
Management of Pheochromocytomas and Paragangliomas: A Case-Based Review of Clinical Aspects and Perspectives


Abstract

1. Introduction
2. Case Report
3. Genetic Background
4. Diagnosis
4.1. Measurement of Metanephrines
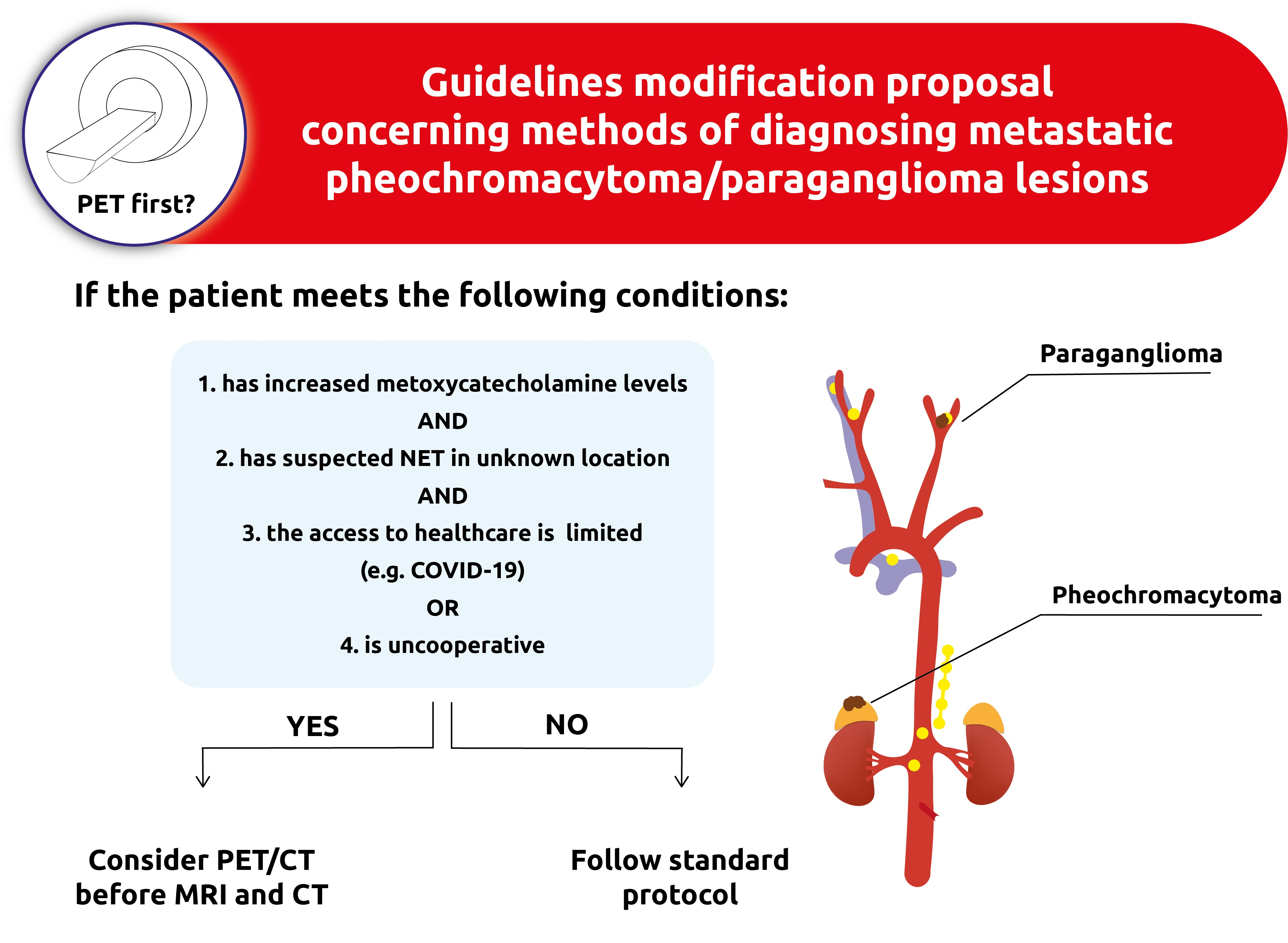
4.2. Imaging
4.3. Clinical Presentation Associated with Genetic Background
4.4. Histopathology
4.5. Differential Diagnosis
5. Treatment
5.1. Local Treatment of Early Disease
5.2. Treatment of Metastatic or Relapsed Disease
6. Future Directions in PGL Treatment
6.1. Molecular Targeted Agents
6.1.1. Everolimus
6.1.2. Axitinib
6.1.3. Cabozantinib
6.1.4. Sunitinib and Sorafenib
6.1.5. HIF Inhibitors
6.2. Anti-PD-1 Immunotherapy
6.3. Cancer Vaccines
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Neumann, H.P.H.; Young, W.F.J.; Eng, C. Pheochromocytoma and Paraganglioma. N. Engl. J. Med. 2019, 0381, 552–565. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. WHO Classification of Tumours of Endocrine Organs, 4th ed.; Lloyd, R., Osamura, R., Klöppel, G., Rosai, J., Eds.; WHO: Geneva, Switzerland, 2017; Volume 10.
- National Cancer Institute. Rare Endocrine Tumors: Paraganglioma. Available online: https://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/rare-endocrine-tumor/paraganglioma (accessed on 1 February 2022).
- Stenström, G.; Svärdsudd, K. Pheochromocytoma in Sweden 1958-1981. An analysis of the National Cancer Registry Data. Acta Med. Scand. 1986, 220, 225–232. [Google Scholar] [CrossRef]
- Ebbehoj, A.; Stochholm, K.; Jacobsen, S.F.; Trolle, C.; Jepsen, P.; Robaczyk, M.G.; Rasmussen, Å.K.; Feldt-Rasmussen, U.; Thomsen, R.W.; Søndergaard, E.; et al. Incidence and Clinical Presentation of Pheochromocytoma and Sympathetic Paraganglioma: A Population-based Study. J. Clin. Endocrinol. Metab. 2021, 106, e2251–e2261. [Google Scholar] [CrossRef] [PubMed]
- Severi, S.; Bongiovanni, A.; Ferrara, M.; Nicolini, S.; Di Mauro, F.; Sansovini, M.; Lolli, I.; Tardelli, E.; Cittanti, C.; Di Iorio, V.; et al. Peptide receptor radionuclide therapy in patients with metastatic progressive pheochromocytoma and paraganglioma: Long-term toxicity, efficacy and prognostic biomarker data of phase II clinical trials. ESMO Open 2021, 6, 100171. [Google Scholar] [CrossRef]
- Leung, A.A.; Pasieka, J.L.; Hyrcza, M.D.; Pacaud, D.; Dong, Y.; Boyd, J.M.; Sadrzadeh, H.; Kline, G.A. Epidemiology of pheochromocytoma and paraganglioma: Population-based cohort study. Eur. J. Endocrinol. 2021, 184, 19–28. [Google Scholar] [CrossRef]
- Gruber, L.M.; Hartman, R.P.; Thompson, G.B.; McKenzie, T.J.; Lyden, M.L.; Dy, B.M.; Young, W.F.; Bancos, I. Pheochromocytoma Characteristics and Behavior Differ Depending on Method of Discovery. J. Clin. Endocrinol. Metab. 2019, 104, 1386–1393. [Google Scholar] [CrossRef]
- Cancer.Net. Pheochromocytoma and Paraganglioma Guide: Risk Factors. Available online: https://www.cancer.net/cancer-types/pheochromocytoma-and-paraganglioma/risk-factors (accessed on 1 February 2022).
- Carney, J.A. Carney triad. Front. Horm. Res. 2013, 41, 92–110. [Google Scholar] [CrossRef]
- Gilbo, P.; Morris, C.G.; Amdur, R.J.; Werning, J.W.; Dziegielewski, P.T.; Ma, J.K.; Mendenhall, W.M. Radiotherapy for benign head and neck paragangliomas: A 45-year experience. Cancer 2014, 120, 3738–3743. [Google Scholar] [CrossRef] [PubMed]
- Przychodni, Ś. Endokrynolog. Available online: https://swiatprzychodni.pl/specjalnosci/endokrynolog/ (accessed on 1 February 2022).
- Taïeb, D.; Pacak, K. Genetic Determinants of Pheochromocytoma and Paraganglioma Imaging Phenotypes. J. Nucl. Med. 2020, 61, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Li, M.; Guan, X.; Yu, A.; Xiao, Q.; Wang, C.; Hu, Y.; Zhu, F.; Yin, H.; Yi, X.; et al. Clinical Syndromes and Genetic Screening Strategies of Pheochromocytoma and Paraganglioma. J. Kidney Cancer VHL 2018, 5, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Ricketts, C.J.; Chowdhury, R.; McDonough, M.A.; Gentle, D.; Kirby, G.; Schlisio, S.; Kenchappa, R.S.; Carter, B.D.; Kaelin, W.G.; et al. Mutation analysis of HIF prolyl hydroxylases (PHD/EGLN) in individuals with features of phaeochromocytoma and renal cell carcinoma susceptibility. Endocr. Relat. Cancer 2011, 18, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zhang, J.; Tan, X.; Huang, Y.; Xu, J.; Silk, N.; Zhang, D.; Liu, Q.; Jiang, J. The VHL/HIF Axis in the Development and Treatment of Pheochromocytoma/Paraganglioma. Front. Endocrinol. 2020, 11, 586857. [Google Scholar] [CrossRef] [PubMed]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Askmalm, M.S.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2019, 285, 187–204. [Google Scholar] [CrossRef]
- Crona, J.; Taïeb, D.; Pacak, K. New perspectives on pheochromocytoma and paraganglioma: Toward a molecular classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.X.; Cui, J.J.; Wang, L.Y.; Yin, J.Y. The role of CSDE1 in translational reprogramming and human diseases. Cell Commun. Signal. 2020, 18, 14. [Google Scholar] [CrossRef]
- Abdallah, A.; Pappo, A.; Reiss, U.; Shulkin, B.L.; Zhuang, Z.; Pacak, K.; Bahrami, A. Clinical manifestations of Pacak-Zhuang syndrome in a male pediatric patient. Pediatr Blood Cancer. 2020, 67, e28096. [Google Scholar] [CrossRef]
- Pillai, S.; Gopalan, V.; Smith, R.A.; Lam, A.K.Y. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit. Rev. Oncol. Hematol. 2016, 100, 190–208. [Google Scholar] [CrossRef]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef]
- Buffet, A.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A.P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101416. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, M.; Foulkes, W.D. Pheochromocytoma and paraganglioma syndromes: Genetics and management update. Curr. Oncol. 2014, 21, e8–e17. [Google Scholar] [CrossRef] [PubMed]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; Van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.-P.; et al. SDHA Immunohistochemistry Detects Germline SDHA Gene Mutations in Apparently Sporadic Paragangliomas and Pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–E1476. [Google Scholar] [CrossRef] [PubMed]
- Oudijk, L.; de Krijger, R.R.; Rapa, I.; Beuschlein, F.; De Cubas, A.A.; Dei Tos, A.P.; Dinjens, W.N.M.; Korpershoek, E.; Mančíková, V.; Mannelli, M.; et al. H-RAS Mutations Are Restricted to Sporadic Pheochromocytomas Lacking Specific Clinical or Pathological Features: Data From a Multi-Institutional Series. J. Clin. Endocrinol. Metab. 2014, 99, E1376–E1380. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Cascón, A.; Schiavi, F.; Morales, N.P.; Comino-Mendez, I.; Abermil, N.; Inglada, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX Mutations Cause Hereditary and Sporadic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef]
- Gaal, J.; Burnichon, N.; Korpershoek, E.; Roncelin, I.; Bertherat, J.; Plouin, P.-F.; De Krijger, R.R.; Gimenez-Roqueplo, A.-P.; Dinjens, W.N.M. Isocitrate Dehydrogenase Mutations Are Rare in Pheochromocytomas and Paragangliomas. J. Clin. Endocrinol. Metab. 2010, 95, 1274–1278. [Google Scholar] [CrossRef]
- Lang, F.; Jha, A.; Meuter, L.; Pacak, K.; Yang, C. Identification of Isocitrate Dehydrogenase 2 (IDH2) Mutation in Carotid Body Paraganglioma. Front. Endocrinol. 2021, 12, 731096. Available online: https://www.frontiersin.org/article/10.3389/fendo.2021.731096 (accessed on 10 January 2022). [CrossRef]
- Lenders, J.W.M.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens. 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Szosland, K.; Kopff, B.; Lewiński, A. Pheochromocytoma-chromaffin cell tumor. Endokrynol. Pol. 2006, 57, 54–62. Available online: https://journals.viamedica.pl/endokrynologia_polska/article/view/25774 (accessed on 10 January 2022).
- Därr, R.; Kuhn, M.; Bode, C.; Bornstein, S.R.; Pacak, K.; Lenders, J.W.; Eisenhofer, G. Accuracy of recommended sampling and assay methods for the determination of plasma-free and urinary fractionated metanephrines in the diagnosis of pheochromocytoma and paraganglioma: A systematic review. Endocrine 2017, 56, 495–503. [Google Scholar] [CrossRef]
- Farrugia, F.A.; Martikos, G.; Tzanetis, P.; Charalampopoulos, A.; Misiakos, E.; Zavras, N.; Sotiropoulos, D. Pheochromocytoma, diagnosis and treatment: Review of the literature. Endocr. Regul. 2017, 51, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Baracco, R.; Kapur, G. Pheochromocytoma and paraganglioma-an update on diagnosis, evaluation, and management. Pediatr. Nephrol. 2020, 35, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Oudijk, L.; Gaal, J.; Koopman, K.; de Krijger, R.R. An Update on the Histology of Pheochromocytomas: How Does it Relate to Genetics? Horm. Metab. Res. 2019, 51, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Zhang, W.Y.; Wu, X.T. Clinicopathological analysis of paraganglioma with literature review. World J. Gastroenterol. 2009, 15, 3003–3008. [Google Scholar] [CrossRef]
- Mannelli, M.; Lenders, J.W.M.; Pacak, K.; Parenti, G.; Eisenhofer, G. Subclinical phaeochromocytoma. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 507–515. [Google Scholar] [CrossRef]
- An, Y.-Y.; Yang, G.Z.; Lin, B.; Zhang, N.; Hou, H.-T.; Zhu, F.-M.; Tian, F.-J.; Wang, J. Differentiation of lipid-poor adenoma from pheochromocytoma on biphasic contrast-enhanced, CT. Abdom. Radiol. 2021, 46, 4353–4361. [Google Scholar] [CrossRef]
- Szolar, D.H.; Korobkin, M.; Reittner, P.; Berghold, A.; Bauernhofer, T.; Trummer, H.; Schoellnast, H.; Preidler, K.W.; Samonigg, H. Adrenocortical Carcinomas and Adrenal Pheochromocytomas: Mass and Enhancement Loss Evaluation at Delayed Contrast-enhanced CT. Radiology 2005, 234, 479–485. [Google Scholar] [CrossRef]
- Jain, S.; Agarwal, L.; Nadkarni, S.; Ameta, A.; Goyal, A.; Kumar, R.; Rao, A.; Gupta, K. Adrenocortical carcinoma posing as a pheochromocytoma: A diagnostic dilemma. J. Surg. Case Rep. 2014, 2014, rju030. [Google Scholar] [CrossRef][Green Version]
- Ni, H.; Htet, A. Adrenal cortical carcinoma masquerading as pheochromocytoma: A case report. Ecancermedicalscience 2012, 6, 277. [Google Scholar] [CrossRef]
- Shah, U.; Giubellino, A.; Pacak, K. Pheochromocytoma: Implications in tumorigenesis and the actual management. Minerva Endocrinol. 2012, 37, 141–156. [Google Scholar] [PubMed]
- Weingarten, T.N.; Cata, J.P.; O’Hara, J.F.; Prybilla, D.J.; Pike, T.L.; Thompson, G.B.; Grant, C.S.; Warner, D.O.; Bravo, E.; Sprung, J. Comparison of two preoperative medical management strategies for laparoscopic resection of pheochromocytoma. Urology 2010, 76, 508.e6–508.e11. [Google Scholar] [CrossRef] [PubMed]
- Randle, R.W.; Balentine, C.J.; Pitt, S.C.; Schneider, D.F.; Sippel, R.S. Selective Versus Non-selective α-Blockade Prior to Laparoscopic Adrenalectomy for Pheochromocytoma. Ann. Surg. Oncol. 2017, 24, 244–250. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Pheochromocytoma and Paraganglioma Treatment: Health Professional Treatment. Available online: https://www.cancer.gov/types/pheochromocytoma/hp/pheochromocytoma-treatment-pdq#_9 (accessed on 14 February 2022).
- Prakash, P.; Ramachandran, R.; Tandon, N.; Kumar, R. Open surgery for pheochromocytoma: Current indications and outcomes from a retrospective cohort. Indian J. Urol. 2020, 36, 21–25. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Matute Teresa, F.; Mercader-Cidoncha, E.; Mitjavila-Casanovas, M.; Robledo, M.; Tena, I.; Alvarez-Escola, C.; Arístegui, M.; Bella-Cueto, M.R.; Ferrer-Albiach, C.; et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin. Transl. Oncol. 2021, 23, 1995–2019. [Google Scholar] [CrossRef]
- Makeieff, M.; Thariat, J.; Reyt, E.; Righini, C.A. Treatment of cervical paragangliomas. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2012, 129, 308–314. [Google Scholar] [CrossRef]
- González, I.D.; Vallejo, A.; Guerrero Lizcano, E.; Pabón Girón, A. Adrenal Pheochromocytoma Treated With Stereotactic Body Radiation Therapy. Cureus 2021, 13, e12456. [Google Scholar] [CrossRef]
- Merzouqi, B.; el Bouhmadi, K.; Oukesou, Y.; Rouadi, S.; Abada, R.L.; Roubal, M.; Mahtar, M. Head and neck paragangliomas: Ten years of experience in a third health center. A cohort study. Ann. Med. Surg 2021, 66, 102412. [Google Scholar] [CrossRef]
- Carrasquillo, J.A.; Pandit-Taskar, N.; Chen, C.C. I-131 Metaiodobenzylguanidine Therapy of Pheochromocytoma and Paraganglioma. Semin. Nucl. Med. 2016, 46, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Van Hulsteijn, L.T.; Niemeijer, N.D.; Dekkers, O.M.; Corssmit, E.P.M. (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 80, 487–501. [Google Scholar] [CrossRef]
- Pryma, D.A.; Chin, B.B.; Noto, R.B.; Dillon, J.S.; Perkins, S.; Solnes, L.; Kostakoglu, L.; Serafini, A.N.; Pampaloni, M.H.; Jensen, J.; et al. Efficacy and Safety of High-Specific-Activity (131)I-MIBG Therapy in Patients with Advanced Pheochromocytoma or Paraganglioma. J. Nucl. Med. 2019, 60, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, M.P.; Kane, A.; Zhu, J.; Morse, M.A.; Wong, T.; Borges-Neto, S. Long-Term Outcomes of 125 Patients With Metastatic Pheochromocytoma or Paraganglioma Treated With 131-I MIBG. J. Clin. Endocrinol. Metab. 2020, 105, e494–e501. [Google Scholar] [CrossRef] [PubMed]
- Taïeb, D.; Hicks, R.J.; Hindié, E.; Guillet, B.A.; Avram, A.; Ghedini, P.; Timmers, H.J.; Scott, A.T.; Elojeimy, S.; Rubello, D.; et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging. 2019, 46, 2112–2137. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Naruse, M. Recent advances in the management of pheochromocytoma and paraganglioma. Hypertens. Res. 2020, 43, 1141–1151. [Google Scholar] [CrossRef]
- Nölting, S.; Maurer, J.; Spöttl, G.; Prada, E.T.A.; Reuther, C.; Young, K.; Korbonits, M.; Göke, B.; Grossman, A.; Auernhammer, C.J. Additive Anti-Tumor Effects of Lovastatin and Everolimus In Vitro through Simultaneous Inhibition of Signaling Pathways. PLoS ONE 2015, 10, e0143830. [Google Scholar] [CrossRef]
- Nölting, S.; Garcia, E.; Alusi, G.; Giubellino, A.; Pacak, K.; Korbonits, M.; Grossman, A.B. Combined blockade of signalling pathways shows marked anti-tumour potential in phaeochromocytoma cell lines. J. Mol. Endocrinol. 2012, 49, 79–96. [Google Scholar] [CrossRef][Green Version]
- Fankhauser, M.; Bechmann, N.; Lauseker, M.; Goncalves, J.; Favier, J.; Klink, B.; William, D.; Gieldon, L.; Maurer, J.; Spöttl, G.; et al. Synergistic Highly Potent Targeted Drug Combinations in Different Pheochromocytoma Models Including Human Tumor Cultures. Endocrinology 2019, 160, 2600–2617. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kim, T.W.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.-K.; Lee, H.J.; Lee, K.-W.; Bang, Y.-J. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef]
- Chaux, A.; Brimo, F.; Gonzalez-Roibon, N.; Shah, S.; Schultz, L.; Rizk, J.-M.; Argani, P.; Hicks, J.; Netto, G.J. Immunohistochemical Evidence of Dysregulation of the Mammalian Target of Rapamycin Pathway in Primary and Metastatic Pheochromocytomas. Urology 2012, 3952, A1 YP-A18. [Google Scholar] [CrossRef]
- Oudijk, L.; Papathomas, T.; de Krijger, R.; Korpershoek, E.; Gimenez-Roqueplo, A.-P.; Favier, J.; Canu, L.; Mannelli, M.; Rapa, I.; Currás-Freixes, M.; et al. The mTORC1 Complex Is Significantly Overactivated in SDHX-Mutated Paragangliomas. Neuroendocrinology 2017, 105, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Tong, A.; Wang, F.; Cui, Y.; Li, C.; Zhang, Y.; Yan, Z. The Roles of PI3K/AKT/mTOR and MAPK/ERK Signaling Pathways in Human Pheochromocytomas. Int. J. Endocrinol. 2016, 2016, 5286972. [Google Scholar] [CrossRef] [PubMed]
- Giubellino, A.; Bullova, P.; Nölting, S.; Turkova, H.; Powers, J.F.; Liu, Q.; Guichard, S.; Tischler, A.S.; Grossman, A.B.; Pacak, K. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: In vitro and in vivo studies in female athymic nude mice. Endocrinology 2013, 154, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Xu, T.; Zhong, S.; Shen, Z. Targeting of mTORC2 may have advantages over selective targeting of mTORC1 in the treatment of malignant pheochromocytoma. Tumor Biol. 2015, 36, 5273–5281. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Qin, L.; Xu, T.; Zhu, Z.; Zhong, S.; Zhang, M.; Shen, Z. The Dual mTORC1 and mTORC2 Inhibitor PP242 Shows Strong Antitumor Activity in a Pheochromocytoma PC12 Cell Tumor Model. Urology 2015, 85, e1–e273. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Zitzmann, K.; von Rüden, J.; Brand, S.; Göke, B.; Lichtl, J.; Spöttl, G.; Auernhammer, C.J. Compensatory activation of Akt in response to mTOR and Raf inhibitors—a rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010, 295, 100–109. [Google Scholar] [CrossRef]
- Nölting, S.; Grossman, A.B. Signaling pathways in pheochromocytomas and paragangliomas: Prospects for future therapies. Endocr. Pathol. 2012, 23, 21–33. [Google Scholar] [CrossRef]
- Saddozai, U.A.K.; Wang, F.; Akbar, M.U.; Zhang, L.; An, Y.; Zhu, W.; Xie, L.; Li, Y.; Ji, X.; Guo, X. Identification of Clinical Relevant Molecular Subtypes of Pheochromocytoma. Front. Endocrinol. 2021, 12, 605797. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Chuk, M.K.; Widemann, B.C.; Minard, C.G.; Liu, X.; Kim, A.; Bernhardt, M.B.; Kudgus, R.A.; Reid, J.M.; Voss, S.D.; Blaney, S.; et al. A phase 1 study of cabozantinib in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1211, a report from the Children’s Oncology Group. Pediatr. Blood Cancer. 2018, 65, e27077. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Centanni, M.; Virili, C.; Miccoli, M.; Ferrari, P.; Ruffilli, I.; Ragusa, F.; Antonelli, A.; Fallahi, P. Sunitinib in the Treatment of Thyroid Cancer. Curr. Med. Chem. 2019, 26, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Tanaka, Y.; Aita, Y.; Ishii, K.-A.; Ikeda, T.; Isobe, K.; Kawakami, Y.; Shimano, H.; Hara, H.; Takekoshi, K. Sunitinib induces apoptosis in pheochromocytoma tumor cells by inhibiting VEGFR2/Akt/mTOR/S6K1 pathways through modulation of Bcl-2 and BAD. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E615–E625. [Google Scholar] [CrossRef]
- Denorme, M.; Yon, L.; Roux, C.; Gonzalez, B.; Baudin, E.; Anouar, Y.; Dubessy, C. Both sunitinib and sorafenib are effective treatments for pheochromocytoma in a xenograft model. Cancer Lett. 2014, 352, 236–244. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Ezzat, S.; Joshua, A.M.; Bourdeau, I.; Leibowitz-Amit, R.; Olney, H.J.; Krzyzanowska, M.; Reuther, D.; Chin, S.; Wang, L.; et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: The SNIPP trial. Br. J. Cancer 2019, 120, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A. New HIF2α inhibitors: Potential implications as therapeutics for advanced pheochromocytomas and paragangliomas. Endocr. Relat. Cancer 2017, 24, C9–C19. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Brière, J.J.; Burnichon, N.; Rivière, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; De Reyniès, A.; Bertherat, J.; Badoual, C.; et al. The Warburg Effect Is Genetically Determined in Inherited Pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar] [CrossRef]
- Morin, A.; Goncalves, J.; Moog, S.; Castro-Vega, L.-J.; Job, S.; Buffet, A.; Fontenille, M.-J.; Woszczyk, J.; Gimenez-Roqueplo, A.-P.; Letouzé, E.; et al. TET-Mediated Hypermethylation Primes SDH-Deficient Cells for HIF2α-Driven Mesenchymal Transition. Cell Rep. 2020, 30, 4551–4566.e7. [Google Scholar] [CrossRef]
- Naing, A.; Meric-Bernstam, F.; Stephen, B.; Karp, D.D.; Hajjar, J.; Ahnert, J.R.; Piha-Paul, S.A.; Colen, R.R.; Jimenez, C.; Raghav, K.P.; et al. Phase 2 study of pembrolizumab in patients with advanced rare cancers. J. Immunother. Cancer 2020, 8, e000347. [Google Scholar] [CrossRef]
- Jimenez, C.; Subbiah, V.; Stephen, B.; Ma, J.; Milton, D.; Xu, M.; Zarifa, A.; Akhmedzhanov, F.O.; Tsimberidou, A.; Habra, M.A.; et al. Phase II Clinical Trial of Pembrolizumab in Patients with Progressive Metastatic Pheochromocytomas and Paragangliomas. Cancers 2020, 12, 2307. [Google Scholar] [CrossRef] [PubMed]
- Woon, L.C.; Xin, L.J.J.; Pin, C.S. Nivolumab for the treatment of hepatocellular carcinoma. Expert Opin. Biol. Ther. 2020, 20, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Fábián, Z.; Töröcsik, B.; Kiss, K.; Csatary, L.K.; Bodey, B.; Tigyi, J.; Csatary, C.; Szeberényi, J. Induction of apoptosis by a Newcastle disease virus vaccine (MTH-68/H) in PC12 rat phaeochromocytoma cells. Anticancer Res. 2001, 21, 125–135. [Google Scholar] [PubMed]
- Szeberényi, J.; Fábián, Z.; Töröcsik, B.; Kiss, K.; Csatary, L.K. Newcastle disease virus-induced apoptosis in PC12 pheochromocytoma cells. Am. J. Ther. 2003, 10, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Papewalis, C.; Kouatchoua, C.; Ehlers, M.; Jacobs, B.; Porwol, D.; Schinner, S.; Willenberg, H.S.; Anlauf, M.; Raffel, A.; Eisenhofer, G.; et al. Chromogranin A as potential target for immunotherapy of malignant pheochromocytoma. Mol. Cell. Endocrinol. 2011, 335, 69–77. [Google Scholar] [CrossRef]
- Gunawardane, P.T.K.; Grossman, A. Phaeochromocytoma and Paraganglioma. Adv. Exp. Med. Biol. 2017, 956, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Corssmit, E.P.M.; Snel, M.; Kapiteijn, E. Malignant pheochromocytoma and paraganglioma: Management options. Curr. Opin. Oncol. 2020, 32, 20–26. Available online: https://journals.lww.com/co-oncology/Fulltext/2020/01000/Malignant_pheochromocytoma_and_paraganglioma_.5.aspx (accessed on 15 February 2022). [CrossRef]
- Kong, G.; Schenberg, T.; Yates, C.J.; Trainer, A.; Sachithanandan, N.; Iravani, A.; Kumar, A.R.; Hofman, M.S.; Akhurst, T.; Michael, M.; et al. The Role of 68Ga-DOTA-Octreotate PET/CT in Follow-Up of SDH-Associated Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2019, 104, 5091–5099. [Google Scholar] [CrossRef]
- Han, S.; Suh, C.H.; Woo, S.; Kim, Y.J.; Lee, J.J. Performance of (68)Ga-DOTA-Conjugated Somatostatin Receptor-Targeting Peptide PET in Detection of Pheochromocytoma and Paraganglioma: A Systematic Review and Metaanalysis. J. Nucl. Med. 2019, 60, 369–376. [Google Scholar] [CrossRef]
- Brown, E.E.; Kumar, S.; Rajji, T.K.; Pollock, B.G.; Mulsant, B.H. Anticipating and Mitigating the Impact of the COVID-19 Pandemic on Alzheimer’s Disease and Related Dementias. Am. J. Geriatr. Psychiatry 2020, 28, 712–721. [Google Scholar] [CrossRef]
- Patt, D.; Gordan, L.; Diaz, M.; Okon, T.; Grady, L.; Harmison, M.; Markward, N.; Sullivan, M.; Peng, J.; Zhou, A. Impact of COVID-19 on Cancer Care: How the Pandemic Is Delaying Cancer Diagnosis and Treatment for American Seniors. JCO Clin. Cancer Inform. 2020, 4, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, M.; Barbi, E.; Apicella, A.; Marchetti, F.; Cardinale, F.; Trobia, G. Delayed access or provision of care in Italy resulting from fear of COVID-19. Lancet Child Adolesc. Health 2020, 4, e10–e11. [Google Scholar] [CrossRef]
- Xie, W.; Yang, X.; Cao, X.; Liu, P. Effects of a comprehensive reservation service for non-emergency registration on appointment registration rate, patient waiting time, patient satisfaction and outpatient volume in a tertiary hospital in China. BMC Health Serv. Res. 2019, 19, 782. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Groß, S.E.; Pfaff, H.; Dresen, A. Waiting time, communication quality, and patient satisfaction: An analysis of moderating influences on the relationship between perceived waiting time and the satisfaction of breast cancer patients during their inpatient stay. Patient Educ. Couns. 2020, 103, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Bleustein, C.; Rothschild, D.B.; Valen, A.; Valatis, E.; Schweitzer, L.; Jones, R. Wait times, patient satisfaction scores, and the perception of care. Am. J. Manag. Care 2014, 20, 393–400. [Google Scholar]
- Lamba, N.; Niemierko, A.; Martinez, R.; Leland, P.; Shih, H.A. The Interaction of Waiting Time and Patient Experience during Radiation Therapy: A Survey of Patients from a Tertiary Cancer Center. J. Med. Imaging Radiat. Sci. 2020, 51, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Westin, A.M.L.; Barksdale, C.L.; Stephan, S.H. The effect of waiting time on youth engagement to evidence based treatments. Community Ment. Health J. 2014, 50, 221–228. [Google Scholar] [CrossRef]
- Treglia, G.; Sadeghi, R.; Giovinazzo, F.; Galiandro, F.; Annunziata, S.; Muoio, B.; Kroiss, A.S. PET with Different Radiopharmaceuticals in Neuroendocrine Neoplasms: An Umbrella Review of Published Meta-Analyses. Cancers 2021, 13, 5172. [Google Scholar] [CrossRef]
- Neumann, H.P.; Young, W.F.J.; Krauss, T.; Bayley, J.-P.; Schiavi, F.; Opocher, G.; Boedeker, C.C.; Tirosh, A.; Castinetti, F.; Ruf, J.; et al. 65 YEARS OF THE DOUBLE HELIX: Genetics informs precision practice in the diagnosis and management of pheochromocytoma. Endocr. Relat. Cancer 2018, 25, T201–T219. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Metoxycatecholamines Tested | Levels of Metoxycatecholamines in 24 h Urine Test | ||
|---|---|---|---|
| January 2020 (µg/Ml) | March 2020 (µg/24 h) | August 2021 (µg/24 h) | |
| Metanephrine | 550 | 88.9 | 65.1 |
| 3-Methoxytyramine | 1047 | 356.5 | 224.8 |
| Normetanephrine | 6902 | 1858.2 | 1180.5 |
| Cluster | Mutated Gene | Frequency of Mutation | Mechanism | Hormonal Activity | Syndrome Name | Preferred Image Diagnostic | Main Localization | Treatment |
|---|---|---|---|---|---|---|---|---|
| Pseudohypoxic cluster | VHL | 7% | Accumulation of HIF-2 α | NE, NMT | VHL | 18F-FDOPA | Abdomen | α or β receptor inhibitors, surgery |
| SDHD | 9% | Accumulation of fumarate and succinate | NE, NMT, DA, 3-MT | PGL 1 | 1st choice: 68Ga-somatostatin analog PET/CT 2nd: F-FDG avidity PET/CT | Head and neck | α or β receptor inhibitors, surgery chemotherapy (cyclophosphamide, vincristine, acarbazine, temozolomide) | |
| SDHAF2 | <1% | PGL 2 | 1st choice: 68Ga-somatostatin analog 2nd: F-FDG avidity | Chest, carotid body | ||||
| SDHC | 0–6.6% | PGL 3 | 1st choice: 68Ga-somatostatin analog 2nd: F-FDG avidity | Chest, head and neck, carotid body | ||||
| SDHB | 10% | PGL 4 | 1st choice: 68Ga-somatostatin analog 2nd: F-FDG avidity | Chest, abdomen | ||||
| SDHA | 3% of sporadic PPGL | PGL 5 | 1st choice: 68Ga-somatostatin analog 2nd: F-FDG avidity | Head and neck, abdomen | ||||
| EGLN1/2/3 | 2 patients | No regulation of the stability of HIF-α by PDH-1,-2,-3 | NE, NMT | – | 18F-FDOPA PET/CT | Abdomen | α or β receptor inhibitors, surgery | |
| HIF2A | 2 patients | Dysregulation of adaptation to hypoxia | NE, NMT | Pacak-Zhuang syndrome | Avid F-FDOPA and F-FDG uptake 18F-FDOPA PET/CT | Abdomen | α or β receptor inhibitors, surgery | |
| IDH | 1 patient | Accumulation of 2-hydroxy, glutarate | NA | NA | NA | NA | α or β receptor inhibitors, surgery | |
| MDH2 | 5 patients | Tumor suppression gene mutations | NE, NMT | NA | NA | Chest, abdomen | α or β receptor inhibitors, surgery | |
| Kinase receptor signaling | RET | 6% | Activation of Ras/MAPK and PI2K/AKT signaling | NE, NMT, EPI, MT, N-methyltransferase | MEN-2 | 18F-FDOPA | Adrenal medulla | α or β receptor inhibitors, surgery |
| FH | NA | Accumulation of fumarate, succinate | NE, NMT | NA | 68Ga-DOTATATE PET/CT | NA | α or β receptor inhibitors, surgery | |
| NF1 | 5–7% | mTOR signaling activation | adrenergic phenotype | NF type 1 | 18F-FDOPA PET-CT | Adrenal | α or β receptor inhibitors, surgery | |
| MAX | 1.1% | Myc signaling activation | NE, NMT | Familial PHEO | 1st choice: 18F-FDOPA PET/CT | Abdomen | α or β receptor inhibitors, surgery | |
| TMEM127 | 2% | mTOR signaling activation | NMT, MT | Familial PHEO | 1st choice: 18F-FDOPA PET/CT | Abdomen | α or β receptor inhibitors, surgery | |
| H-RAS | 5.2% (small group of patients) | Ras mutation | Adrenal, adrenergic phenotype | NA | 1st choice: 18F-FDOPA PET/CT | Adrenal | α or β receptor inhibitors, surgery | |
| K-RAS | NA | NA | 1st choice: 18F-FDOPA PET/CT | Adrenal | α or β receptor inhibitors, surgery | |||
| ATRX | 1 patient | Loss of function of ATRX | Noradrenergic phenotype | NA | NA | Adrenal | α or β receptor inhibitors, surgery | |
| Wnt signaling cluster | CSDE1 | 4 patients | Loss of function of CSDE1 | Adrenal, adrenergic phenotype | NA | NA | Adrenal | α or β receptor inhibitors, surgery |
| MAML3 | NA | Increased Wnt and Hedgehog signaling | NE, NMT, EPI, MT | NA | NA | Adrenal | α or β receptor inhibitors, surgery |
| Recommended Material and Method | Catecholamine | Example of a Reference Norm |
|---|---|---|
| 24 h urine test, HPLC | Noradrenaline | 15–80 µg/24 h |
| 24 h urine test, HPLC | Adrenaline | 0–20 µg/24 h |
| Urine, free metanephrines, spectrophotometrically | Metoxyadrenaline | 0–12 µg/24 h |
| Urine, spectrophotometrically | Vanillinmandelic acid | 0–7.9 mg/24 h |
| Plasma, HPLC | Noradrenaline | 80–498 pg/mL |
| Plasma, HPLC | Adrenaline | 4–83 pg/mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobocki, B.K.; Perdyan, A.; Szot, O.; Rutkowski, J. Management of Pheochromocytomas and Paragangliomas: A Case-Based Review of Clinical Aspects and Perspectives. J. Clin. Med. 2022, 11, 2591. https://doi.org/10.3390/jcm11092591
Sobocki BK, Perdyan A, Szot O, Rutkowski J. Management of Pheochromocytomas and Paragangliomas: A Case-Based Review of Clinical Aspects and Perspectives. Journal of Clinical Medicine. 2022; 11(9):2591. https://doi.org/10.3390/jcm11092591
Chicago/Turabian StyleSobocki, Bartosz Kamil, Adrian Perdyan, Olga Szot, and Jacek Rutkowski. 2022. "Management of Pheochromocytomas and Paragangliomas: A Case-Based Review of Clinical Aspects and Perspectives" Journal of Clinical Medicine 11, no. 9: 2591. https://doi.org/10.3390/jcm11092591
APA StyleSobocki, B. K., Perdyan, A., Szot, O., & Rutkowski, J. (2022). Management of Pheochromocytomas and Paragangliomas: A Case-Based Review of Clinical Aspects and Perspectives. Journal of Clinical Medicine, 11(9), 2591. https://doi.org/10.3390/jcm11092591