
Activation of Sigma-1 Receptor Alleviates ER-Associated Cell Death and Microglia Activation in Traumatically Injured Mice

Abstract
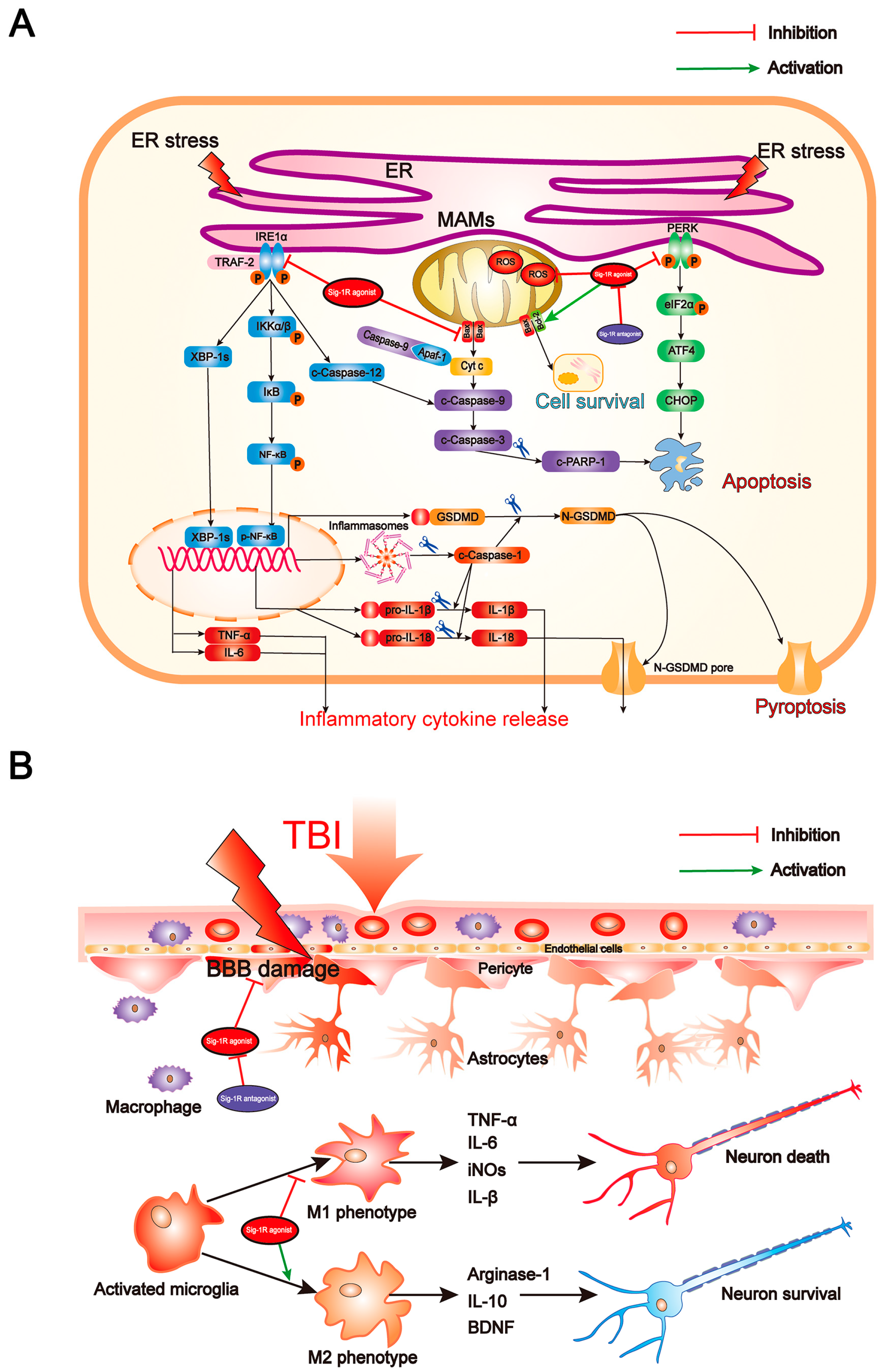
:1. Introduction
2. Materials and Methods
2.1. Human Brain Tissues
2.2. Experimental Design
2.3. Mouse Model of TBI
2.4. Sig-1R Gene Knockout
2.5. Neurological Assessment
2.6. Morris Water Maze
2.7. Measurement of Lesion Volume
2.8. Brain Water Content
2.9. Cerebral Cortical Perfusion
2.10. Evans Blue Extravasation Assay
2.11. Immunofluorescence Staining
2.12. Western Blot Analysis
2.13. Preparation of Single Cells and Mitochondrial Reactive Oxygen Species Content
2.14. Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling (TUNEL) Assay
2.15. Statistical Analysis
3. Results
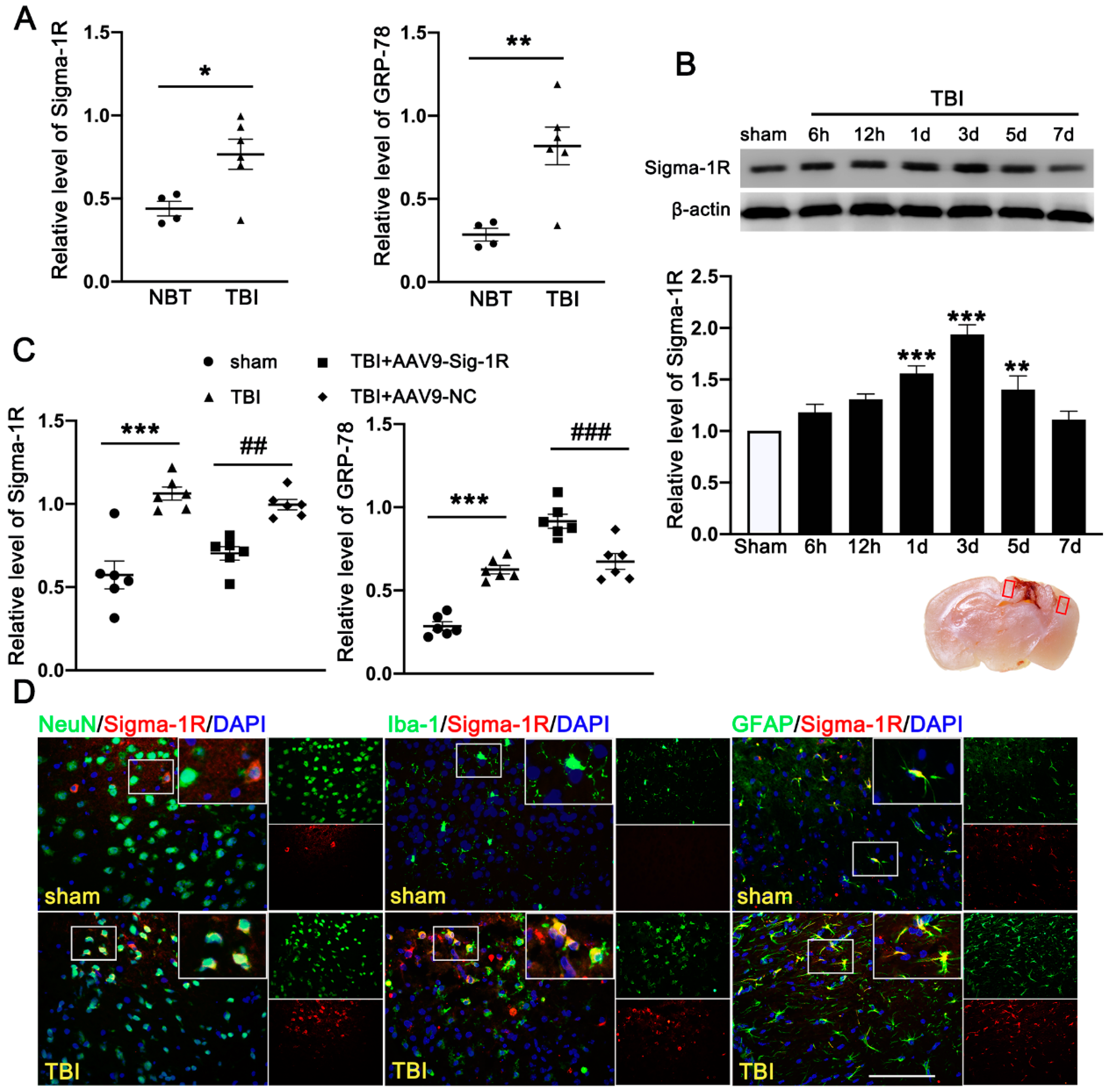
3.1. TBI Upregulated the Expression of Sig-1R and GRP78 in TBI Patients and Mice Subjected to TBI
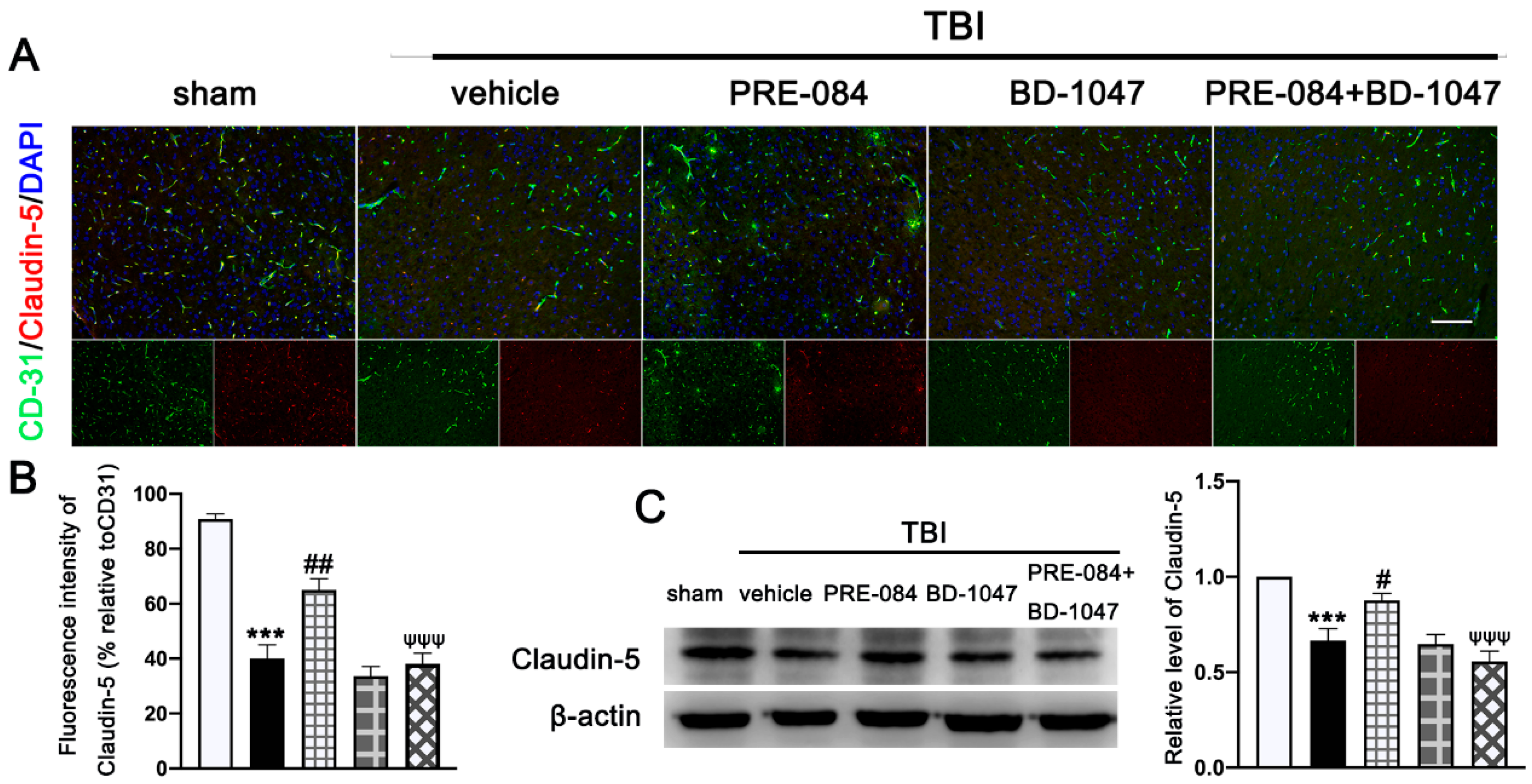
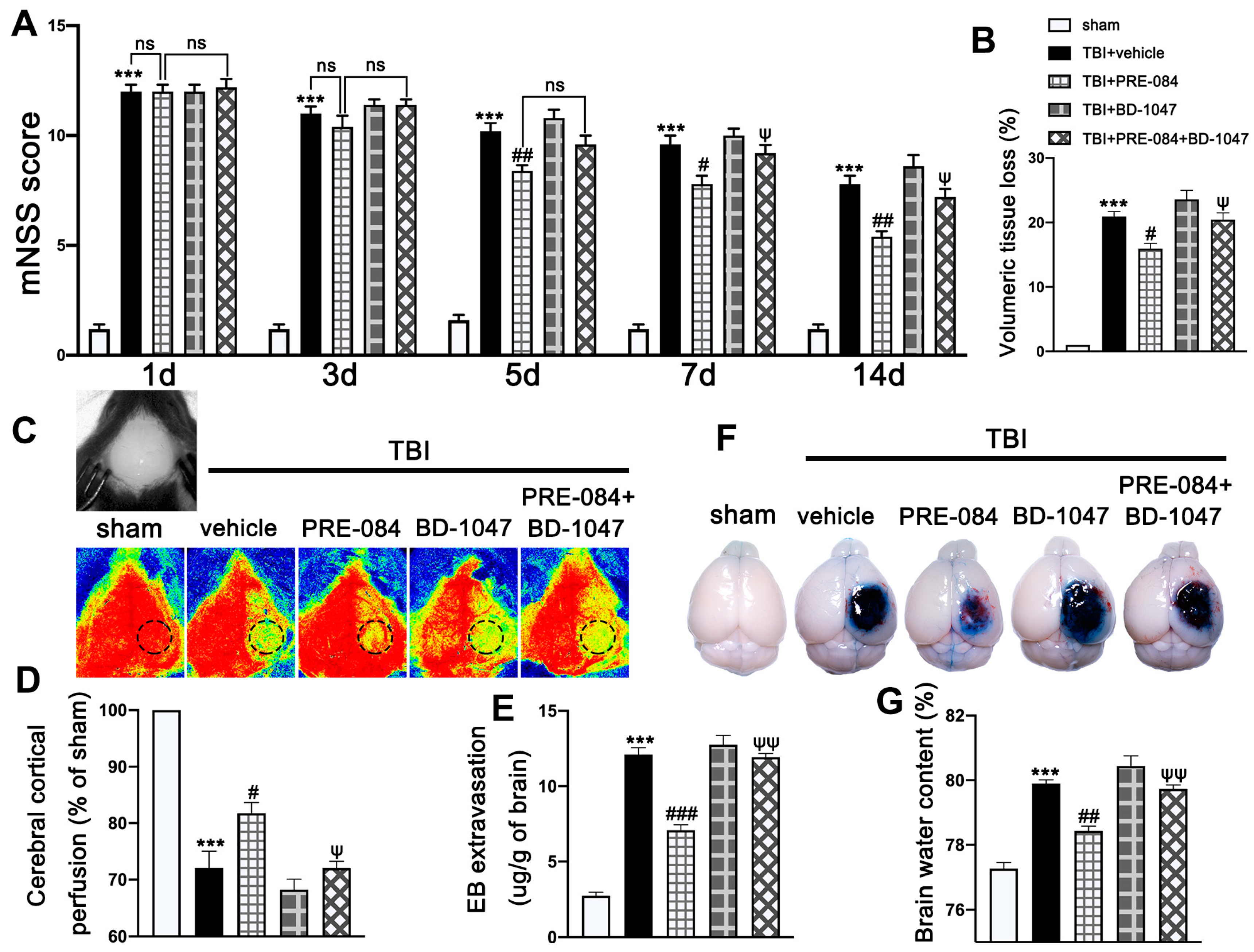
3.2. Activation of Sig-1R Improved Neurological Outcomes and Cerebrovascular Function after TBI
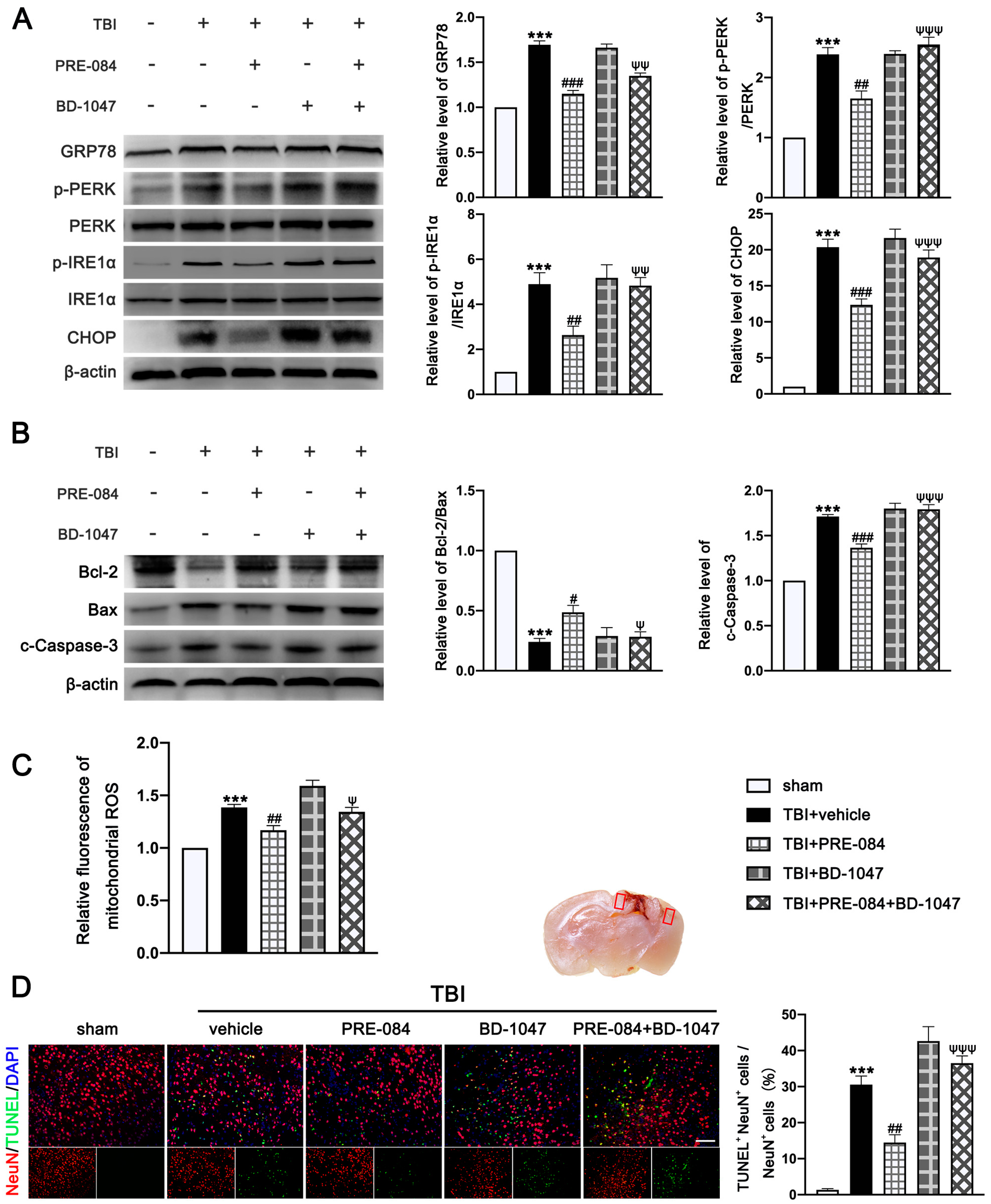
3.3. PRE-084 Suppressed Mitochondrial Dysfunction and ER Stress-Mediated Neuronal Apoptosis after TBI
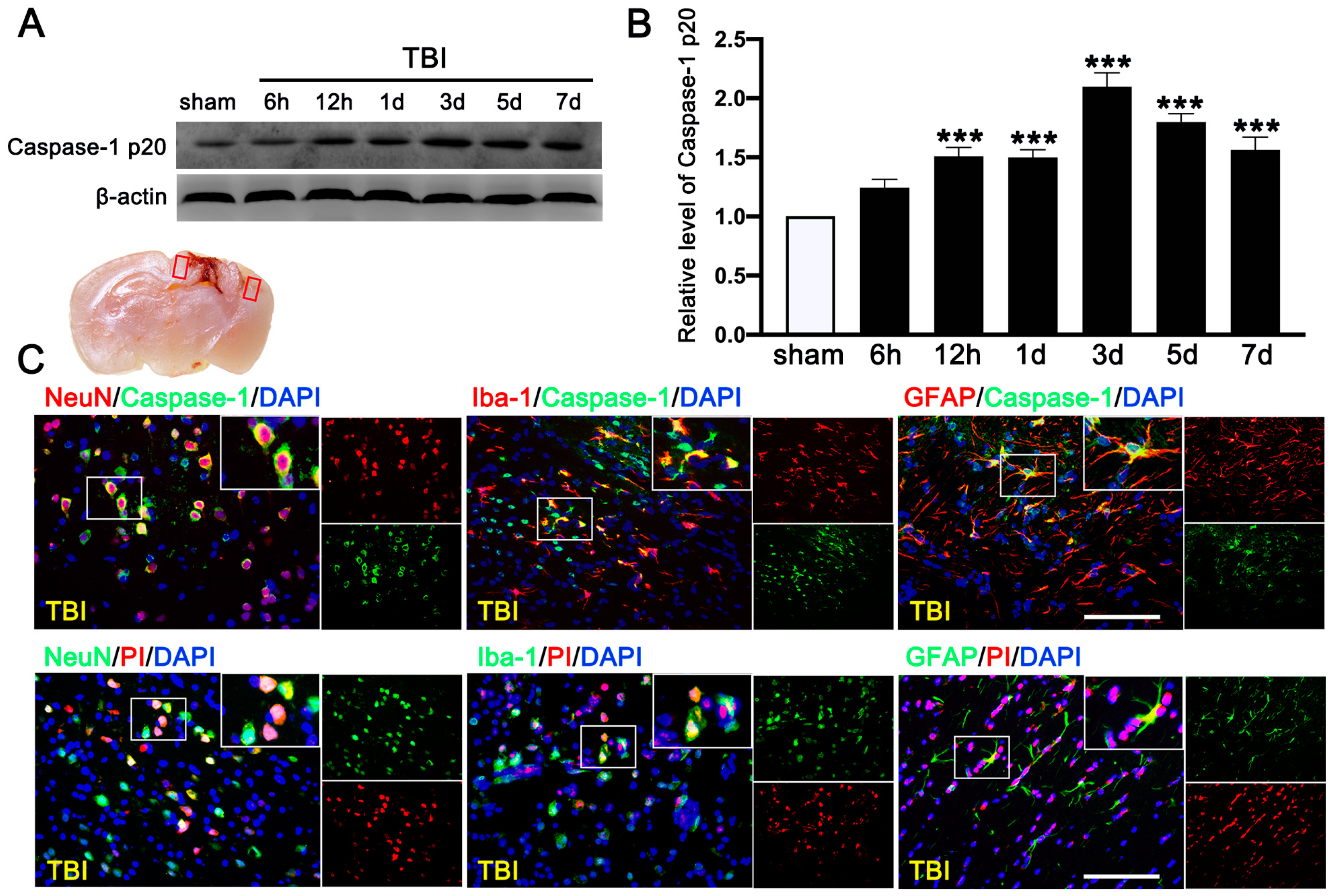
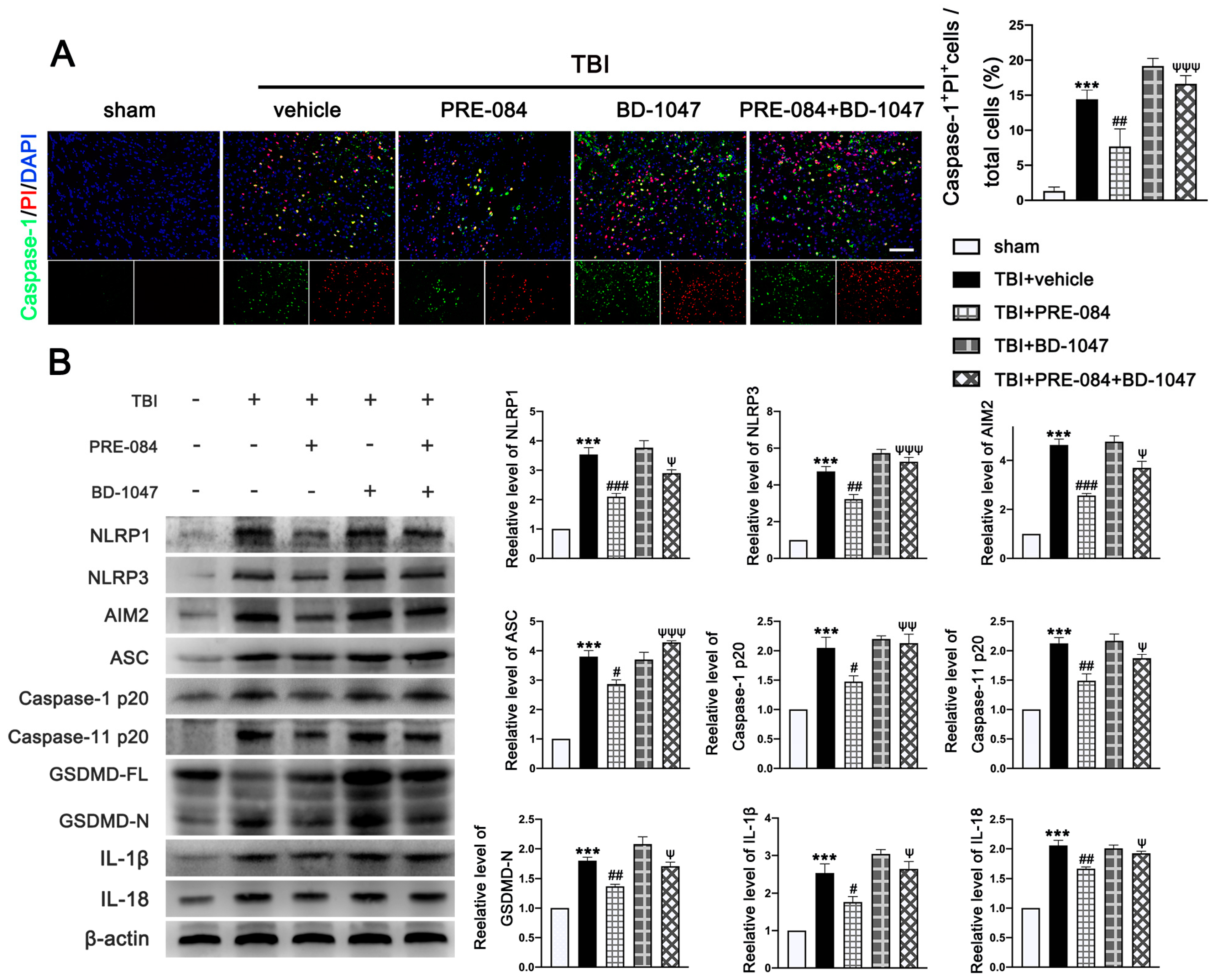
3.4. PRE-084 Attenuated Inflammasome-Mediated Pyroptosis
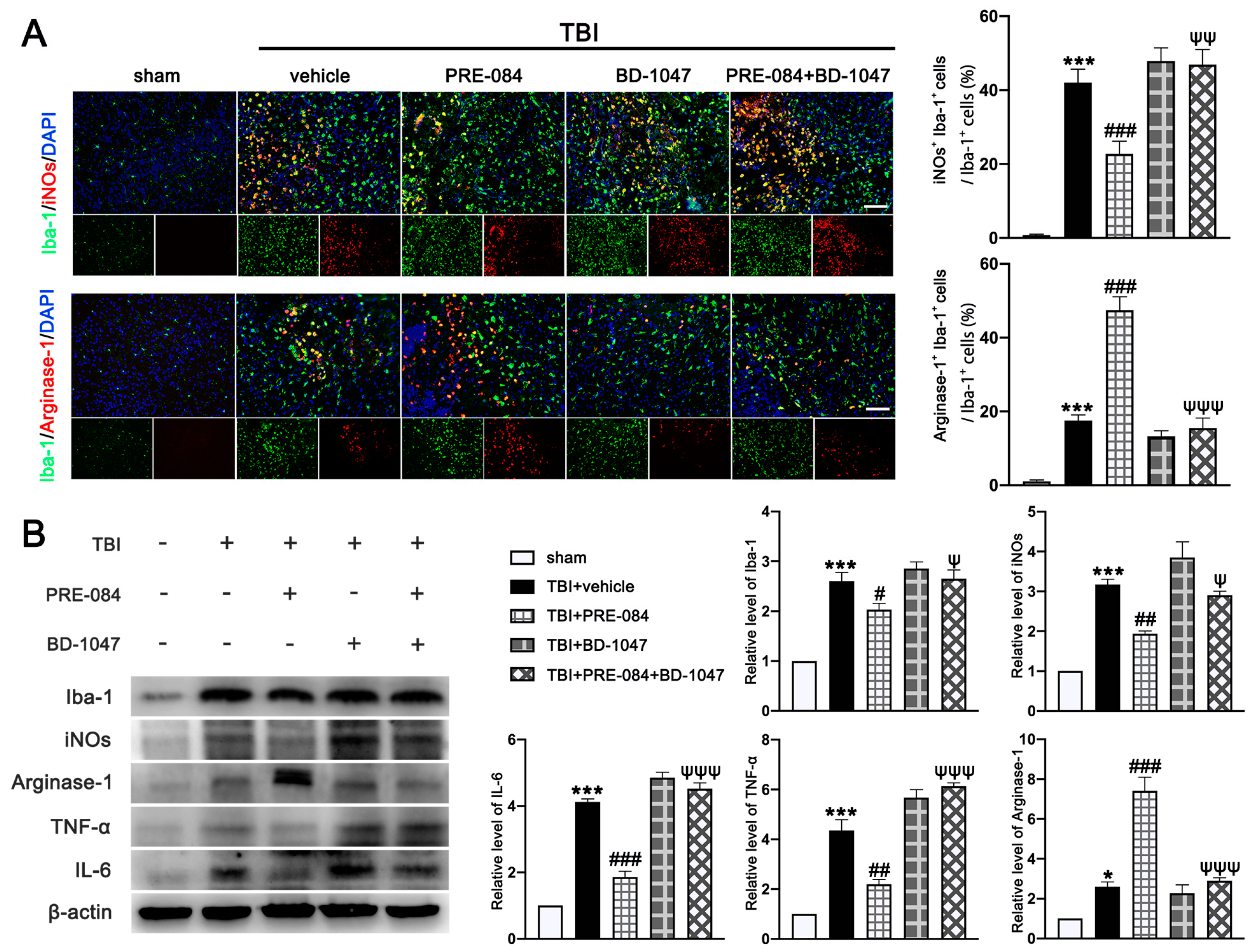
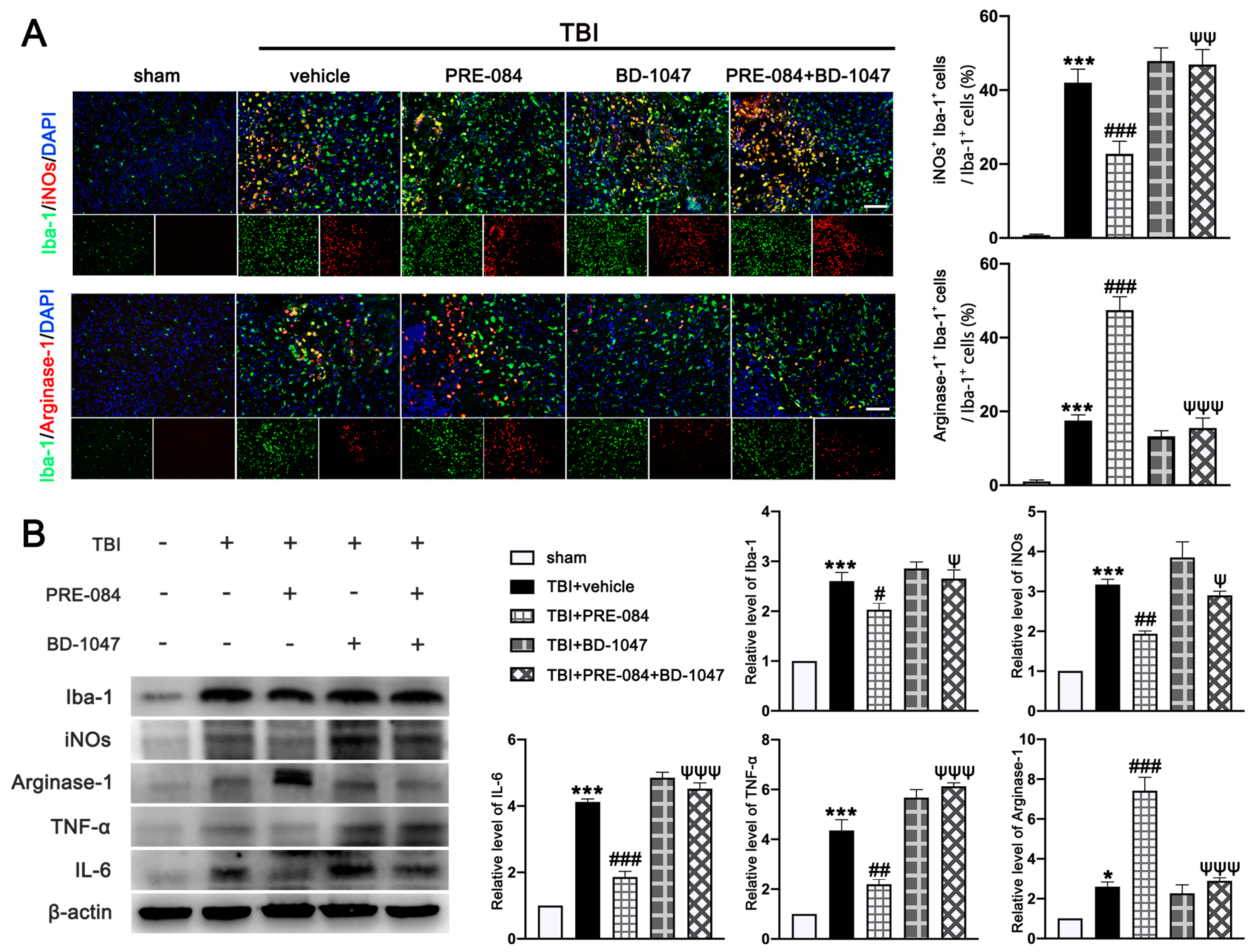
3.5. PRE-084 Promoted Microglia/Macrophages Activation and Inhibited Release of Inflammatory Cytokines following TBI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TBI | Traumatic brain injury |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| Sig-1R | Sigma-1 receptor |
| MAMs | Mitochondria-associated membranes |
| IRE1α | Inositol-requiring enzyme 1α |
| GRP78 | 78 kDa glucose-regulated protein |
| CNS | Central nervous system |
| AD | Alzheimer’s Disease |
| HD | Huntington’s Disease |
| PD | Parkinson’s Disease |
| ALS | Amyotrophic lateral sclerosis |
| IRE1α | Inositol-requiring enzyme 1α |
| AAV9 | Adeno-associated virus serotype 9 |
| mNSS | Modified neurological severity score |
| CCI | Controlled cortical impact |
References
- Chen, X.; Zhang, B.; Chai, Y.; Dong, B.; Lei, P.; Jiang, R.; Zhang, J. Methylprednisolone exacerbates acute critical illness-related corticosteroid insufficiency associated with traumatic brain injury in rats. Brain Res. 2011, 1382, 298–307. [Google Scholar] [CrossRef]
- Raghupathi, R.; Graham, D.I.; McIntosh, T.K. Apoptosis after traumatic brain injury. J. Neurotrauma 2000, 17, 927–938. [Google Scholar] [CrossRef]
- Tehse, J.; Taghibiglou, C. The overlooked aspect of excitotoxicity: Glutamate-independent excitotoxicity in traumatic brain injuries. Eur. J. Neurosci. 2019, 49, 1157–1170. [Google Scholar] [CrossRef]
- Xie, B.S.; Wang, Y.Q.; Lin, Y.; Mao, Q.; Feng, J.F.; Gao, G.Y.; Jiang, J.Y. Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci. Ther. 2019, 25, 465–475. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 inflammasome in traumatic brain injury: Potential as a biomarker and therapeutic target. J. Neuroinflamm. 2020, 17, 104. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Semple, B.D.; Hellewell, S.C.; Bye, N.; Ziebell, J.M. The complexity of neuroinflammation consequent to traumatic brain injury: From research evidence to potential treatments. Acta Neuropathol. 2019, 137, 731–755. [Google Scholar] [CrossRef]
- Janowitz, T.; Menon, D.K. Exploring new routes for neuroprotective drug development in traumatic brain injury. Sci. Transl. Med. 2010, 2, 27rv1. [Google Scholar] [CrossRef]
- Sen, T.; Saha, P.; Gupta, R.; Foley, L.M.; Jiang, T.; Abakumova, O.S.; Hitchens, T.K.; Sen, N. Aberrant ER Stress Induced Neuronal-IFNbeta Elicits White Matter Injury Due to Microglial Activation and T-Cell Infiltration after TBI. J. Neurosci. 2020, 40, 424–446. [Google Scholar] [CrossRef]
- Sun, D.; Gu, G.; Wang, J.; Chai, Y.; Fan, Y.; Yang, M.; Xu, X.; Gao, W.; Li, F.; Yin, D.; et al. Administration of Tauroursodeoxycholic Acid Attenuates Early Brain Injury via Akt Pathway Activation. Front. Cell. Neurosci. 2017, 11, 193. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [PubMed] [Green Version]
- Lahmy, V.; Meunier, J.; Malmstrom, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of Tau hyperphosphorylation and Aβ1_42 generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2013, 38, 1706–1723. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.; Wu, J.; Geva, M.; Kusko, R.; Grossman, I.; Hayden, M.; Bezprozvanny, I. The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease. Neurobiol. Dis. 2017, 97, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francardo, V.; Bez, F.; Wieloch, T.; Nissbrandt, H.; Ruscher, K.; Cenci, M.A. Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 2014, 137, 1998–2014. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Tanaka, H.; Takata, M.; Nagahara, Y.; Noda, Y.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Hara, H. SA4503, a sigma-1 receptor agonist, suppresses motor neuron damage in in vitro and in vivo amyotrophic lateral sclerosis models. Neurosci. Lett. 2014, 559, 174–178. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, L.; Liu, D.; Chi, T.; Ji, X.; Liu, P.; Yang, X.; Tian, X.; Zou, L. Sigma-1 receptor protects against endoplasmic reticulum stress-mediated apoptosis in mice with cerebral ischemia/reperfusion injury. Apoptosis 2019, 24, 157–167. [Google Scholar] [CrossRef]
- Rosen, D.A.; Seki, S.M.; Fernandez-Castaneda, A.; Beiter, R.M.; Eccles, J.D.; Woodfolk, J.A.; Gaultier, A. Modulation of the sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci. Transl. Med. 2019, 11, eaau5266. [Google Scholar] [CrossRef]
- Dong, H.; Ma, Y.; Ren, Z.; Xu, B.; Zhang, Y.; Chen, J.; Yang, B. Sigma-1 Receptor Modulates Neuroinflammation After Traumatic Brain Injury. Cell. Mol. Neurobiol. 2016, 36, 639–645. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yin, D.; Ren, H.; Gao, W.; Li, F.; Sun, D.; Wu, Y.; Zhou, S.; Lyu, L.; Yang, M.; et al. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol. Dis. 2018, 117, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Donkin, J.J.; Nimmo, A.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 1388–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, J.; Gao, X.; Wu, Y.; Gu, G.; Shi, M.; Chai, Y.; Yue, S.; Zhang, J. Tauroursodeoxycholic acid prevents ER stress-induced apoptosis and improves cerebral and vascular function in mice subjected to subarachnoid hemorrhage. Brain Res. 2020, 1727, 146566. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Z.; Chai, Y.; Luo, L.; Jiang, R.; Dong, J.; Zhang, J. Stress-dose hydrocortisone reduces critical illness-related corticosteroid insufficiency associated with severe traumatic brain injury in rats. Crit. Care 2013, 17, R241. [Google Scholar] [CrossRef] [Green Version]
- Allahtavakoli, M.; Jarrott, B. Sigma-1 receptor ligand PRE-084 reduced infarct volume, neurological deficits, pro-inflammatory cytokines and enhanced anti-inflammatory cytokines after embolic stroke in rats. Brain Res. Bull. 2011, 85, 219–224. [Google Scholar] [CrossRef]
- Peviani, M.; Salvaneschi, E.; Bontempi, L.; Petese, A.; Manzo, A.; Rossi, D.; Salmona, M.; Collina, S.; Bigini, P.; Curti, D. Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol. Dis. 2014, 62, 218–232. [Google Scholar] [CrossRef]
- Hu, X.; Chen, H.; Xu, H.; Wu, Y.; Wu, C.; Jia, C.; Li, Y.; Sheng, S.; Xu, C.; Xu, H.; et al. Role of Pyroptosis in Traumatic Brain and Spinal Cord Injuries. Int. J. Biol. Sci. 2020, 16, 2042–2050. [Google Scholar] [CrossRef]
- Chen, G.; Gao, C.; Yan, Y.; Wang, T.; Luo, C.; Zhang, M.; Chen, X.; Tao, L. Inhibiting ER Stress Weakens Neuronal Pyroptosis in a Mouse Acute Hemorrhagic Stroke Model. Mol. Neurobiol. 2020, 57, 5324–5335. [Google Scholar] [CrossRef]
- Le, X.; Mu, J.; Peng, W.; Tang, J.; Xiang, Q.; Tian, S.; Feng, Y.; He, S.; Qiu, Z.; Ren, G.; et al. DNA methylation downregulated ZDHHC1 suppresses tumor growth by altering cellular metabolism and inducing oxidative/ER stress-mediated apoptosis and pyroptosis. Theranostics 2020, 10, 9495–9511. [Google Scholar] [CrossRef]
- Huang, J.; Lu, W.; Doycheva, D.M.; Gamdzyk, M.; Hu, X.; Liu, R.; Zhang, J.H.; Tang, J. IRE1alpha inhibition attenuates neuronal pyroptosis via miR-125/NLRP1 pathway in a neonatal hypoxic-ischemic encephalopathy rat model. J. Neuroinflamm. 2020, 17, 152. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K. Multiple roles of macrophage in skin. J. Derm. Sci. 2021, 104, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef]
- Liberale, L.; Dallegri, F.; Montecucco, F.; Carbone, F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb. Haemost. 2017, 117, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Dun, Y.; Thangaraju, M.; Duplantier, J.; Dong, Z.; Liu, K.; Ganapathy, V.; Smith, S.B. Sigma receptor 1 modulates endoplasmic reticulum stress in retinal neurons. Investig. Ophthalmol. Vis. Sci. 2011, 52, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Morihara, R.; Yamashita, T.; Liu, X.; Nakano, Y.; Fukui, Y.; Sato, K.; Ohta, Y.; Hishikawa, N.; Shang, J.; Abe, K. Protective effect of a novel sigma-1 receptor agonist is associated with reduced endoplasmic reticulum stress in stroke male mice. J. Neurosci. Res. 2018, 96, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Omi, T.; Tanimukai, H.; Kanayama, D.; Sakagami, Y.; Tagami, S.; Okochi, M.; Morihara, T.; Sato, M.; Yanagida, K.; Kitasyoji, A.; et al. Fluvoxamine alleviates ER stress via induction of Sigma-1 receptor. Cell Death Dis. 2014, 5, e1332. [Google Scholar] [CrossRef]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age | Gender | Cause of Injury | Other Injuries | Time Post- Injury (h) | Region of Surgery | GCS |
|---|---|---|---|---|---|---|---|
| Traumatic brain injury | 40 | Male | Falling injury | None | 20 | Left temporal lobe | 6 |
| Traumatic brain injury | 62 | Male | Traffic accident | None | 16 | Right temporal lobe | 8 |
| Traumatic brain injury | 37 | Female | Traffic accident | None | 18 | Right temporal lobe | 6 |
| Traumatic brain injury | 75 | Male | Traffic accident | None | 25 | Right parietal lobe | 5 |
| Traumatic brain injury | 59 | Male | Traffic accident | None | 29 | Left frontal lobe | 8 |
| Traumatic brain injury | 66 | Female | Struck by object | None | 22 | Left parietal lobe | 7 |
| Arteriovenous malformation | 48 | Male | - | None | - | Right temporal lobe | - |
| Arteriovenous malformation | 33 | Female | - | None | - | Right parietal lobe | - |
| Arteriovenous malformation | 42 | Female | - | None | - | Left temporal lobe | - |
| Arteriovenous malformation | 38 | Male | - | None | - | Right frontal lobe | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, M.; Liu, L.; Min, X.; Mi, L.; Chai, Y.; Chen, F.; Wang, J.; Yue, S.; Zhang, J.; Deng, Q.; et al. Activation of Sigma-1 Receptor Alleviates ER-Associated Cell Death and Microglia Activation in Traumatically Injured Mice. J. Clin. Med. 2022, 11, 2348. https://doi.org/10.3390/jcm11092348
Shi M, Liu L, Min X, Mi L, Chai Y, Chen F, Wang J, Yue S, Zhang J, Deng Q, et al. Activation of Sigma-1 Receptor Alleviates ER-Associated Cell Death and Microglia Activation in Traumatically Injured Mice. Journal of Clinical Medicine. 2022; 11(9):2348. https://doi.org/10.3390/jcm11092348
Chicago/Turabian StyleShi, Mingming, Liang Liu, Xiaobin Min, Liang Mi, Yan Chai, Fanglian Chen, Jianhao Wang, Shuyuan Yue, Jianning Zhang, Quanjun Deng, and et al. 2022. "Activation of Sigma-1 Receptor Alleviates ER-Associated Cell Death and Microglia Activation in Traumatically Injured Mice" Journal of Clinical Medicine 11, no. 9: 2348. https://doi.org/10.3390/jcm11092348
APA StyleShi, M., Liu, L., Min, X., Mi, L., Chai, Y., Chen, F., Wang, J., Yue, S., Zhang, J., Deng, Q., & Chen, X. (2022). Activation of Sigma-1 Receptor Alleviates ER-Associated Cell Death and Microglia Activation in Traumatically Injured Mice. Journal of Clinical Medicine, 11(9), 2348. https://doi.org/10.3390/jcm11092348