holland, A.J.; Webb, T. Ageing in people with Prader-Willi syndrome: Mortality in the UK population cohort and morbidity in an older sample of adults. Psychol. Med. 2015, 45, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.V.; Whittington, J.E.; Holland, A.J.; Boer, H.; Clarke, D.; Webb, T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: A population-based study. Dev. Med. Child Neurol. 2002, 44, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, A.J.P.; LaSalle, J.M. Epigenetics in Prader-Willi syndrome. Front. Genet. 2021, 12, 624581. [Google Scholar] [CrossRef]
- Cheon, C.K. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 126–135. [Google Scholar] [CrossRef]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398–402. [Google Scholar] [CrossRef]
- Gunay-Aygun, M.; Schwartz, S.; Heeger, S.; O’Riordan, M.A.; Cassidy, S.B. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001, 108, E92. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Whittington, J.E.; Butler, J.V.; Holland, A.J. Changing rates of genetic subtypes of Prader-Willi syndrome in the UK. Eur. J. Hum. Genet. 2007, 15, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Hartin, S.N.; Hossain, W.A.; Manzardo, A.M.; Kimonis, V.; Dykens, E.; Gold, J.A.; Kim, S.J.; Weisensel, N.; Tamura, R.; et al. Molecular genetic classification in Prader-Willi syndrome: A multisite cohort study. J. Med. Genet. 2019, 56, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Thompson, T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004, 113, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Zarcone, J.; Napolitano, D.; Peterson, C.; Breidbord, J.; Ferraioli, S.; Caruso-Anderson, M.; Holsen, L.; Butler, M.G.; Thompson, T. The relationship between compulsive behaviour and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J. Intellect. Disabil. Res. 2007, 51, 478–487. [Google Scholar] [CrossRef]
- Bittel, D.C.; Kibiryeva, N.; Butler, M.G. Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pediatrics 2006, 118, e1276–e1283. [Google Scholar] [CrossRef]
- Schwartz, L.; Caixàs, A.; Dimitropoulos, A.; Dykens, E.; Duis, J.; Einfeld, S.; Gallagher, L.; Holland, A.; Rice, L.; Roof, E.; et al. Behavioral features in Prader-Willi syndrome (PWS): Consensus paper from the International PWS Clinical Trial Consortium. J. Neurodev. Disord. 2021, 13, 25. [Google Scholar] [CrossRef]
- Vogels, A.; Matthijs, G.; Legius, E.; Devriendt, K.; Fryns, J.P. Chromosome 15 maternal uniparental disomy and psychosis in Prader-Willi syndrome. J. Med. Genet. 2003, 40, 72–73. [Google Scholar] [CrossRef]
- Morgan, V.A.; Leonard, H.; Bourke, J.; Jablensky, A. Intellectual disability co-occurring with schizophrenia and other psychiatric illness: Population-based study. Br. J. Psychiatry 2008, 193, 364–372. [Google Scholar] [CrossRef]
- Kim, Y.; Wang, S.E.; Jiang, Y.H. Epigenetic therapy of Prader-Willi syndrome. Transl. Res. 2019, 208, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef]
- Correa-da-Silva, F.; Fliers, E.; Swaab, D.F.; Yi, C. Hypothalamic neuropeptides and neurocircuitries in Prader Willi syndrome. J. Neuroendocrinol. 2021, 33, e12994. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.M.; Swanson, L.W. Comparison of the spatial distribution of seven types of neuroendocrine neurons in the rat paraventricular nucleus: Toward a global 3D model. J. Comp. Neurol. 2009, 516, 423–441. [Google Scholar] [CrossRef]
- Ishunina, T.A.; Swaab, D.F. Vasopressin and oxytocin neurons of the human supraoptic and paraventricular nucleus: Size changes in relation to age and sex. J. Clin. Endocrinol. Metab. 1999, 84, 4637–4644. [Google Scholar] [CrossRef] [PubMed]
- Arase, K.; York, D.A.; Shimizu, H.; Shargill, N.; Bray, G.A. Effects of corticotropin-releasing factor on food intake and brown adipose tissue thermogenesis in rats. Am. J. Physiol. 1988, 255, E255–E259. [Google Scholar] [CrossRef]
- Campbell, R.E.; Grove, K.L.; Smith, M.S. Distribution of corticotropin releasing hormone receptor immunoreactivity in the rat hypothalamus: Coexpression in neuropeptide Y and dopamine neurons in the arcuate nucleus. Brain Res. 2003, 973, 223–232. [Google Scholar] [CrossRef]
- Alves, C.; Franco, R.R. Prader-Willi syndrome: Endocrine manifestations and management. Arch. Endocrinol. Metab. 2020, 64, 223–234. [Google Scholar] [CrossRef]
- Coll, A.P.; Farooqi, I.S.; O’Rahilly, S. The hormonal control of food intake. Cell 2007, 129, 251–262. [Google Scholar] [CrossRef]
- Greenswag, L.R. Adults with Prader-Willi syndrome: A survey of 232 cases. Dev. Med. Child Neurol. 1987, 29, 145–152. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.U. Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.P. Prader-Willi syndrome: Advances in genetics, pathophysiology and treatment. Trends Endocrinol. Metab. 2004, 15, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Cassidy, S.B. Prader-Willi syndrome: Genetic, behavioral, and treatment issues. Child Adolesc. Psychiatr. Clin. 1996, 5, 913–927. [Google Scholar] [CrossRef]
- Dykens, E.; Shah, B. Psychiatric disorders in Prader-Willi syndrome: Epidemiology and management. CNS Drugs 2003, 17, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Meguro, M.; Mitsuya, K.; Sui, H.; Shigenami, K.; Kugoh, H.; Nakao, M.; Oshimura, M. Evidence for uniparental, paternal expression of the human GABAA receptor subunit genes, using microcell-mediated chromosome transfer. Hum. Mol. Genet. 1997, 6, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Shapira, N.A.; Lessig, M.C.; Murphy, T.K.; Driscoll, D.J.; Goodman, W.K. Topiramate attenuates self-injurious behaviour in Prader-Willi Syndrome. Int. J. Neuropsychopharmacol. 2002, 5, 141–145. [Google Scholar] [CrossRef]
- Lucignani, G.; Panzacchi, A.; Bosio, L.; Moresco, R.M.; Ravasi, L.; Coppa, I.; Chiumello, G.; Frey, K.; Koeppe, R.; Fazio, F. GABA A receptor abnormalities in Prader-Willi syndrome assessed with positron emission tomography and [11C]flumazenil. Neuroimage 2004, 22, 22–28. [Google Scholar] [CrossRef]
- Smith, A.; Hung, D. The dilemma of diagnostic testing for Prader-Willi syndrome. Transl. Pediatr. 2017, 6, 46–56. [Google Scholar] [CrossRef]
- Butler, M.G. Management of obesity in Prader-Willi syndrome. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 592–593. [Google Scholar] [CrossRef]
- Miller, J.L.; Lynn, C.H.; Driscoll, D.C.; Goldstone, A.P.; Gold, J.A.; Kimonis, V.; Dykens, E.; Butler, M.G.; Shuster, J.J.; Driscoll, D.J. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. A 2011, 155A, 1040–1049. [Google Scholar] [CrossRef]
- Butler, M.G.; Miller, J.L.; Forster, J.L. Prader-Willi syndrome—Clinical genetics, diagnosis and treatment approaches: An update. Curr. Pediatr. Rev. 2019, 15, 207–244. [Google Scholar] [CrossRef] [PubMed]
- Bekx, M.T.; Carrel, A.L.; Shriver, T.C.; Li, Z.; Allen, D.B. Decreased energy expenditure is caused by abnormal body composition in infants with Prader-Willi syndrome. J. Pediatr. 2003, 143, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Grugni, G.; Crinò, A.; Pagani, S.; Meazza, C.; Buzi, F.; De Toni, T.; Gargantini, L.; Pilotta, A.; Pozzan, G.B.; Radetti, G.; et al. Growth hormone secretory pattern in non-obese children and adolescents with Prader-Willi syndrome. J. Pediatr. Endocrinol. Metab. 2011, 24, 477–481. [Google Scholar] [CrossRef]
- Holland, A.J.; Aman, L.C.S.; Whittington, J.E. Defining mental and behavioural disorders in genetically determined neurodevelopmental syndromes with particular reference to Prader-Willi syndrome. Genes 2019, 10, 1025. [Google Scholar] [CrossRef]
- Salles, J.; Lacassagne, E.; Eddiry, S.; Franchitto, N.; Salles, J.P.; Tauber, M. What can we learn from PWS and SNORD116 genes about the pathophysiology of addictive disorders? Mol. Psychiatry 2021, 26, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.M.; Haqq, A.M.; Wismer, W.V. A review of chemosensory perceptions, food preferences and food-related behaviours in subjects with Prader-Willi Syndrome. Appetite 2016, 99, 17–24. [Google Scholar] [CrossRef]
- Tauber, M.; Diene, G.; Mimoun, E.; Çabal-Berthoumieu, S.; Mantoulan, C.; Molinas, C.; Muscatelli, F.; Salles, J.P. Prader-Willi syndrome as a model of human hyperphagia. Front. Horm. Res. 2014, 42, 93–106. [Google Scholar]
- Lindgren, A.C.; Barkeling, B.; Hägg, A.; Ritzén, E.M.; Marcus, C.; Rössner, S. Eating behavior in Prader-Willi syndrome, normal weight, and obese control groups. J. Pediatr. 2000, 137, 50–55. [Google Scholar] [CrossRef]
- Whittington, J.; Holland, A.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. Academic underachievement by people with Prader-Willi syndrome. J. Intellect. Disabil. Res. 2004, 48, 188–200. [Google Scholar] [CrossRef]
- Ho, A.Y.; Dimitropoulos, A. Clinical management of behavioral characteristics of Prader-Willi syndrome. Neuropsychiatr. Dis. Treat. 2010, 6, 107–118. [Google Scholar]
- Michaud, A.; Vainik, U.; Garcia-Garcia, I.; Dagher, A. Overlapping neural endophenotypes in addiction and obesity. Front. Endocrinol. 2017, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.S.; Dolan, R.J. Involvement of human amygdala and orbitofrontal cortex in hunger-enhanced memory for food stimuli. J. Neurosci. 2001, 21, 5304–5310. [Google Scholar] [CrossRef]
- Rolls, E.T. Taste, olfactory, and food texture processing in the brain, and the control of food intake. Physiol. Behav. 2005, 85, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Petrovich, G.D.; Setlow, B.; Holland, P.C.; Gallagher, M. Amygdalo-hypothalamic circuit allows learned cues to override satiety and promote eating. J. Neurosci. 2002, 22, 8748–8753. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, Y.; von Deneen, K.M.; Zhu, H.; Gao, J.H. Brain structural alterations in obese children with and without Prader-Willi syndrome. Hum. Brain Mapp. 2017, 38, 4228–4238. [Google Scholar] [CrossRef]
- Craig, A.D. Interoception: The sense of the physiological condition of the body. Curr. Opin. Neurobiol. 2003, 13, 500–505. [Google Scholar] [CrossRef]
- Manning, K.E.; Holland, A.J. Puzzle pieces: Neural structure and function in Prader-Willi syndrome. Diseases 2015, 3, 382–415. [Google Scholar] [CrossRef]
- Guerreiro, M.M.; Hage, S.R.; Guimarães, C.A.; Abramides, D.V.; Fernandes, W.; Pacheco, P.S.; Piovesana, A.M.; Montenegro, M.A.; Cendes, F. Developmental language disorder associated with polymicrogyria. Neurology 2002, 59, 245–250. [Google Scholar] [CrossRef]
- Winklewski, P.J.; Sabisz, A.; Naumczyk, P.; Jodzio, K.; Szurowska, E.; Szarmach, A. Understanding the physiopathology behind axial and radial diffusivity changes-what do we know? Front. Neurol. 2018, 9, 92. [Google Scholar] [CrossRef]
- Iughetti, L.; Bosio, L.; Corrias, A.; Gargantini, L.; Ragusa, L.; Livieri, C.; Predieri, B.; Bruzzi, P.; Caselli, G.; Grugni, G. Pituitary height and neuroradiological alterations in patients with Prader-Labhart-Willi syndrome. Eur. J. Pediatr. 2008, 167, 701–702. [Google Scholar] [CrossRef]
- Emerick, J.E.; Vogt, K.S. Endocrine manifestations and management of Prader-Willi syndrome. Int. J. Pediatr. Endocrinol. 2013, 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Fujii, T.; Abe, N.; Hosokai, Y.; Shinohara, M.; Takahashi, S.; Mori, E. Small gray matter volume in orbitofrontal cortex in Prader-Willi syndrome: A voxel-based MRI study. Hum. Brain Mapp. 2011, 32, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Honea, R.A.; Holsen, L.M.; Lepping, R.J.; Perea, R.; Butler, M.G.; Brooks, W.M.; Savage, C.R. The neuroanatomy of genetic subtype differences in Prader-Willi syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Lukoshe, A.; White, T.; Schmidt, M.N.; van der Lugt, A.; Hokken-Koelega, A.C. Divergent structural brain abnormalities between different genetic subtypes of children with Prader-Willi syndrome. J. Neurodev. Disord. 2013, 5, 31. [Google Scholar] [CrossRef]
- Lukoshe, A.; Hokken-Koelega, A.C.; van der Lugt, A.; White, T. Reduced cortical complexity in children with Prader-Willi Syndrome and its association with cognitive impairment and developmental delay. PLoS ONE 2014, 9, e107320. [Google Scholar] [CrossRef]
- Manning, K.E.; Tait, R.; Suckling, J.; Holland, A.J. Grey matter volume and cortical structure in Prader-Willi syndrome compared to typically developing young adults. Neuroimage Clin. 2018, 17, 899–909. [Google Scholar] [CrossRef]
- Ge, M.M.; Gao, Y.Y.; Wu, B.B.; Yan, K.; Qin, Q.; Wang, H.; Zhou, W.; Yang, L. Relationship between phenotype and genotype of 102 Chinese newborns with Prader-Willi syndrome. Mol. Biol. Rep. 2019, 46, 4717–4724. [Google Scholar] [CrossRef]
- Yamada, K.; Watanabe, M.; Suzuki, K.; Suzuki, Y. Cerebellar volumes associate with behavioral phenotypes in Prader-Willi syndrome. Cerebellum 2020, 19, 778–787. [Google Scholar] [CrossRef]
- Caixàs, A.; Blanco-Hinojo, L.; Pujol, J.; Deus, J.; Giménez-Palop, O.; Torrents-Rodas, D.; Coronas, R.; Novell, R.; Esteba-Castillo, S. Altered gesture imitation and brain anatomy in adult Prader-Willi syndrome patients. J. Int. Neuropsychol. Soc. 2021, 27, 1024–1036. [Google Scholar] [CrossRef]
- Royall, D.R.; Lauterbach, E.C.; Cummings, J.L.; Reeve, A.; Rummans, T.A.; Kaufer, D.I.; LaFrance, W.C., Jr.; Coffey, C.E. Executive control function: A review of its promise and challenges for clinical research. A report from the committee on research of the American Neuropsychiatric Association. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 377–405. [Google Scholar] [CrossRef]
- Wang, G.J.; Yang, J.; Volkow, N.D.; Telang, F.; Ma, Y.; Zhu, W.; Wong, C.T.; Tomasi, D.; Thanos, P.K.; Fowler, J.S. Gastric stimulation in obese subjects activates the hippocampus and other regions involved in brain reward circuitry. Proc. Natl. Acad. Sci. USA 2006, 103, 15641–15645. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; Barrea, L.; Faggiano, F.; Maiorino, M.I.; Parrillo, M.; Pugliese, G.; Ruggeri, R.M.; Scarano, E.; Savastano, S.; Colao, A. Obesity in Prader-Willi syndrome: Physiopathological mechanisms, nutritional and pharmacological approaches. J. Endocrinol. Investig. 2021, 44, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Y.; Yu, C.; Lin, L.; Li, C.; Jiang, T. Reduced cortical folding in mental retardation. Am. J. Neuroradiol. 2010, 31, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Titomanlio, L.; De Brasi, D.; Romano, A.; Genesio, R.; Diano, A.A.; Del Giudice, E. Partial cerebellar hypoplasia in a patient with Prader-Willi syndrome. Acta Paediatr. 2006, 95, 861–863. [Google Scholar] [CrossRef] [PubMed]
- Caligiore, D.; Pezzulo, G.; Baldassarre, G.; Bostan, A.C.; Strick, P.L.; Doya, K.; Helmich, R.C.; Dirkx, M.; Houk, J.; Jörntell, H.; et al. Consensus paper: Towards a systems-level view of cerebellar function: The interplay between cerebellum, basal ganglia, and cortex. Cerebellum 2017, 16, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108, 511–533. [Google Scholar]
- Yamada, K.; Matsuzawa, H.; Uchiyama, M.; Kwee, I.L.; Nakada, T. Brain developmental abnormalities in Prader-Willi syndrome detected by diffusion tensor imaging. Pediatrics 2006, 118, e442–e448. [Google Scholar] [CrossRef]
- Rice, L.J.; Lagopoulos, J.; Brammer, M.; Einfeld, S.L. Microstructural white matter tract alteration in Prader-Willi syndrome: A diffusion tensor imaging study. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 362–367. [Google Scholar] [CrossRef]
- Lukoshe, A.; van den Bosch, G.E.; van der Lugt, A.; Kushner, S.A.; Hokken-Koelega, A.C.; White, T. Aberrant white matter microstructure in children and adolescents with the subtype of Prader-Willi syndrome at high risk for psychosis. Schizophr. Bull. 2017, 43, 1090–1099. [Google Scholar] [CrossRef]
- Sach, M.; Winkler, G.; Glauche, V.; Liepert, J.; Heimbach, B.; Koch, M.A.; Büchel, C.; Weiller, C. Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain 2004, 127, 340–350. [Google Scholar] [CrossRef]
- Shapira, N.A.; Lessig, M.C.; He, A.G.; James, G.A.; Driscoll, D.J.; Liu, Y. Satiety dysfunction in Prader-Willi syndrome demonstrated by fMRI. J. Neurol. Neurosurg. Psychiatry 2005, 76, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Zarcone, J.R.; Brooks, W.M.; Butler, M.G.; Thompson, T.I.; Ahluwalia, J.S.; Nollen, N.L.; Savage, C.R. Neural mechanisms underlying hyperphagia in Prader-Willi syndrome. Obesity 2006, 14, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; James, G.A.; Goldstone, A.P.; Couch, J.A.; He, G.; Driscoll, D.J.; Liu, Y. Enhanced activation of reward mediating prefrontal regions in response to food stimuli in Prader-Willi syndrome. J. Neurol. Neurosurg. Psychiatry 2017, 78, 615–619. [Google Scholar] [CrossRef]
- Dimitropoulos, A.; Schultz, R.T. Food-related neural circuitry in Prader-Willi syndrome: Response to high- versus low-calorie foods. J. Autism Dev. Disord. 2008, 38, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Zarcone, J.R.; Chambers, R.; Butler, M.G.; Bittel, D.C.; Brooks, W.M.; Thompson, T.I.; Savage, C.R. Genetic subtype differences in neural circuitry of food motivation in Prader-Willi syndrome. Int. J. Obes. 2009, 33, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Savage, C.R.; Martin, L.E.; Bruce, A.S.; Lepping, R.J.; Ko, E.; Brooks, W.M.; Butler, M.G.; Zarcone, J.R.; Goldstein, J.M. Importance of reward and prefrontal circuitry in hunger and satiety: Prader-Willi syndrome vs simple obesity. Int. J. Obes. 2012, 36, 638–647. [Google Scholar] [CrossRef]
- Klabunde, M.; Saggar, M.; Hustyi, K.M.; Hammond, J.L.; Reiss, A.L.; Hall, S.S. Neural correlates of self-injurious behavior in Prader-Willi syndrome. Hum. Brain Mapp. 2015, 36, 4135–4143. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, H.; Qiu, S.; Tian, J.; Wen, X.; Miller, J.L.; von Deneen, K.M.; Zhou, Z.; Gold, M.S.; Liu, Y. Altered functional brain networks in Prader-Willi syndrome. NMR Biomed. 2013, 26, 622–629. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Zhang, G.; Zhu, Q.; Cai, W.; Tian, J.; Zhang, Y.E.; Miller, J.L.; Wen, X.; Ding, M.; et al. The neurobiological drive for overeating implicated in Prader-Willi syndrome. Brain Res. 2015, 1620, 72–80. [Google Scholar] [CrossRef]
- Pujol, J.; Blanco-Hinojo, L.; Esteba-Castillo, S.; Caixàs, A.; Harrison, B.J.; Bueno, M.; Deus, J.; Rigla, M.; Macià, D.; Llorente-Onaindia, J.; et al. Anomalous basal ganglia connectivity and obsessive-compulsive behaviour in patients with Prader Willi syndrome. J. Psychiatry Neurosci. 2016, 41, 261–271. [Google Scholar] [CrossRef]
- Tauber, M.; Boulanouar, K.; Diene, G.; Çabal-Berthoumieu, S.; Ehlinger, V.; Fichaux-Bourin, P.; Molinas, C.; Faye, S.; Valette, M.; Pourrinet, J.; et al. The use of oxytocin to improve feeding and social skills in infants with Prader-Willi syndrome. Pediatrics 2017, 139, e20162976. [Google Scholar] [CrossRef]
- Taylor, R.L.; Caldwell, M.L. Type and strength of food preferences of individuals with Prader-Willi syndrome. J. Intellect. Disabil. Res. 1985, 29, 109–112. [Google Scholar] [CrossRef]
- Hinton, E.C.; Holland, A.J.; Gellatly, M.S.; Soni, S.; Owen, A.M. An investigation into food preferences and the neural basis of food-related incentive motivation in Prader-Willi syndrome. J. Intellect. Disabil. Res. 2006, 50, 633–642. [Google Scholar] [CrossRef]
- Hinton, E.C.; Holland, A.J.; Gellatly, M.S.; Soni, S.; Patterson, M.; Ghatei, M.A.; Owen, A.M. Neural representations of hunger and satiety in Prader-Willi syndrome. Int. J. Obes. 2006, 30, 313–321. [Google Scholar] [CrossRef]
- Aman, L.C.S.; Manning, K.E.; Whittington, J.E.; Holland, A.J. Mechanistic insights into the genetics of affective psychosis from Prader-Willi syndrome. Lancet Psychiatry 2018, 5, 370–378. [Google Scholar] [CrossRef]


{kind=link}
| Phase | Period | Clinical Characteristics |
|---|---|---|
| Phase 0 | Before birth | Decreased fetal movements and low birth weight |
| Phase 1a | 0–9 months | Feeding difficulties (poor sucking and hypotonia) requiring gastric or nasogastric tubes Decreased appetite |
| Phase 1b | 9–24 months | Feeding difficulties have lessened and weight might increase at a normal rate |
| Phase 2a | 2–4.5 years old | Begin to gain weight without changes in appetite or feeding Restriction to 60–80% of recommended daily allowance for calories is needed to prevent obesity |
| Phase 2b | 4.5–8 years old | Appetite and weight acceleration Global developmental delay |
| Phase 3 | Starting from 8 years old | Lack of satiety is obvious and classic gluttony also becomes prominent Onset of mild intellectual disability and food-related temper tantrums |
| Phase 4 | After being an adult | Only some adult patients with PWS control food seeking behaviors and temper tantrums |
| Number | Author | Country and Equipment | MRI Assessment Method | Subjects (n) Total/PWS | PWS Mean Age (Years) | Key Findings |
|---|---|---|---|---|---|---|
| 1 | Ogura et al. [62] | Japan GE 1.5 T | VBM | 25/12 | 23.7 |
|
| 2 | Honea et al. [63] | USA Siemens 3.0 T | VBM/ GLM | 48/23 5 DEL I 10 DEL II 8 mUPD | 25.2 (DEL) 17.4 (mUPD) |
|
| 3 | Lukoshe et al. [64] | The Netherlands GE 3.0 T | Cortical reconstruction and volumetric segmentation | 31/20 11 DEL 9 mUPD | 12.3(DEL) 10.6(mUPD) |
|
| 4 | Lukoshe et al. [65] | The Netherlands GE 3.0 T | SBM | 35/24 12 DEL 12 mUPD | 12.6 (DEL) 11.5 (mUPD) |
|
| 5 | Xu et al. [55] | China Siemens 3.0 T | Structure covariant | 48/12 12 PWS 18 OB 18 HC | 7.2 (PWS) 9.0 (OB) |
|
| 6 | Manning et al. [66] | UK -- | VBM/ GLM | 60/20 | 23.10 |
|
| 7 | Ge et al. [67] | China -- | VBM | 102 75 DEL 27 mUPD | newborns |
|
| 8 | Yamada et al. [68] | Japan GE 3.0 T | —— | 61/21 | 21.0 |
|
| 9 | Caixàs et al. [69] | Spain 1.5 T | VBM | 162/30 30 PWS 132 ID | 27.5 |
|
| Number | Author | Country and Equipment | MRI Assessment Method | Subjects (n) Total/PWS | PWS Mean Age (Years) | Key Findings |
|---|---|---|---|---|---|---|
| 1 | Yamada et al. [77] | Japan GE 3.0 T | DTI | 16/8 | 19 |
|
| 2 | Rice et al. [78] | Australia GE 3.0 T | DTI TBSS | 30/15 | 21 |
|
| 3 | Xu et al. [55] | China Siemens 3.0 T | DTI 24 directions | 48/12 12 PWS 18 OB 18 HC | 7.2 (PWS) 9.0 (OB) |
|
| 4 | Lukoshe et al. [79] | The Netherlands GE 3.0 T | DTI TBSS | 89/28 15 DEL 13 mUPD | 14.4 (DEL) 11.8 (mUPD) |
|
| Number | Author | Country and Equipment | MRI Measures and Analysis | Subjects (n) Total/PWS | PWS Mean Age (Years) | Key Findings |
|---|---|---|---|---|---|---|
| 1 | Shapira et al. [81] | USA GE 3.0 T | TCA; glucose ingestion overnight | 3 PWS | 36.7 |
|
| 2 | Holsen et al. [82] | USA Siemens 3.0 T | Visual food stimuli | 18/9 5 PWS with pre-meal 4 PWS with pos-meal | 14.7 |
|
| 3 | Miller et al. [83] | Ingestion of an oral glucose load; Viewing pictures | 16/8 | 25 |
| |
| 4 | Dimitropoulos et al. [84] | USA Siemens 3.0 T | Visual food stimuli | 19/9 | 21.6 |
|
| 5 | Holsen et al. [85] | USA Siemens 3.0 T | Visual food stimuli | 18 PWS 9 mUPD 9 DEL II | 24.4 (DEL) 20.3 (mUPD) |
|
| 6 | Holsen et al. [86] | USA Siemens 3.0 T | Visual food stimuli | 43/14 14 PWS 14 OB 15 HC | 24.3 |
|
| 7 | Klabunde et al. [87] | USA GE 3.0 T | Record skin picking episodes | 17 PWS 10 with episodes | 15.7 |
|
| Number | Author | Country and Equipment | MRI Measures and Analysis | Subjects (n) Total/PWS | PWS Mean Age (Years) | Key Findings |
|---|---|---|---|---|---|---|
| 1 | Zhang et al. [88] | China Siemens 3.0 T | ALFF | 39/21 | 7.3 |
|
| 2 | Zhang et al. [89] | China Siemens 3.0 T | ALFF GCA | 39/21 | 9.3 |
|
| 3 | Pujol et al. [90] | Spain Siemens 1.5 T | seed based | 53/24 | 26.3 |
|
| 4 | Tauber et al. [91] | France -- | ICA | 18 PWS with OXT 18 PWS without OXT | newborns |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Z.; Cai, J. Progress in Brain Magnetic Resonance Imaging of Individuals with Prader–Willi Syndrome. J. Clin. Med. 2023, 12, 1054. https://doi.org/10.3390/jcm12031054
Huang Z, Cai J. Progress in Brain Magnetic Resonance Imaging of Individuals with Prader–Willi Syndrome. Journal of Clinical Medicine. 2023; 12(3):1054. https://doi.org/10.3390/jcm12031054
Chicago/Turabian StyleHuang, Zhongxin, and Jinhua Cai. 2023. "Progress in Brain Magnetic Resonance Imaging of Individuals with Prader–Willi Syndrome" Journal of Clinical Medicine 12, no. 3: 1054. https://doi.org/10.3390/jcm12031054
APA StyleHuang, Z., & Cai, J. (2023). Progress in Brain Magnetic Resonance Imaging of Individuals with Prader–Willi Syndrome. Journal of Clinical Medicine, 12(3), 1054. https://doi.org/10.3390/jcm12031054