
Factors Modulating COVID-19: A Mechanistic Understanding Based on the Adverse Outcome Pathway Framework

 ,
,  , , ,
, , ,  , ,
, ,  , , , ,
, , , ,
Abstract
1. Introduction
2. Factors Modulating COVID-19: Epidemiological and Clinical Data
2.1. Biological (Intrinsic)
2.2. Pre-Existing Co-Morbidities
2.3. Lifestyle Factors
2.4. Environmental Factors
2.5. Therapeutic Intervention against COVID-19
3. Factors Modulating COVID-19: A Mechanistic Understanding via the AOP
3.1. Key Event Relationships Related to Initial Viral Infection
3.1.1. Binding to ACE2 Leads to Viral Entry and Coronavirus Production (KER2056-KER2310)
3.1.2. S-Protein Binding to ACE2 Induces ACE2 Dysregulation (KER2311)
3.2. Key Event Relationships Related to Central Inflammatory Processes
3.2.1. Decreased Fibrinolysis Increases Secretion of Proinflammatory Mediators (KER2356)
3.2.2. TLR Dysregulation Increases Secretion of Proinflammatory Mediators (KER2303)
3.2.3. Excessive Secretion of Proinflammatory Mediators and Accumulated Recruitment of Inflammatory Cells Lead to Hyperinflammation (KER1703-KER2354)
3.2.4. Increased Intestinal Permeability Fuels Hyperinflammation (KER2495)
4. Lessons Learned
4.1. Identification of Knowledge Gaps Guiding Further Research
4.2. Biomarkers Related to MFs as Predictive Tools
4.3. Risk Factors Impact on Therapeutic Interventions Effects
4.4. Interplay between Factors Modulating COVID-19
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed 2020, 91, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Nymark, P.; Sachana, M.; Leite, S.B.; Sund, J.; Krebs, C.E.; Sullivan, K.; Edwards, S.; Viviani, L.; Willett, C.; Landesmann, B.; et al. Systematic Organization of COVID-19 Data Supported by the Adverse Outcome Pathway Framework. Front. Public Health 2021, 9, 638605. [Google Scholar] [CrossRef] [PubMed]
- Wittwehr, C.; Amorim, M.J.; Clerbaux, L.A.; Krebs, C.; Landesmann, B.; Macmillan, D.S.; Nymark, P.; Ram, R.; Garcia-Reyero, N.; Sachana, M.; et al. Understanding COVID-19 through adverse outcome pathways—2nd CIAO AOP Design Workshop. ALTEX 2021, 38, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. COVID-19 and the liver: An adverse outcome pathway perspective. Toxicology 2021, 455, 152765. [Google Scholar] [CrossRef]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar] [CrossRef]
- Villeneuve, D.L.; Crump, D.; Garcia-Reyero, N.; Hecker, M.; Hutchinson, T.H.; LaLone, C.A.; Landesmann, B.; Lettieri, T.; Munn, S.; Nepelska, M.; et al. Adverse outcome pathway (AOP) development I: Strategies and principles. Toxicol. Sci. 2014, 142, 312–320. [Google Scholar] [CrossRef]
- Leist, M.; Ghallab, A.; Graepel, R.; Marchan, R.; Hassan, R.; Bennekou, S.H.; Limonciel, A.; Vinken, M.; Schildknecht, S.; Waldmann, T.; et al. Adverse outcome pathways: Opportunities, limitations and open questions. Arch. Toxicol. 2017, 91, 3477–3505. [Google Scholar] [CrossRef]
- Marshall, L.J.; Austin, C.P.; Casey, W.; Fitzpatrick, S.C.; Willett, C. Recommendations toward a human pathway-based approach to disease research. Drug Discov. Today 2018, 23, 1824–1832. [Google Scholar] [CrossRef]
- AOP-Wiki. Available online: https://aopwiki.org/ (accessed on 16 March 2022).
- Kleinstreuer, N.C.; Sullivan, K.; Allen, D.; Edwards, S.; Mendrick, D.L.; Embry, M.; Matheson, J.; Rowlands, J.C.; Munn, S.; Maull, E.; et al. Adverse outcome pathways: From research to regulation scientific workshop report. Regul. Toxicol. Pharmacol. 2016, 76, 39–50. [Google Scholar] [CrossRef]
- Terron, A.; Bal-Price, A.; Paini, A.; Monnet-Tschudi, F.; Bennekou, S.H.; Members, E.W.E.; Leist, M.; Schildknecht, S. An adverse outcome pathway for parkinsonian motor deficits associated with mitochondrial complex I inhibition. Arch. Toxicol. 2018, 92, 41–82. [Google Scholar] [CrossRef]
- Korn, D.; Thieme, A.J.; Alves, V.M.; Yeakey, M.; Borba, J.; Capuzzi, S.J.; Fecho, K.; Bizon, C.; Edwards, S.W.; Chirkova, R.; et al. Defining clinical outcome pathways. Drug Discov. Today 2022, 27, 1671–1678. [Google Scholar] [CrossRef]
- Kim, Y.; Park, C.G.; Lim, S.R.; Jun, I.; Lee, Y.O. Advanced Adverse Outcome Pathways Potentially Bridging the Pathogenesis of COVID-19. Preprints 2021. [Google Scholar] [CrossRef]
- Modelling the Pathogenesis of COVID-19 Using the Adverse Outcome Pathway Framework. Available online: www.ciao-covid.net (accessed on 16 March 2022).
- OECD. Users’ Handbook Supplement to the Guidance Document for Developing and Assessing Adverse Outcome Pathways. 2018. Available online: https://www.oecd-ilibrary.org/docserver/5jlv1m9d1g32-en.pdf?expires=1659523666&id=id&accname=guest&checksum=A77C7633D48B3B36547F25CC76CAE409 (accessed on 16 March 2022).
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; the Northwell COVID-19 Research Consortium. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Jordan, R.E.; Adab, P.; Cheng, K.K. Covid-19: Risk factors for severe disease and death. BMJ 2020, 368, m1198. [Google Scholar] [CrossRef]
- Farshbafnadi, M.; Kamali Zonouzi, S.; Sabahi, M.; Dolatshahi, M.; Aarabi, M.H. Aging & COVID-19 susceptibility, disease severity, and clinical outcomes: The role of entangled risk factors. Exp. Gerontol. 2021, 154, 111507. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef]
- Cohen, J.F.; Korevaar, D.A.; Matczak, S.; Chalumeau, M.; Allali, S.; Toubiana, J. COVID-19-Related Fatalities and Intensive-Care-Unit Admissions by Age Groups in Europe: A Meta-Analysis. Front. Med. 2020, 7, 560685. [Google Scholar] [CrossRef]
- Sepulveda, E.R.; Stall, N.M.; Sinha, S.K. A Comparison of COVID-19 Mortality Rates Among Long-Term Care Residents in 12 OECD Countries. J. Am. Med. Dir. Assoc. 2020, 21, 1572–1574.e1573. [Google Scholar] [CrossRef]
- ECDC Public Health Emergency Team; Danis, K.; Fonteneau, L.; Georges, S.; Daniau, C.; Bernard-Stoecklin, S.; Domegan, L.; O‘Donnell, J.; Hauge, S.H.; Dequeker, S.; et al. High impact of COVID-19 in long-term care facilities, suggestion for monitoring in the EU/EEA, May 2020. Eurosurveillance 2020, 25, 2000956. [Google Scholar] [CrossRef]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A.; Axfors, C.; Contopoulos-Ioannidis, D.G. Population-level COVID-19 mortality risk for non-elderly individuals overall and for non-elderly individuals without underlying diseases in pandemic epicenters. Environ. Res. 2020, 188, 109890. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Peckham, H.; de Gruijter, N.M.; Raine, C.; Radziszewska, A.; Ciurtin, C.; Wedderburn, L.R.; Rosser, E.C.; Webb, K.; Deakin, C.T. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun. 2020, 11, 6317. [Google Scholar] [CrossRef] [PubMed]
- The COVID-19 Sex-Disaggregated Data Tracker. Available online: https://globalhealth5050.org/the-sex-gender-and-covid-19-project/the-data-tracker/ (accessed on 16 March 2022).
- Takahashi, T.; Iwasaki, A. Sex differences in immune responses. Science 2021, 371, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Pathak, G.A.; Singh, K.; Miller-Fleming, T.W.; Wendt, F.R.; Ehsan, N.; Hou, K.; Johnson, R.; Lu, Z.; Gopalan, S.; Yengo, L.; et al. Integrative analyses identify susceptibility genes underlying COVID-19 hospitalization. medRxiv 2020. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Y.; Li, X.; Li, W.; Liu, X.; Xue, X. The Impact of ACE2 Polymorphisms on COVID-19 Disease: Susceptibility, Severity, and Therapy. Front. Cell Infect. Microbiol. 2021, 11, 753721. [Google Scholar] [CrossRef]
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 475. [Google Scholar] [CrossRef]
- Solanich, X.; Vargas-Parra, G.; van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Antoli, A.; Del Valle, J.; Rocamora-Blanch, G.; Setien, F.; Esteller, M.; et al. Genetic Screening for TLR7 Variants in Young and Previously Healthy Men With Severe COVID-19. Front. Immunol. 2021, 12, 719115. [Google Scholar] [CrossRef]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef]
- Jacob, C.O. On the genetics and immunopathogenesis of COVID-19. Clin. Immunol. 2020, 220, 108591. [Google Scholar] [CrossRef]
- Tal, Y.; Adini, A.; Eran, A.; Adini, I. Racial disparity in Covid-19 mortality rates—A plausible explanation. Clin. Immunol. 2020, 217, 108481. [Google Scholar] [CrossRef]
- Bezemer, G.F.G.; Garssen, J. TLR9 and COVID-19: A Multidisciplinary Theory of a Multifaceted Therapeutic Target. Front. Pharmacol. 2020, 11, 601685. [Google Scholar] [CrossRef]
- Chatzi, M.; Papanikolaou, J.; Makris, D.; Papathanasiou, I.; Tsezou, A.; Karvouniaris, M.; Zakynthinos, E. Toll-like receptor 2, 4 and 9 polymorphisms and their association with ICU-acquired infections in Central Greece. J. Crit. Care 2018, 47, 1–8. [Google Scholar] [CrossRef]
- Zhao, P.; Lu, G.; Cai, L. Polymorphisms in the toll-like receptor 9 gene associated with sepsis and multiple organ dysfunction after major blunt trauma (Br J Surg 2011; 98: 1252–1259). Br. J. Surg. 2012, 99, 145, author reply 145. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef]
- Ji, W.; Huh, K.; Kang, M.; Hong, J.; Bae, G.H.; Lee, R.; Na, Y.; Choi, H.; Gong, S.Y.; Choi, Y.H.; et al. Effect of Underlying Comorbidities on the Infection and Severity of COVID-19 in Korea: A Nationwide Case-Control Study. J. Korean Med. Sci. 2020, 35, e237. [Google Scholar] [CrossRef]
- Choi, G.J.; Kim, H.M.; Kang, H. The Potential Role of Dyslipidemia in COVID-19 Severity: An Umbrella Review of Systematic Reviews. J. Lipid Atheroscler. 2020, 9, 435–448. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, K.; Misra-Hebert, A.; Bena, J.; Kashyap, S.R. Impact of Metabolic Syndrome on Severity of COVID-19 Illness. Metab. Syndr. Relat. Disord. 2022, 20, 191–198. [Google Scholar] [CrossRef]
- Ghoneim, S.; Butt, M.U.; Hamid, O.; Shah, A.; Asaad, I. The incidence of COVID-19 in patients with metabolic syndrome and non-alcoholic steatohepatitis: A population-based study. Metabol. Open 2020, 8, 100057. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, D.; Wu, L.; He, G.; Ye, W. Declined serum high density lipoprotein cholesterol is associated with the severity of COVID-19 infection. Clin. Chim. Acta 2020, 510, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Chen, D.; Wu, L.; He, G.; Ye, W. Low Serum Cholesterol Level Among Patients with COVID-19 Infection in Wenzhou, China. SSRN Electron. J. 2020. [Google Scholar] [CrossRef]
- Masana, L.; Correig, E.; Ibarretxe, D.; Anoro, E.; Arroyo, J.A.; Jerico, C.; Guerrero, C.; Miret, M.; Naf, S.; Pardo, A.; et al. Low HDL and high triglycerides predict COVID-19 severity. Sci. Rep. 2021, 11, 7217. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Q.; Zhao, X.; Dong, H.; Wu, C.; Wu, F.; Yu, B.; Lv, J.; Zhang, S.; Wu, G.; et al. Low high-density lipoprotein level is correlated with the severity of COVID-19 patients: An observational study. Lipids Health Dis. 2020, 19, 204. [Google Scholar] [CrossRef]
- Hariyanto, T.I.; Kurniawan, A. Dyslipidemia is associated with severe coronavirus disease 2019 (COVID-19) infection. Diabetes Metab. Syndr. 2020, 14, 1463–1465. [Google Scholar] [CrossRef]
- Ho, J.S.Y.; Fernando, D.I.; Chan, M.Y.; Sia, C.H. Obesity in COVID-19: A Systematic Review and Meta-analysis. Ann. Acad. Med. Singap. 2020, 49, 996–1008. [Google Scholar] [CrossRef]
- Demeulemeester, F.; de Punder, K.; van Heijningen, M.; van Doesburg, F. Obesity as a Risk Factor for Severe COVID-19 and Complications: A Review. Cells 2021, 10, 933. [Google Scholar] [CrossRef]
- Gao, M.; Piernas, C.; Astbury, N.M.; Hippisley-Cox, J.; O’Rahilly, S.; Aveyard, P.; Jebb, S.A. Associations between body-mass index and COVID-19 severity in 6·9 million people in England: A prospective, community-based, cohort study. Lancet Diabetes Endocrinol. 2021, 9, 350–359. [Google Scholar] [CrossRef]
- Molina-Mora, J.A.; Gonzalez, A.; Jimenez-Morgan, S.; Cordero-Laurent, E.; Brenes, H.; Soto-Garita, C.; Sequeira-Soto, J.; Duarte-Martinez, F. Clinical Profiles at the Time of Diagnosis of SARS-CoV-2 Infection in Costa Rica During the Pre-vaccination Period Using a Machine Learning Approach. Phenomics 2022, 1–11. [Google Scholar] [CrossRef]
- Verity, R.; Okell, L.C.; Dorigatti, I.; Winskill, P.; Whittaker, C.; Imai, N.; Cuomo-Dannenburg, G.; Thompson, H.; Walker, P.G.T.; Fu, H.; et al. Estimates of the severity of coronavirus disease 2019: A model-based analysis. Lancet Infect. Dis. 2020, 20, 669–677. [Google Scholar] [CrossRef]
- Inciardi, R.M.; Adamo, M.; Lupi, L.; Cani, D.S.; Di Pasquale, M.; Tomasoni, D.; Italia, L.; Zaccone, G.; Tedino, C.; Fabbricatore, D.; et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur. Heart J. 2020, 41, 1821–1829. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Tomasoni, D.; Inciardi, R.M.; Lombardi, C.M.; Tedino, C.; Agostoni, P.; Ameri, P.; Barbieri, L.; Bellasi, A.; Camporotondo, R.; Canale, C.; et al. Impact of heart failure on the clinical course and outcomes of patients hospitalized for COVID-19. Results of the Cardio-COVID-Italy multicentre study. Eur. J. Heart Fail. 2020, 22, 2238–2247. [Google Scholar] [CrossRef]
- Belarte-Tornero, L.C.; Valdivielso-More, S.; Vicente Elcano, M.; Sole-Gonzalez, E.; Ruiz-Bustillo, S.; Calvo-Fernandez, A.; Subinara, I.; Cabero, P.; Soler, C.; Cubero-Gallego, H.; et al. Prognostic Implications of Chronic Heart Failure and Utility of NT-proBNP Levels in Heart Failure Patients with SARS-CoV-2 Infection. J. Clin. Med. 2021, 10, 323. [Google Scholar] [CrossRef]
- Lala, A.; Johnson, K.W.; Januzzi, J.L.; Russak, A.J.; Paranjpe, I.; Richter, F.; Zhao, S.; Somani, S.; Van Vleck, T.; Vaid, A.; et al. Prevalence and Impact of Myocardial Injury in Patients Hospitalized With COVID-19 Infection. J. Am. Coll. Cardiol. 2020, 76, 533–546. [Google Scholar] [CrossRef]
- Yonas, E.; Alwi, I.; Pranata, R.; Huang, I.; Lim, M.A.; Gutierrez, E.J.; Yamin, M.; Siswanto, B.B.; Virani, S.S. Effect of heart failure on the outcome of COVID-19—A meta analysis and systematic review. Am. J. Emerg. Med. 2021, 46, 204–211. [Google Scholar] [CrossRef]
- Sokol, H.; Contreras, V.; Maisonnasse, P.; Desmons, A.; Delache, B.; Sencio, V.; Machelart, A.; Brisebarre, A.; Humbert, L.; Deryuter, L.; et al. SARS-CoV-2 infection in nonhuman primates alters the composition and functional activity of the gut microbiota. Gut Microbes 2021, 13, 1893113. [Google Scholar] [CrossRef]
- Sencio, V.; Machelart, A.; Robil, C.; Benech, N.; Hoffmann, E.; Galbert, C.; Deryuter, L.; Heumel, S.; Hantute-Ghesquier, A.; Flourens, A.; et al. Alteration of the gut microbiota following SARS-CoV-2 infection correlates with disease severity in hamsters. Gut Microbes 2022, 14, 2018900. [Google Scholar] [CrossRef]
- Venzon, M.; Bernard-Raichon, L.; Klein, J.; Axelrad, J.; Hussey, G.; Sullivan, A.; Casanovas-Massana, A.; Noval, M.; Valero-Jimenez, A.; Gago, J.; et al. Gut microbiome dysbiosis during COVID-19 is associated with increased risk for bacteremia and microbial translocation. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.; Tso, E.Y.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K.; et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 2021, 70, 276–284. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients with COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef]
- Zhang, F.; Wan, Y.; Zuo, T.; Yeoh, Y.K.; Liu, Q.; Zhang, L.; Zhan, H.; Lu, W.; Xu, W.; Lui, G.C.Y.; et al. Prolonged Impairment of Short-Chain Fatty Acid and L-Isoleucine Biosynthesis in Gut Microbiome in Patients With COVID-19. Gastroenterology 2022, 162, 548–561.e544. [Google Scholar] [CrossRef]
- Moreira-Rosario, A.; Marques, C.; Pinheiro, H.; Araujo, J.R.; Ribeiro, P.; Rocha, R.; Mota, I.; Pestana, D.; Ribeiro, R.; Pereira, A.; et al. Gut Microbiota Diversity and C-Reactive Protein Are Predictors of Disease Severity in COVID-19 Patients. Front. Microbiol. 2021, 12, 705020. [Google Scholar] [CrossRef]
- Sarkar, A.; Harty, S.; Moeller, A.H.; Klein, S.L.; Erdman, S.E.; Friston, K.J.; Carmody, R.N. The gut microbiome as a biomarker of differential susceptibility to SARS-CoV-2. Trends Mol. Med. 2021, 27, 1115–1134. [Google Scholar] [CrossRef]
- Bousquet, J.; Anto, J.M.; Czarlewski, W.; Haahtela, T.; Fonseca, S.C.; Iaccarino, G.; Blain, H.; Vidal, A.; Sheikh, A.; Akdis, C.A.; et al. Cabbage and fermented vegetables: From death rate heterogeneity in countries to candidates for mitigation strategies of severe COVID-19. Allergy 2021, 76, 735–750. [Google Scholar] [CrossRef]
- Losso, J.N.; Losso, M.N.; Toc, M.; Inungu, J.N.; Finley, J.W. The Young Age and Plant-Based Diet Hypothesis for Low SARS-CoV-2 Infection and COVID-19 Pandemic in Sub-Saharan Africa. Plant Foods Hum. Nutr. 2021, 76, 270–280. [Google Scholar] [CrossRef]
- Bousquet, J.; Anto, J.M.; Iaccarino, G.; Czarlewski, W.; Haahtela, T.; Anto, A.; Akdis, C.A.; Blain, H.; Canonica, G.W.; Cardona, V.; et al. Is diet partly responsible for differences in COVID-19 death rates between and within countries? Clin. Transl. Allergy 2020, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Ponzo, V.; Pellegrini, M.; D’Eusebio, C.; Bioletto, F.; Goitre, I.; Buscemi, S.; Frea, S.; Ghigo, E.; Bo, S. Mediterranean Diet and SARS-COV-2 Infection: Is There Any Association? A Proof-of-Concept Study. Nutrients 2021, 13, 1721. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.W.; Roberts, A.P.; Fruge, A.D. Negative Association Between Mediterranean Diet Adherence and COVID-19 Cases and Related Deaths in Spain and 23 OECD Countries: An Ecological Study. Front. Nutr. 2021, 8, 591964. [Google Scholar] [CrossRef] [PubMed]
- Merino, J.; Joshi, A.D.; Nguyen, L.H.; Leeming, E.R.; Mazidi, M.; Drew, D.A.; Gibson, R.; Graham, M.S.; Lo, C.H.; Capdevila, J.; et al. Diet quality and risk and severity of COVID-19: A prospective cohort study. Gut 2021, 70, 2096–2104. [Google Scholar] [CrossRef]
- Holick, M.F. The vitamin D deficiency pandemic: Approaches for diagnosis, treatment and prevention. Rev. Endocr. Metab. Disord. 2017, 18, 153–165. [Google Scholar] [CrossRef]
- Tangpricha, V.; Pearce, E.N.; Chen, T.C.; Holick, M.F. Vitamin D insufficiency among free-living healthy young adults. Am. J. Med. 2002, 112, 659–662. [Google Scholar] [CrossRef]
- Akbar, M.R.; Wibowo, A.; Pranata, R.; Setiabudiawan, B. Low Serum 25-hydroxyvitamin D (Vitamin D) Level Is Associated With Susceptibility to COVID-19, Severity, and Mortality: A Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 660420. [Google Scholar] [CrossRef]
- Vanegas-Cedillo, P.E.; Bello-Chavolla, O.Y.; Ramirez-Pedraza, N.; Rodriguez Encinas, B.; Perez Carrion, C.I.; Jasso-Avila, M.I.; Valladares-Garcia, J.C.; Hernandez-Juarez, D.; Vargas-Vazquez, A.; Antonio-Villa, N.E.; et al. Serum Vitamin D Levels Are Associated With Increased COVID-19 Severity and Mortality Independent of Whole-Body and Visceral Adiposity. Front. Nutr. 2022, 9, 813485. [Google Scholar] [CrossRef]
- Ghelani, D.; Alesi, S.; Mousa, A. Vitamin D and COVID-19: An Overview of Recent Evidence. Int. J. Mol. Sci. 2021, 22, 10559. [Google Scholar] [CrossRef]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Dietary reference values for vitamin D. EFSA J. 2016, 14, e04547. [Google Scholar] [CrossRef]
- Szarpak, L.; Filipiak, K.J.; Gasecka, A.; Gawel, W.; Koziel, D.; Jaguszewski, M.J.; Chmielewski, J.; Gozhenko, A.; Bielski, K.; Wroblewski, P.; et al. Vitamin D supplementation to treat SARS-CoV-2 positive patients. Evidence from meta-analysis. Cardiol. J. 2021, 29, 188–196. [Google Scholar] [CrossRef]
- Tentolouris, N.; Samakidou, G.; Eleftheriadou, I.; Tentolouris, A.; Jude, E.B. The effect of vitamin D supplementation on mortality and intensive care unit admission of COVID-19 patients. A systematic review, meta-analysis and meta-regression. Diabetes Metab. Res. Rev. 2021, 38, e3517. [Google Scholar] [CrossRef]
- Cara, K.C.; Beauchesne, A.R.; Li, R.; Chung, M. Cochrane Review Summary on “Vitamin D Supplementation for the Treatment of COVID-19: A Living Systematic Review”. J. Diet. Suppl. 2022, 19, 143–145. [Google Scholar] [CrossRef]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef]
- Paez-Osuna, F.; Valencia-Castaneda, G.; Rebolledo, U.A. The link between COVID-19 mortality and PM2.5 emissions in rural and medium-size municipalities considering population density, dust events, and wind speed. Chemosphere 2022, 286, 131634. [Google Scholar] [CrossRef]
- Ismail, I.M.I.; Rashid, M.I.; Ali, N.; Altaf, B.A.S.; Munir, M. Temperature, humidity and outdoor air quality indicators influence COVID-19 spread rate and mortality in major cities of Saudi Arabia. Environ. Res. 2022, 204, 112071. [Google Scholar] [CrossRef]
- Fattorini, D.; Regoli, F. Role of the chronic air pollution levels in the COVID-19 outbreak risk in Italy. Environ. Pollut. 2020, 264, 114732. [Google Scholar] [CrossRef]
- Wu, X.; Nethery, R.C.; Sabath, M.B.; Braun, D.; Dominici, F. Air pollution and COVID-19 mortality in the United States: Strengths and limitations of an ecological regression analysis. Sci. Adv. 2020, 6, eabd4049. [Google Scholar] [CrossRef]
- Ishmatov, A. “SARS-CoV-2 is transmitted by particulate air pollution”: Misinterpretations of statistical data, skewed citation practices, and misuse of specific terminology spreading the misconception. Environ. Res. 2022, 204, 112116. [Google Scholar] [CrossRef]
- Conticini, E.; Frediani, B.; Caro, D. Can atmospheric pollution be considered a co-factor in extremely high level of SARS-CoV-2 lethality in Northern Italy? Environ. Pollut. 2020, 261, 114465. [Google Scholar] [CrossRef]
- Tsai, D.H.; Riediker, M.; Berchet, A.; Paccaud, F.; Waeber, G.; Vollenweider, P.; Bochud, M. Effects of short- and long-term exposures to particulate matter on inflammatory marker levels in the general population. Environ. Sci. Pollut. Res. Int. 2019, 26, 19697–19704. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, B.Z.; Sidell, M.A.; Chow, T.; Eckel, S.P.; Pavlovic, N.; Martinez, M.P.; Lurmann, F.; Thomas, D.C.; Gilliland, F.D.; et al. Near-roadway air pollution associated with COVID-19 severity and mortality—Multiethnic cohort study in Southern California. Environ. Int. 2021, 157, 106862. [Google Scholar] [CrossRef] [PubMed]
- The Consequences of Inaction. OECD Environmental Outlook to 2050. 2012. Available online: https://www.oecd.org/g20/topics/energy-environment-green-growth/oecdenvironmentaloutlookto2050theconsequencesofinaction.htm (accessed on 11 May 2022).
- Quinete, N.; Hauser-Davis, R.A. Drinking water pollutants may affect the immune system: Concerns regarding COVID-19 health effects. Environ. Sci. Pollut. Res. Int. 2021, 28, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Chain, E.; Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Leblanc, J.C.; et al. Risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J. 2020, 18, e06223. [Google Scholar] [CrossRef]
- Catelan, D.; Biggeri, A.; Russo, F.; Gregori, D.; Pitter, G.; Da Re, F.; Fletcher, T.; Canova, C. Exposure to Perfluoroalkyl Substances and Mortality for COVID-19: A Spatial Ecological Analysis in the Veneto Region (Italy). Int. J. Environ. Res. Public Health 2021, 18, 2734. [Google Scholar] [CrossRef]
- Ji, J.; Song, L.; Wang, J.; Yang, Z.; Yan, H.; Li, T.; Yu, L.; Jian, L.; Jiang, F.; Li, J.; et al. Association between urinary per- and poly-fluoroalkyl substances and COVID-19 susceptibility. Environ. Int. 2021, 153, 106524. [Google Scholar] [CrossRef]
- Grandjean, P.; Timmermann, C.A.G.; Kruse, M.; Nielsen, F.; Vinholt, P.J.; Boding, L.; Heilmann, C.; Molbak, K. Severity of COVID-19 at elevated exposure to perfluorinated alkylates. PLoS ONE 2020, 15, e0244815. [Google Scholar] [CrossRef]
- Nielsen, C.; Joud, A. Susceptibility to COVID-19 after High Exposure to Perfluoroalkyl Substances from Contaminated Drinking Water: An Ecological Study from Ronneby, Sweden. Int. J. Environ. Res. Public Health 2021, 18, 10702. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Bardi, G.; Duraes, L. Adverse outcome pathway in immunotoxicity of perfluoroalkyls. Curr. Opin. Toxicol. 2021, 25, 23–29. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernandez Garcia, C.; Kieny, M.P.; et al. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef]
- Serra, A.; Fratello, M.; Federico, A.; Ojha, R.; Provenzani, R.; Tasnadi, E.; Cattelani, L.; Del Giudice, G.; Kinaret, P.A.S.; Saarimaki, L.A.; et al. Computationally prioritized drugs inhibit SARS-CoV-2 infection and syncytia formation. Brief Bioinform. 2022, 23, bbab507. [Google Scholar] [CrossRef]
- Morselli Gysi, D.; do Valle, I.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Ghiassian, S.D.; Patten, J.J.; Davey, R.A.; Loscalzo, J.; et al. Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2025581118. [Google Scholar] [CrossRef]
- Pavel, A.; Del Giudice, G.; Federico, A.; Di Lieto, A.; Kinaret, P.A.S.; Serra, A.; Greco, D. Integrated network analysis reveals new genes suggesting COVID-19 chronic effects and treatment. Brief Bioinform. 2021, 22, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- RECOVERY Collaborative Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar] [CrossRef]
- Salama, C.; Han, J.; Yau, L.; Reiss, W.G.; Kramer, B.; Neidhart, J.D.; Criner, G.J.; Kaplan-Lewis, E.; Baden, R.; Pandit, L.; et al. Tocilizumab in Patients Hospitalized with Covid-19 Pneumonia. N. Engl. J. Med. 2021, 384, 20–30. [Google Scholar] [CrossRef] [PubMed]
- REMAP-CAP Investigators; Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar] [CrossRef]
- Clinicians encouraged to consider tocilizumab or sarilumab in treatment of hospitalised COVID-19 patients. Pharm. J. 2021, 54, 524–540. [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Kalil, A.C.; Stebbing, J. Baricitinib: The first immunomodulatory treatment to reduce COVID-19 mortality in a placebo-controlled trial. Lancet Respir. Med. 2021, 9, 1349–1351. [Google Scholar] [CrossRef]
- REMAP-CAP Investigators; ACTIV-4a Investigators; ATTACC Investigators; Goligher, E.C.; Bradbury, C.A.; McVerry, B.J.; Lawler, P.R.; Berger, J.S.; Gong, M.N.; Carrier, M.; et al. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with Covid-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar] [CrossRef]
- Spyropoulos, A.C.; Goldin, M.; Giannis, D.; Diab, W.; Wang, J.; Khanijo, S.; Mignatti, A.; Gianos, E.; Cohen, M.; Sharifova, G.; et al. Efficacy and Safety of Therapeutic-Dose Heparin vs Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients With COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 1612–1620. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group. Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2022, 399, 665–676. [Google Scholar] [CrossRef]
- RECOVERY Trial Regeneron’s Monoclonal Antibody Combination Reduces Deaths for Hospitalised COVID-19 Patients. 2021. Available online: https://www.recoverytrial.net/news/recovery-trial-finds-regeneron2019s-monoclonal-antibody-combination-reduces-deaths-for-hospitalised-covid-19-patients-who-have-not-mounted-their-own-immune-response-1 (accessed on 11 May 2022).
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Xiao, J.; Hooper, A.T.; Hamilton, J.D.; Musser, B.J.; et al. REGEN-COV Antibody Combination and Outcomes in Outpatients with COVID-19. N. Engl. J. Med. 2021, 385, e81. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef]
- O’Brien, M.P.; Forleo-Neto, E.; Sarkar, N.; Isa, F.; Hou, P.; Chan, K.C.; Musser, B.J.; Bar, K.J.; Barnabas, R.V.; Barouch, D.H.; et al. Effect of Subcutaneous Casirivimab and Imdevimab Antibody Combination vs Placebo on Development of Symptomatic COVID-19 in Early Asymptomatic SARS-CoV-2 Infection: A Randomized Clinical Trial. JAMA 2022, 327, 432–441. [Google Scholar] [CrossRef]
- Gupta, A.; Gonzalez-Rojas, Y.; Juarez, E.; Crespo Casal, M.; Moya, J.; Falci, D.R.; Sarkis, E.; Solis, J.; Zheng, H.; Scott, N.; et al. Early Treatment for Covid-19 with SARS-CoV-2 Neutralizing Antibody Sotrovimab. N. Engl. J. Med. 2021, 385, 1941–1950. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Ali, K.; Azher, T.; Baqi, M.; Binnie, A.; Borgia, S.; Carrier, F.M.; Cavayas, Y.A.; Chagnon, N.; Cheng, M.P.; Conly, J.; et al. Remdesivir for the treatment of patients in hospital with COVID-19 in Canada: A randomized controlled trial. CMAJ 2022, 194, E242–E251. [Google Scholar] [CrossRef]
- Olender, S.A.; Perez, K.K.; Go, A.S.; Balani, B.; Price-Haywood, E.G.; Shah, N.S.; Wang, S.; Walunas, T.L.; Swaminathan, S.; Slim, J.; et al. Remdesivir for Severe Coronavirus Disease 2019 (COVID-19) Versus a Cohort Receiving Standard of Care. Clin. Infect. Dis 2021, 73, e4166–e4174. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Vaca, C.E.; Paredes, R.; Mera, J.; Webb, B.J.; Perez, G.; Oguchi, G.; Ryan, P.; Nielsen, B.U.; Brown, M.; et al. Early Remdesivir to Prevent Progression to Severe Covid-19 in Outpatients. N. Engl. J. Med. 2022, 386, 305–315. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Merck and Ridgeback Biotherapeutics Provide Update on Results from MOVe-OUT Study of Molnupiravir, an Investigational Oral Antiviral Medicine, in At Risk Adults With Mild-to-Moderate COVID-19. 2021. Available online: https://www.merck.com/news/merck-and-ridgeback-biotherapeutics-provide-update-on-results-from-move-out-study-of-molnupiravir-an-investigational-oral-antiviral-medicine-in-at-risk-adults-with-mild-to-moderate-covid-19/ (accessed on 11 May 2022).
- Robinson, J. Oral antiviral may cut risk of COVID-19 hospitalisation by nearly 90%, manufacturer claims. Pharm. J. 2021, 307. [Google Scholar] [CrossRef]
- Pfizer Announces Additional Phase 2/3 Study Results Confirming Robust Efficacy of Novel COVID-19 Oral Antiviral Treatment Candidate in Reducing Risk of Hospitalization or Death. 2021. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-additional-phase-23-study-results (accessed on 11 May 2022).
- Pfizer’s Novel COVID-19 Oral Antiviral Treatment Candidate Reduced Risk of Hospitalization or Death by 89% in Interim Analysis of Phase 2/3 EPIC-HR Study. 2021. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate?linkId=138863812 (accessed on 11 May 2022).
- Knapen, D.; Angrish, M.M.; Fortin, M.C.; Katsiadaki, I.; Leonard, M.; Margiotta-Casaluci, L.; Munn, S.; O’Brien, J.M.; Pollesch, N.; Smith, L.C.; et al. Adverse outcome pathway networks I: Development and applications. Environ. Toxicol. Chem. 2018, 37, 1723–1733. [Google Scholar] [CrossRef]
- Bernard, I.; Limonta, D.; Mahal, L.K.; Hobman, T.C. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses 2020, 13, 29. [Google Scholar] [CrossRef]
- D’Alonzo, D.; De Fenza, M.; Pavone, V. COVID-19 and pneumonia: A role for the uPA/uPAR system. Drug Discov. Today 2020, 25, 1528–1534. [Google Scholar] [CrossRef]
- Villeneuve, D.L.; Landesmann, B.; Allavena, P.; Ashley, N.; Bal-Price, A.; Corsini, E.; Halappanavar, S.; Hussell, T.; Laskin, D.; Lawrence, T.; et al. Representing the Process of Inflammation as Key Events in Adverse Outcome Pathways. Toxicol. Sci. 2018, 163, 346–352. [Google Scholar] [CrossRef]
- Chidambaram, V.; Tun, N.L.; Haque, W.Z.; Majella, M.G.; Sivakumar, R.K.; Kumar, A.; Hsu, A.T.; Ishak, I.A.; Nur, A.A.; Ayeh, S.K.; et al. Factors associated with disease severity and mortality among patients with COVID-19: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0241541. [Google Scholar] [CrossRef]
- Mudatsir, M.; Fajar, J.K.; Wulandari, L.; Soegiarto, G.; Ilmawan, M.; Purnamasari, Y.; Mahdi, B.A.; Jayanto, G.D.; Suhendra, S.; Setianingsih, Y.A.; et al. Predictors of COVID-19 severity: A systematic review and meta-analysis. F1000Research 2020, 9, 1107. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Mulchandani, R.; Lyngdoh, T.; Kakkar, A.K. Deciphering the COVID-19 cytokine storm: Systematic review and meta-analysis. Eur. J. Clin. Investig. 2021, 51, e13429. [Google Scholar] [CrossRef]
- Song, J.; Hu, W.; Yu, Y.; Shen, X.; Wang, Y.; Yan, J.; Yang, X.; Gong, S.; Wang, M. A Comparison of Clinical Characteristics and Outcomes in Elderly and Younger Patients with COVID-19. Med. Sci. Monit. 2020, 26, e925047. [Google Scholar] [CrossRef]
- Sward, P.; Edsfeldt, A.; Reepalu, A.; Jehpsson, L.; Rosengren, B.E.; Karlsson, M.K. Age and sex differences in soluble ACE2 may give insights for COVID-19. Crit. Care 2020, 24, 221. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28, 586–601.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gheblawi, M.; Nikhanj, A.; Munan, M.; MacIntyre, E.; O’Neil, C.; Poglitsch, M.; Colombo, D.; Del Nonno, F.; Kassiri, Z.; et al. Dysregulation of ACE (Angiotensin-Converting Enzyme)-2 and Renin-Angiotensin Peptides in SARS-CoV-2 Mediated Mortality and End-Organ Injuries. Hypertension 2022, 79, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.L.; Teng, J.L.L.; Jia, L.; Zhang, C.; Huang, C.; Cai, J.P.; Zhou, R.; Chan, K.H.; Zhao, H.; Zhu, L.; et al. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228.e2212. [Google Scholar] [CrossRef]
- Nagy, B., Jr.; Fejes, Z.; Szentkereszty, Z.; Suto, R.; Varkonyi, I.; Ajzner, E.; Kappelmayer, J.; Papp, Z.; Toth, A.; Fagyas, M. A dramatic rise in serum ACE2 activity in a critically ill COVID-19 patient. Int. J. Infect. Dis. 2021, 103, 412–414. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Miller, A.; Leach, A.; Thomas, J.; McAndrew, C.; Bentley, E.; Mattiuzzo, G.; John, L.; Mirazimi, A.; Harris, G.; Gamage, N.; et al. A super-potent tetramerized ACE2 protein displays enhanced neutralization of SARS-CoV-2 virus infection. Sci. Rep. 2021, 11, 10617. [Google Scholar] [CrossRef]
- Baker, S.A.; Kwok, S.; Berry, G.J.; Montine, T.J. Angiotensin-converting enzyme 2 (ACE2) expression increases with age in patients requiring mechanical ventilation. PLoS ONE 2021, 16, e0247060. [Google Scholar] [CrossRef]
- Sepe, S.; Rossiello, F.; Cancila, V.; Iannelli, F.; Matti, V.; Cicio, G.; Cabrini, M.; Marinelli, E.; Alabi, B.R.; di Lillo, A.; et al. DNA damage response at telomeres boosts the transcription of SARS-CoV-2 receptor ACE2 during aging. EMBO Rep. 2022, 23, e53658. [Google Scholar] [CrossRef]
- Chen, K.; Bi, J.; Su, Y.; Chappell, M.C.; Rose, J.C. Sex-Specific Changes in Renal Angiotensin-Converting Enzyme and Angiotensin-Converting Enzyme 2 Gene Expression and Enzyme Activity at Birth and Over the First Year of Life. Reprod. Sci. 2016, 23, 200–210. [Google Scholar] [CrossRef]
- Mompeon, A.; Lazaro-Franco, M.; Bueno-Beti, C.; Perez-Cremades, D.; Vidal-Gomez, X.; Monsalve, E.; Gironacci, M.M.; Hermenegildo, C.; Novella, S. Estradiol, acting through ERalpha, induces endothelial non-classic renin-angiotensin system increasing angiotensin 1-7 production. Mol. Cell Endocrinol. 2016, 422, 1–8. [Google Scholar] [CrossRef]
- Mahmoud, I.S.; Jarrar, Y.B. Targeting the intestinal TMPRSS2 protease to prevent SARS-CoV-2 entry into enterocytes-prospects and challenges. Mol. Biol. Rep. 2021, 48, 4667–4675. [Google Scholar] [CrossRef]
- Shabbir, S.; Hafeez, A.; Rafiq, M.A.; Khan, M.J. Estrogen shields women from COVID-19 complications by reducing ER stress. Med. Hypotheses 2020, 143, 110148. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, X.M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.M.; Tien, J.C.; et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA 2020, 118. [Google Scholar] [CrossRef]
- Mukherjee, S.; Pahan, K. Is COVID-19 Gender-sensitive? J. Neuroimmune Pharmacol. 2021, 16, 38–47. [Google Scholar] [CrossRef]
- Gebhard, C.; Regitz-Zagrosek, V.; Neuhauser, H.K.; Morgan, R.; Klein, S.L. Impact of sex and gender on COVID-19 outcomes in Europe. Biol. Sex Differ. 2020, 11, 29. [Google Scholar] [CrossRef]
- Haitao, T.; Vermunt, J.V.; Abeykoon, J.; Ghamrawi, R.; Gunaratne, M.; Jayachandran, M.; Narang, K.; Parashuram, S.; Suvakov, S.; Garovic, V.D. COVID-19 and Sex Differences: Mechanisms and Biomarkers. Mayo Clin. Proc. 2020, 95, 2189–2203. [Google Scholar] [CrossRef]
- Ragia, G.; Manolopoulos, V.G. Assessing COVID-19 susceptibility through analysis of the genetic and epigenetic diversity of ACE2-mediated SARS-CoV-2 entry. Pharmacogenomics 2020, 21, 1311–1329. [Google Scholar] [CrossRef]
- Sehailia, M.; Chemat, S. Antimalarial-agent artemisinin and derivatives portray more potent binding to Lys353 and Lys31-binding hotspots of SARS-CoV-2 spike protein than hydroxychloroquine: Potential repurposing of artenimol for COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 6184–6194. [Google Scholar] [CrossRef]
- Cruz, J.O.; Conceicao, I.; Sousa, S.M.B.; Luizon, M.R. Functional prediction and frequency of coding variants in human ACE2 at binding sites with SARS-CoV-2 spike protein on different populations. J. Med. Virol. 2021, 93, 71–73. [Google Scholar] [CrossRef]
- Gibson, W.T.; Evans, D.M.; An, J.; Jones, S.J.M. ACE 2 Coding Variants: A Potential X-linked Risk Factor for COVID-19 Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gemmati, D.; Bramanti, B.; Serino, M.L.; Secchiero, P.; Zauli, G.; Tisato, V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int. J. Mol. Sci. 2020, 21, 3474. [Google Scholar] [CrossRef]
- Asselta, R.; Paraboschi, E.M.; Mantovani, A.; Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 2020, 12, 10087–10098. [Google Scholar] [CrossRef]
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef]
- Molina-Mora, J.A.; Cordero-Laurent, E.; Calderon-Osorno, M.; Chacon-Ramirez, E.; Duarte-Martinez, F. Metagenomic pipeline for identifying co-infections among distinct SARS-CoV-2 variants of concern: Study cases from Alpha to Omicron. Sci. Rep. 2022, 12, 9377. [Google Scholar] [CrossRef]
- Franke, K.R.; Isett, R.; Robbins, A.; Paquette-Straub, C.; Shapiro, C.A.; Lee, M.M.; Crowgey, E.L. Genomic surveillance of SARS-CoV-2 in the state of Delaware reveals tremendous genomic diversity. PLoS ONE 2022, 17, e0262573. [Google Scholar] [CrossRef]
- Nagpal, S.; Pal, R.; Ashima; Tyagi, A.; Tripathi, S.; Nagori, A.; Ahmad, S.; Mishra, H.P.; Malhotra, R.; Kutum, R.; et al. Genomic Surveillance of COVID-19 Variants With Language Models and Machine Learning. Front. Genet. 2022, 13, 858252. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Sorice, M.; Misasi, R.; Riitano, G.; Manganelli, V.; Martellucci, S.; Longo, A.; Garofalo, T.; Mattei, V. Targeting Lipid Rafts as a Strategy Against Coronavirus. Front. Cell Dev. Biol. 2020, 8, 618296. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Ballout, R.A.; Sviridov, D.; Bukrinsky, M.I.; Remaley, A.T. The lysosome: A potential juncture between SARS-CoV-2 infectivity and Niemann-Pick disease type C, with therapeutic implications. FASEB J. 2020, 34, 7253–7264. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yuan, Z.; Pavel, M.A.; Jablonski, S.M.; Jablonski, J.; Hobson, R.; Valente, S.; Reddy, C.B.; Hansen, S.B. The role of high cholesterol in age-related COVID19 lethality. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Kocar, E.; Rezen, T.; Rozman, D. Cholesterol, lipoproteins, and COVID-19: Basic concepts and clinical applications. Biochim Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158849. [Google Scholar] [CrossRef]
- Al-Benna, S. Association of high level gene expression of ACE2 in adipose tissue with mortality of COVID-19 infection in obese patients. Obes. Med. 2020, 19, 100283. [Google Scholar] [CrossRef]
- Jia, X.; Yin, C.; Lu, S.; Chen, Y.; Liu, Q.; Bai, J.; Lu, Y. Two Things about COVID-19 Might Need Attention. Preprints 2020, 2020020315. [Google Scholar] [CrossRef]
- Al Heialy, S.; Hachim, M.Y.; Senok, A.; Gaudet, M.; Abou Tayoun, A.; Hamoudi, R.; Alsheikh-Ali, A.; Hamid, Q. Regulation of Angiotensin- Converting Enzyme 2 in Obesity: Implications for COVID-19. Front. Physiol. 2020, 11, 555039. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Thanaraj, T.A.; Qaddoumi, M.G.; Hashem, A.; Abubaker, J.; Al-Mulla, F. The Role of Lipid Metabolism in COVID-19 Virus Infection and as a Drug Target. Int. J. Mol. Sci. 2020, 21, 3544. [Google Scholar] [CrossRef]
- Wan, L.; Fan, C.; Zhang, P.; Wang, X.; Sun, J.; Zhang, Y.; Yan, Q.; Gong, J.; Yang, H.; Yang, X.; et al. Cholesterol Metabolism—Impacts on SARS-CoV-2 Infection Prognosis. medRxiv 2020. [Google Scholar] [CrossRef]
- Goulter, A.B.; Goddard, M.J.; Allen, J.C.; Clark, K.L. ACE2 gene expression is up-regulated in the human failing heart. BMC Med. 2004, 2, 19. [Google Scholar] [CrossRef]
- Zisman, L.S.; Keller, R.S.; Weaver, B.; Lin, Q.; Speth, R.; Bristow, M.R.; Canver, C.C. Increased angiotensin-(1-7)-forming activity in failing human heart ventricles: Evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation 2003, 108, 1707–1712. [Google Scholar] [CrossRef]
- Khoury, E.E.; Knaney, Y.; Fokra, A.; Kinaneh, S.; Azzam, Z.; Heyman, S.N.; Abassi, Z. Pulmonary, cardiac and renal distribution of ACE2, furin, TMPRSS2 and ADAM17 in rats with heart failure: Potential implication for COVID-19 disease. J. Cell Mol. Med. 2021, 25, 3840–3855. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Bedi, K.C., Jr.; Papangeli, I.; Akkad, A.D.; Arduini, A.; Hayat, S.; Eraslan, G.; Muus, C.; Bhattacharyya, R.P.; et al. Myocyte-Specific Upregulation of ACE2 in Cardiovascular Disease: Implications for SARS-CoV-2-Mediated Myocarditis. Circulation 2020, 142, 708–710. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef]
- Nicin, L.; Abplanalp, W.T.; Mellentin, H.; Kattih, B.; Tombor, L.; John, D.; Schmitto, J.D.; Heineke, J.; Emrich, F.; Arsalan, M.; et al. Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 2020, 41, 1804–1806. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef]
- Margo Daems, L.L.; Ilona Cuijpers, R.; Liesenborghs, L.; Cuijpers, I.; Boudewijns, R.; Raman, J.; Simmonds, S.; Geuens, N.; Lox, M.; Verhamme, P.; et al. SARS-CoV-2 infection leads to cardiac pericyte loss, fibrosis, cardiomyocyte hypertrophy, and diastolic dysfunction. Res. Sq. Prepr. 2020. [Google Scholar] [CrossRef]
- Bristow, M.R.; Zisman, L.S.; Altman, N.L.; Gilbert, E.M.; Lowes, B.D.; Minobe, W.A.; Slavov, D.; Schwisow, J.A.; Rodriguez, E.M.; Carroll, I.A.; et al. Dynamic Regulation of SARS-Cov-2 Binding and Cell Entry Mechanisms in Remodeled Human Ventricular Myocardium. JACC Basic Transl. Sci. 2020, 5, 871–883. [Google Scholar] [CrossRef]
- Uri, K.; Fagyas, M.; Kertesz, A.; Borbely, A.; Jenei, C.; Bene, O.; Csanadi, Z.; Paulus, W.J.; Edes, I.; Papp, Z.; et al. Circulating ACE2 activity correlates with cardiovascular disease development. J. Renin. Angiotensin. Aldosterone Syst. 2016, 17, 1470320316668435. [Google Scholar] [CrossRef]
- Wallentin, L.; Lindback, J.; Eriksson, N.; Hijazi, Z.; Eikelboom, J.W.; Ezekowitz, M.D.; Granger, C.B.; Lopes, R.D.; Yusuf, S.; Oldgren, J.; et al. Angiotensin-converting enzyme 2 (ACE2) levels in relation to risk factors for COVID-19 in two large cohorts of patients with atrial fibrillation. Eur. Heart J. 2020, 41, 4037–4046. [Google Scholar] [CrossRef]
- Lundstrom, A.; Ziegler, L.; Havervall, S.; Rudberg, A.S.; von Meijenfeldt, F.; Lisman, T.; Mackman, N.; Sanden, P.; Thalin, C. Soluble angiotensin-converting enzyme 2 is transiently elevated in COVID-19 and correlates with specific inflammatory and endothelial markers. J. Med. Virol. 2021, 93, 5908–5916. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hasan, M.; Ahmed, A. Potential detrimental role of soluble ACE2 in severe COVID-19 comorbid patients. Rev. Med. Virol. 2021, 31, 1–12. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Edwinson, A.; Yang, L.; Chen, J.; Grover, M. Colonic expression of Ace2, the SARS-CoV-2 entry receptor, is suppressed by commensal human microbiota. Gut Microbes 2021, 13, 1984105. [Google Scholar] [CrossRef]
- Yang, T.; Chakraborty, S.; Saha, P.; Mell, B.; Cheng, X.; Yeo, J.Y.; Mei, X.; Zhou, G.; Mandal, J.; Golonka, R.; et al. Gnotobiotic Rats Reveal That Gut Microbiota Regulates Colonic mRNA of Ace2, the Receptor for SARS-CoV-2 Infectivity. Hypertension 2020, 76, e1–e3. [Google Scholar] [CrossRef]
- Geva-Zatorsky, N.; Sefik, E.; Kua, L.; Pasman, L.; Tan, T.G.; Ortiz-Lopez, A.; Yanortsang, T.B.; Yang, L.; Jupp, R.; Mathis, D.; et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell 2017, 168, 928–943.e11. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Lin, W.; Tang, W.; Chan, F.K.L.; Ng, S.C. Review article: Probiotics, prebiotics and dietary approaches during COVID-19 pandemic. Trends Food Sci. Technol. 2021, 108, 187–196. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Guo, M.; Tao, W.; Flavell, R.A.; Zhu, S. Potential intestinal infection and faecal-oral transmission of SARS-CoV-2. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Kopetz, S.; Vilar, E.; Shen, J.P.; Chen, K.; Maitra, A. Relative Abundance of SARS-CoV-2 Entry Genes in the Enterocytes of the Lower Gastrointestinal Tract. Genes 2020, 11, 645. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Beumer, J.; Geurts, M.H.; Lamers, M.M.; Puschhof, J.; Zhang, J.; van der Vaart, J.; Mykytyn, A.Z.; Breugem, T.I.; Riesebosch, S.; Schipper, D.; et al. A CRISPR/Cas9 genetically engineered organoid biobank reveals essential host factors for coronaviruses. Nat. Commun. 2021, 12, 5498. [Google Scholar] [CrossRef]
- AOP 428. Available online: https://aopwiki.org/aops/428 (accessed on 16 March 2022).
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Rodriguez-Pineiro, A.M.; Schutte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Backhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef]
- Sahu, S.; Patil, C.R.; Kumar, S.; Apparsundaram, S.; Goyal, R.K. Role of ACE2-Ang (1-7)-Mas axis in post-COVID-19 complications and its dietary modulation. Mol. Cell Biochem. 2022, 477, 225–240. [Google Scholar] [CrossRef]
- Senthil Kumar, K.J.; Gokila Vani, M.; Wang, C.S.; Chen, C.C.; Chen, Y.C.; Lu, L.P.; Huang, C.H.; Lai, C.S.; Wang, S.Y. Geranium and Lemon Essential Oils and Their Active Compounds Downregulate Angiotensin-Converting Enzyme 2 (ACE2), a SARS-CoV-2 Spike Receptor-Binding Domain, in Epithelial Cells. Plants 2020, 9, 770. [Google Scholar] [CrossRef]
- Thuy, B.T.P.; My, T.T.A.; Hai, N.T.T.; Hieu, L.T.; Hoa, T.T.; Thi Phuong Loan, H.; Triet, N.T.; Anh, T.T.V.; Quy, P.T.; Tat, P.V.; et al. Investigation into SARS-CoV-2 Resistance of Compounds in Garlic Essential Oil. ACS Omega 2020, 5, 8312–8320. [Google Scholar] [CrossRef]
- Maurya, V.K.; Kumar, S.; Prasad, A.K.; Bhatt, M.L.B.; Saxena, S.K. Structure-based drug designing for potential antiviral activity of selected natural products from Ayurveda against SARS-CoV-2 spike glycoprotein and its cellular receptor. Virusdisease 2020, 31, 179–193. [Google Scholar] [CrossRef]
- Kumar, A.; Choudhir, G.; Shukla, S.K.; Sharma, M.; Tyagi, P.; Bhushan, A.; Rathore, M. Identification of phytochemical inhibitors against main protease of COVID-19 using molecular modeling approaches. J. Biomol. Struct. Dyn. 2021, 39, 3760–3770. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors—an in silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2021, 39, 4362–4374. [Google Scholar] [CrossRef]
- Sargiacomo, C.; Sotgia, F.; Lisanti, M.P. COVID-19 and chronological aging: Senolytics and other anti-aging drugs for the treatment or prevention of corona virus infection? Aging 2020, 12, 6511–6517. [Google Scholar] [CrossRef]
- Biagioli, M.; Marchiano, S.; Roselli, R.; Di Giorgio, C.; Bellini, R.; Bordoni, M.; Gidari, A.; Sabbatini, S.; Francisci, D.; Fiorillo, B.; et al. Discovery of a AHR pelargonidin agonist that counter-regulates Ace2 expression and attenuates ACE2-SARS-CoV-2 interaction. Biochem. Pharmacol. 2021, 188, 114564. [Google Scholar] [CrossRef]
- Liu, X.; Raghuvanshi, R.; Ceylan, F.D.; Bolling, B.W. Quercetin and Its Metabolites Inhibit Recombinant Human Angiotensin-Converting Enzyme 2 (ACE2) Activity. J. Agric. Food Chem. 2020, 68, 13982–13989. [Google Scholar] [CrossRef]
- Tamama, K. Potential benefits of dietary seaweeds as protection against COVID-19. Nutr. Rev. 2021, 79, 814–823. [Google Scholar] [CrossRef]
- De Ligt, M.; Hesselink, M.K.C.; Jorgensen, J.; Hoebers, N.; Blaak, E.E.; Goossens, G.H. Resveratrol supplementation reduces ACE2 expression in human adipose tissue. Adipocyte 2021, 10, 408–411. [Google Scholar] [CrossRef]
- Yang, M.; Wei, J.; Huang, T.; Lei, L.; Shen, C.; Lai, J.; Yang, M.; Liu, L.; Yang, Y.; Liu, G.; et al. Resveratrol inhibits the replication of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in cultured Vero cells. Phytother. Res. 2021, 35, 1127–1129. [Google Scholar] [CrossRef]
- Wen, C.C.; Kuo, Y.H.; Jan, J.T.; Liang, P.H.; Wang, S.Y.; Liu, H.G.; Lee, C.K.; Chang, S.T.; Kuo, C.J.; Lee, S.S.; et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef]
- Yang, P.; Du, D.; Zhou, Z.; Lu, N.; Fu, Q.; Ma, J.; Zhao, L.; Chen, A. 3D printing-assisted osteotomy treatment for the malunion of lateral tibial plateau fracture. Injury 2016, 47, 2816–2821. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Peng, M.Y.; Liu, W.C.; Zheng, J.Q.; Lu, C.L.; Hou, Y.C.; Zheng, C.M.; Song, J.Y.; Lu, K.C.; Chao, Y.C. Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D. Int. J. Mol. Sci. 2021, 22, 5251. [Google Scholar] [CrossRef]
- Ismailova, A.; White, J.H. Vitamin D, infections and immunity. Rev. Endocr. Metab. Disord. 2022, 23, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Gilani, S.J.; Bin-Jumah, M.N.; Nadeem, M.S.; Kazmi, I. Vitamin D attenuates COVID-19 complications via modulation of proinflammatory cytokines, antiviral proteins, and autophagy. Expert Rev. Anti Infect. Ther. 2022, 20, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Aztatzi-Aguilar, O.G.; Uribe-Ramirez, M.; Arias-Montano, J.A.; Barbier, O.; De Vizcaya-Ruiz, A. Acute and subchronic exposure to air particulate matter induces expression of angiotensin and bradykinin-related genes in the lungs and heart: Angiotensin-II type-I receptor as a molecular target of particulate matter exposure. Part Fibre Toxicol. 2015, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Yilin, Z.; Yandong, N.; Faguang, J. Role of angiotensin-converting enzyme (ACE) and ACE2 in a rat model of smoke inhalation induced acute respiratory distress syndrome. Burns 2015, 41, 1468–1477. [Google Scholar] [CrossRef]
- Sagawa, T.; Tsujikawa, T.; Honda, A.; Miyasaka, N.; Tanaka, M.; Kida, T.; Hasegawa, K.; Okuda, T.; Kawahito, Y.; Takano, H. Exposure to particulate matter upregulates ACE2 and TMPRSS2 expression in the murine lung. Environ. Res. 2021, 195, 110722. [Google Scholar] [CrossRef]
- Borro, M.; Di Girolamo, P.; Gentile, G.; De Luca, O.; Preissner, R.; Marcolongo, A.; Ferracuti, S.; Simmaco, M. Evidence-Based Considerations Exploring Relations between SARS-CoV-2 Pandemic and Air Pollution: Involvement of PM2.5-Mediated Up-Regulation of the Viral Receptor ACE-2. Int. J. Environ. Res. Public Health 2020, 17, 5573. [Google Scholar] [CrossRef]
- Li, H.H.; Liu, C.C.; Hsu, T.W.; Lin, J.H.; Hsu, J.W.; Li, A.F.; Yeh, Y.C.; Hung, S.C.; Hsu, H.S. Upregulation of ACE2 and TMPRSS2 by particulate matter and idiopathic pulmonary fibrosis: A potential role in severe COVID-19. Part Fibre Toxicol. 2021, 18, 11. [Google Scholar] [CrossRef]
- Vo, T.; Paudel, K.; Choudhary, I.; Patial, S.; Saini, Y. Ozone exposure upregulates the expression of host susceptibility protein TMPRSS2 to SARS-CoV-2. Sci. Rep. 2022, 12, 1357. [Google Scholar] [CrossRef]
- Ahmad, S.; Wen, Y.; Irudayaraj, J.M.K. PFOA induces alteration in DNA methylation regulators and SARS-CoV-2 targets Ace2 and Tmprss2 in mouse lung tissues. Toxicol. Rep. 2021, 8, 1892–1898. [Google Scholar] [CrossRef]
- EMA Starts Rolling Review of REGN-COV2 Antibody Combination (Casirivimab/Imdevimab). 2021. Available online: https://www.ema.europa.eu/en/news/ema-starts-rolling-review-regn-cov2-antibody-combination-casirivimab-imdevimab (accessed on 11 May 2022).
- EMA Starts Rolling Review of Sotrovimab (VIR-7831) for COVID-19. 2021. Available online: https://www.ema.europa.eu/en/news/ema-starts-rolling-review-sotrovimab-vir-7831-covid-19 (accessed on 11 May 2022).
- Braz-de-Melo, H.A.; Faria, S.S.; Pasquarelli-do-Nascimento, G.; Santos, I.O.; Kobinger, G.P.; Magalhaes, K.G. The Use of the Anticoagulant Heparin and Corticosteroid Dexamethasone as Prominent Treatments for COVID-19. Front. Med. 2021, 8, 615333. [Google Scholar] [CrossRef]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb Haemost. 2020, 120, 1700–1715. [Google Scholar] [CrossRef]
- (EMA), E.M.A. Veklury. 2021. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/veklury (accessed on 11 May 2022).
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Hobartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef]
- Frediansyah, A.; Nainu, F.; Dhama, K.; Mudatsir, M.; Harapan, H. Remdesivir and its antiviral activity against COVID-19: A systematic review. Clin. Epidemiol. Glob. Health 2021, 9, 123–127. [Google Scholar] [CrossRef]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Perez-Perez, L.; et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef]
- Toots, M.; Yoon, J.J.; Hart, M.; Natchus, M.G.; Painter, G.R.; Plemper, R.K. Quantitative efficacy paradigms of the influenza clinical drug candidate EIDD-2801 in the ferret model. Transl. Res. 2020, 218, 16–28. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Yoon, J.J.; Toots, M.; Lee, S.; Lee, M.E.; Ludeke, B.; Luczo, J.M.; Ganti, K.; Cox, R.M.; Sticher, Z.M.; Edpuganti, V.; et al. Orally Efficacious Broad-Spectrum Ribonucleoside Analog Inhibitor of Influenza and Respiratory Syncytial Viruses. Antimicrob. Agents Chemother. 2018, 62, e00766-18. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Gotte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles’ heel”: Current effective inhibitor targeting a 3C-like protease. Protein Cell 2013, 4, 248–250. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Ionescu, M.I. An Overview of the Crystallized Structures of the SARS-CoV-2. Protein J. 2020, 39, 600–618. [Google Scholar] [CrossRef]
- Muramatsu, T.; Takemoto, C.; Kim, Y.T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef]
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What coronavirus 3C-like protease tells us: From structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 2021, 41, 1965–1998. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.; Karnik, S.; Saef, J.; Bergmann, C.; Barnard, J.; Lederman, M.M.; Tilton, J.; Cheng, F.; Harding, C.V.; Young, J.B.; et al. SARS-CoV-2 and ACE2: The biology and clinical data settling the ARB and ACEI controversy. EBioMedicine 2020, 58, 102907. [Google Scholar] [CrossRef] [PubMed]
- Peron, J.P.S.; Nakaya, H. Susceptibility of the Elderly to SARS-CoV-2 Infection: ACE-2 Overexpression, Shedding, and Antibody-dependent Enhancement (ADE). Clinics 2020, 75, e1912. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Tong, X.; Wang, Y.; Wang, H.; Wang, L.; Xu, X. Coagulopathy in elderly patients with coronavirus disease 2019. Aging Med. 2020, 3, 260–265. [Google Scholar] [CrossRef]
- AlGhatrif, M.; Cingolani, O.; Lakatta, E.G. The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease: Insights From Cardiovascular Aging Science. JAMA Cardiol. 2020, 5, 747–748. [Google Scholar] [CrossRef]
- Viana, S.D.; Nunes, S.; Reis, F. ACE2 imbalance as a key player for the poor outcomes in COVID-19 patients with age-related comorbidities—Role of gut microbiota dysbiosis. Ageing Res. Rev. 2020, 62, 101123. [Google Scholar] [CrossRef]
- Fernandez-Atucha, A.; Izagirre, A.; Fraile-Bermudez, A.B.; Kortajarena, M.; Larrinaga, G.; Martinez-Lage, P.; Echevarria, E.; Gil, J. Sex differences in the aging pattern of renin-angiotensin system serum peptidases. Biol. Sex Differ. 2017, 8, 5. [Google Scholar] [CrossRef]
- Zipeto, D.; Palmeira, J.D.F.; Arganaraz, G.A.; Arganaraz, E.R. ACE2/ADAM17/TMPRSS2 Interplay May Be the Main Risk Factor for COVID-19. Front. Immunol. 2020, 11, 576745. [Google Scholar] [CrossRef]
- La Vignera, S.; Cannarella, R.; Condorelli, R.A.; Torre, F.; Aversa, A.; Calogero, A.E. Sex-Specific SARS-CoV-2 Mortality: Among Hormone-Modulated ACE2 Expression, Risk of Venous Thromboembolism and Hypovitaminosis D. Int. J. Mol. Sci. 2020, 21, 2948. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Möhlendick, B.S.K.; Breuckmann, K.; Elsner, C.; Babel, N.; Balfanz, P.; Dahl, E.; Dreher, M.; Fistera, D.; Herbstreit, F.; Hölzer, B.; et al. ACE2 polymorphism and susceptibility for SARS-CoV-2 infection and severity of COVID-19. Pharm. Genom. 2021, 31, 165–171. [Google Scholar] [CrossRef]
- Augustine, R.; Abhilash, S.; Nayeem, A.; Salam, S.A.; Augustine, P.; Dan, P.; Maureira, P.; Mraiche, F.; Gentile, C.; Hansbro, P.M.; et al. Increased complications of COVID-19 in people with cardiovascular disease: Role of the renin-angiotensin-aldosterone system (RAAS) dysregulation. Chem. Biol. Interact. 2022, 351, 109738. [Google Scholar] [CrossRef]
- Avolio, E.; Carrabba, M.; Milligan, R.; Kavanagh Williamson, M.; Beltrami, A.P.; Gupta, K.; Elvers, K.T.; Gamez, M.; Foster, R.R.; Gillespie, K.; et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: A potential non-infective mechanism of COVID-19 microvascular disease. Clin. Sci. 2021, 135, 2667–2689. [Google Scholar] [CrossRef]
- Camargo, S.M.R.; Vuille-Dit-Bille, R.N.; Meier, C.F.; Verrey, F. ACE2 and gut amino acid transport. Clin. Sci. 2020, 134, 2823–2833. [Google Scholar] [CrossRef]
- Camargo, S.M.; Singer, D.; Makrides, V.; Huggel, K.; Pos, K.M.; Wagner, C.A.; Kuba, K.; Danilczyk, U.; Skovby, F.; Kleta, R.; et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 2009, 136, 872–882. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Shoaie, S.; Bergentall, M.; Ghaffari, P.; Zhang, C.; Larsson, E.; Backhed, F.; Nielsen, J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef]
- Amraei, R.; Rahimi, N. COVID-19, Renin-Angiotensin System and Endothelial Dysfunction. Cells 2020, 9, 1652. [Google Scholar] [CrossRef]
- Xu, J.; Yang, J.; Chen, J.; Luo, Q.; Zhang, Q.; Zhang, H. Vitamin D alleviates lipopolysaccharideinduced acute lung injury via regulation of the reninangiotensin system. Mol. Med. Rep. 2017, 16, 7432–7438. [Google Scholar] [CrossRef]
- Malek Mahdavi, A. A brief review of interplay between vitamin D and angiotensin-converting enzyme 2: Implications for a potential treatment for COVID-19. Rev. Med. Virol. 2020, 30, e2119. [Google Scholar] [CrossRef]
- Butler, A.E.; Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L. Vitamin D association with the renin angiotensin system in polycystic ovary syndrome. J. Steroid Biochem. Mol. Biol. 2021, 214, 105965. [Google Scholar] [CrossRef]
- Smadja, D.M.; Mentzer, S.J.; Fontenay, M.; Laffan, M.A.; Ackermann, M.; Helms, J.; Jonigk, D.; Chocron, R.; Pier, G.B.; Gendron, N.; et al. COVID-19 is a systemic vascular hemopathy: Insight for mechanistic and clinical aspects. Angiogenesis 2021, 24, 755–788. [Google Scholar] [CrossRef]
- Lupi, L.; Adamo, M.; Inciardi, R.M.; Metra, M. ACE2 down-regulation may contribute to the increased thrombotic risk in COVID-19. Eur. Heart J. 2020, 41, 3200. [Google Scholar] [CrossRef]
- Mari, D.; Coppola, R.; Provenzano, R. Hemostasis factors and aging. Exp. Gerontol. 2008, 43, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M. Hemostasis and aging. Crit. Rev. Oncol. Hematol. 2006, 60, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Oberholzer, H.M.; van der Spuy, W.J.; Meiring, J.H. Age-related changes in fibrin networks and platelets of individuals over 75: A scanning electron microscopy study showing “thrombotic preparedness”. J. Thromb. Thrombolysis 2010, 29, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Medcalf, R.L.; Keragala, C.B. The Fibrinolytic System: Mysteries and Opportunities. Hemasphere 2021, 5, e570. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler Thromb Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Li, G.; Luna, C.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Modulation of inflammatory markers by miR-146a during replicative senescence in trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 2010, 51, 2976–2985. [Google Scholar] [CrossRef]
- O’Donnell, J.; Laffan, M.A. The relationship between ABO histo-blood group, factor VIII and von Willebrand factor. Transfus. Med. 2001, 11, 343–351. [Google Scholar] [CrossRef]
- Salabei, J.K.; Fishman, T.J.; Asnake, Z.T.; Ali, A.; Iyer, U.G. COVID-19 Coagulopathy: Current knowledge and guidelines on anticoagulation. Heart Lung 2021, 50, 357–360. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Al-Sabah, S.; Hammad, M.M.; Hebbar, P.; Channanath, A.M.; John, S.E.; Taher, I.; Almaeen, A.; Ghazy, A.; Mohammad, A.; et al. Prognostic Genetic Markers for Thrombosis in COVID-19 Patients: A Focused Analysis on D-Dimer, Homocysteine and Thromboembolism. Front. Pharmacol. 2020, 11, 587451. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [CrossRef]
- Morelli, V.M.; Lijfering, W.M.; Bos, M.H.A.; Rosendaal, F.R.; Cannegieter, S.C. Lipid levels and risk of venous thrombosis: Results from the MEGA-study. Eur. J. Epidemiol. 2017, 32, 669–681. [Google Scholar] [CrossRef]
- Brill, A.; Yesilaltay, A.; De Meyer, S.F.; Kisucka, J.; Fuchs, T.A.; Kocher, O.; Krieger, M.; Wagner, D.D. Extrahepatic high-density lipoprotein receptor SR-BI and apoA-I protect against deep vein thrombosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1841–1847. [Google Scholar] [CrossRef]
- Griffin, J.H.; Fernandez, J.A.; Deguchi, H. Plasma lipoproteins, hemostasis and thrombosis. Thromb. Haemost. 2001, 86, 386–394. [Google Scholar]
- Henry, B.M.; Szergyuk, I.; de Oliveira, M.H.S.; Abosamak, M.F.; Benoit, S.W.; Benoit, J.L.; Lippi, G. Alterations in the lipid profile associate with a dysregulated inflammatory, prothrombotic, anti-fibrinolytic state and development of severe acute kidney injury in coronavirus disease 2019 (COVID-19): A study from Cincinnati, USA. Diabetes Metab. Syndr. 2021, 15, 863–868. [Google Scholar] [CrossRef]
- Mohammed, Y.; Kootte, R.S.; Kopatz, W.F.; Borchers, C.H.; Buller, H.R.; Versteeg, H.H.; Nieuwdorp, M.; van Mens, T.E. The intestinal microbiome potentially affects thrombin generation in human subjects. J. Thromb. Haemost. 2020, 18, 642–650. [Google Scholar] [CrossRef]
- Famodu, A.A.; Osilesi, O.; Makinde, Y.O.; Osonuga, O.A.; Fakoya, T.A.; Ogunyemi, E.O.; Egbenehkhuere, I.E. The Influence of a Vegetarian Diet on Haemostatic Risk Factors for Cardiovascular Disease in Africans. Thromb. Res. 1999, 95, 31–36. [Google Scholar] [CrossRef]
- Marckmann, P.; Sandstrom, B.; Jespersen, J. Low-fat, high-fiber diet favorably affects several independent risk markers of ischemic heart disease: Observations on blood lipids, coagulation, and fibrinolysis from a trial of middle-aged Danes. Am. J. Clin. Nutr. 1994, 59, 935–939. [Google Scholar] [CrossRef]
- Kim, Y.; Keogh, J.B.; Clifton, P.M. Effects of Two Different Dietary Patterns on Inflammatory Markers, Advanced Glycation End Products and Lipids in Subjects without Type 2 Diabetes: A Randomised Crossover Study. Nutrients 2017, 9, 336. [Google Scholar] [CrossRef]
- Liese, A.D.; Weis, K.E.; Schulz, M.; Tooze, J.A. Food intake patterns associated with incident type 2 diabetes: The Insulin Resistance Atherosclerosis Study. Diabetes Care 2009, 32, 263–268. [Google Scholar] [CrossRef]
- Pieters, M.; Swanepoel, A.C. The effect of plant-based diets on thrombotic risk factors. Pol. Arch. Intern. Med. 2021, 131, 16123. [Google Scholar] [CrossRef]
- Kim, D.H.; Meza, C.A.; Clarke, H.; Kim, J.S.; Hickner, R.C. Vitamin D and Endothelial Function. Nutrients 2020, 12, 575. [Google Scholar] [CrossRef]
- Garcia-Carrasco, M.; Jimenez-Herrera, E.A.; Galvez-Romero, J.L.; Mendoza-Pinto, C.; Mendez-Martinez, S.; Etchegaray-Morales, I.; Munguia-Realpozo, P.; Vazquez de Lara-Cisneros, L.; Santa Cruz, F.J.; Cervera, R. The anti-thrombotic effects of vitamin D and their possible relationship with antiphospholipid syndrome. Lupus 2018, 27, 2181–2189. [Google Scholar] [CrossRef]
- Han, R.; Zhang, F.; Wan, C.; Liu, L.; Zhong, Q.; Ding, W. Effect of perfluorooctane sulphonate-induced Kupffer cell activation on hepatocyte proliferation through the NF-kappaB/TNF-alpha/IL-6-dependent pathway. Chemosphere 2018, 200, 283–294. [Google Scholar] [CrossRef]
- Yang, J.; Wang, C.; Nie, X.; Shi, S.; Xiao, J.; Ma, X.; Dong, X.; Zhang, Y.; Han, J.; Li, T.; et al. Perfluorooctane sulfonate mediates microglial activation and secretion of TNF-alpha through Ca(2)(+)-dependent PKC-NF-small ka, CyrillicB signaling. Int. Immunopharmacol. 2015, 28, 52–60. [Google Scholar] [CrossRef]
- Zhu, J.; Qian, W.; Wang, Y.; Gao, R.; Wang, J.; Xiao, H. Involvement of mitogen-activated protein kinase and NF-kappaB signaling pathways in perfluorooctane sulfonic acid-induced inflammatory reaction in BV2 microglial cells. J. Appl. Toxicol. 2015, 35, 1539–1549. [Google Scholar] [CrossRef]
- Guo, J.; Wu, P.; Cao, J.; Luo, Y.; Chen, J.; Wang, G.; Guo, W.; Wang, T.; He, X. The PFOS disturbed immunomodulatory functions via nuclear Factor-kappaB signaling in liver of zebrafish (Danio rerio). Fish Shellfish Immunol. 2019, 91, 87–98. [Google Scholar] [CrossRef]
- Hirsh, J.; Anand, S.S.; Halperin, J.L.; Fuster, V. Mechanism of action and pharmacology of unfractionated heparin. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1094–1096. [Google Scholar] [CrossRef]
- Akira, S. Toll-like receptor signaling. J. Biol. Chem. 2003, 278, 38105–38108. [Google Scholar] [CrossRef]
- Akira, S.; Hemmi, H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 2003, 85, 85–95. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef]
- Pasare, C.; Medzhitov, R. Toll-like receptors: Linking innate and adaptive immunity. Adv. Exp. Med. Biol. 2005, 560, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Das, N.C.; Patra, R.; Mukherjee, S. In silico analyses on the comparative sensing of SARS-CoV-2 mRNA by the intracellular TLRs of humans. J. Med. Virol. 2021, 93, 2476–2486. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Hibberd, M.L.; Ooi, E.E.; Tang, K.F.; Neo, S.Y.; Tan, J.; Murthy, K.R.; Vega, V.B.; Chia, J.M.; Liu, E.T.; et al. A human in vitro model system for investigating genome-wide host responses to SARS coronavirus infection. BMC Infect. Dis. 2004, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Digard, P.; Lee, H.M.; Sharp, C.; Grey, F.; Gaunt, E. Intra-genome variability in the dinucleotide composition of SARS-CoV-2. Virus Evol. 2020, 6, veaa057. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Sariol, A.; Perlman, S. SARS-CoV-2 takes its Toll. Nat. Immunol. 2021, 22, 801–802. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef]
- Costa, T.J.; Potje, S.R.; Fraga-Silva, T.F.C.; da Silva-Neto, J.A.; Barros, P.R.; Rodrigues, D.; Machado, M.R.; Martins, R.B.; Santos-Eichler, R.A.; Benatti, M.N.; et al. Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vascul. Pharmacol. 2022, 142, 106946. [Google Scholar] [CrossRef]
- Andargie, T.E.; Tsuji, N.; Seifuddin, F.; Jang, M.K.; Yuen, P.S.; Kong, H.; Tunc, I.; Singh, K.; Charya, A.; Wilkins, K.; et al. Cell-free DNA maps COVID-19 tissue injury and risk of death and can cause tissue injury. JCI Insight 2021, 6, e147610. [Google Scholar] [CrossRef]
- Bailey, K.L.; Smith, L.M.; Heires, A.J.; Katafiasz, D.M.; Romberger, D.J.; LeVan, T.D. Aging leads to dysfunctional innate immune responses to TLR2 and TLR4 agonists. Aging Clin. Exp. Res. 2019, 31, 1185–1193. [Google Scholar] [CrossRef]
- Panda, A.; Qian, F.; Mohanty, S.; van Duin, D.; Newman, F.K.; Zhang, L.; Chen, S.; Towle, V.; Belshe, R.B.; Fikrig, E.; et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J. Immunol. 2010, 184, 2518–2527. [Google Scholar] [CrossRef]
- Boehmer, E.D.; Meehan, M.J.; Cutro, B.T.; Kovacs, E.J. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech. Ageing Dev. 2005, 126, 1305–1313. [Google Scholar] [CrossRef]
- Renshaw, M.; Rockwell, J.; Engleman, C.; Gewirtz, A.; Katz, J.; Sambhara, S. Cutting edge: Impaired Toll-like receptor expression and function in aging. J. Immunol. 2002, 169, 4697–4701. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Prattichizzo, F.; Babini, L.; Graciotti, L.; Recchioni, R.; Procopio, A.D. Toll like receptor signaling in “inflammaging”: MicroRNA as new players. Immun. Ageing 2013, 10, 11. [Google Scholar] [CrossRef]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejia, J.E.; Guery, J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef]
- Gao, Z.; Dosman, J.A.; Rennie, D.C.; Schwartz, D.A.; Yang, I.V.; Beach, J.; Senthilselvan, A. Gender-specific associations between polymorphisms in the Toll-like receptor (TLR) genes and lung function among workers in swine operations. J. Toxicol. Environ. Health A 2018, 81, 1186–1198. [Google Scholar] [CrossRef]
- Elsherif, R.H.; Algebaly, H.A.F.; Ismail, D.K.; Meligy, B.; Aziz, M.M.; Ghaith, D.M.; Salah, E. Toll-like receptors 2 and 9 gene polymorphisms in severe sepsis and septic shock: A single center study in the pediatric intensive care unit. Int. J. Clin. Exp. Med. 2019, 12, 4381–4386. [Google Scholar]
- Rettew, J.A.; Huet-Hudson, Y.M.; Marriott, I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol. Reprod. 2008, 78, 432–437. [Google Scholar] [CrossRef]
- Debnath, M.; Banerjee, M.; Berk, M. Genetic gateways to COVID-19 infection: Implications for risk, severity, and outcomes. FASEB J. 2020, 34, 8787–8795. [Google Scholar] [CrossRef]
- Diaz, O.E.; Sorini, C.; Morales, R.A.; Luo, X.; Frede, A.; Krais, A.M.; Chavez, M.N.; Wincent, E.; Das, S.; Villablanca, E.J. Perfluorooctanesulfonic acid modulates barrier function and systemic T-cell homeostasis during intestinal inflammation. Dis. Model. Mech. 2021, 14, dmm049104. [Google Scholar] [CrossRef]
- Thomalla, M.; Schmid, A.; Neumann, E.; Pfefferle, P.I.; Muller-Ladner, U.; Schaffler, A.; Karrasch, T. Evidence of an anti-inflammatory toll-like receptor 9 (TLR 9) pathway in adipocytes. J. Endocrinol. 2019, 240, 325–343. [Google Scholar] [CrossRef]
- Nishimoto, S.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Murata, C.; Kim-Kaneyama, J.R.; Sato, F.; Bando, M.; Yagi, S.; et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci. Adv. 2016, 2, e1501332. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol. Pharm. Bull. 2005, 28, 886–892. [Google Scholar] [CrossRef]
- Frantz, S.; Kobzik, L.; Kim, Y.D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef]
- Foldes, G.; von Haehling, S.; Okonko, D.O.; Jankowska, E.A.; Poole-Wilson, P.A.; Anker, S.D. Fluvastatin reduces increased blood monocyte Toll-like receptor 4 expression in whole blood from patients with chronic heart failure. Int. J. Cardiol. 2008, 124, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 9874109. [Google Scholar] [CrossRef] [PubMed]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.; Holt, M.R.; Heads, R.J. SARS-CoV-2 Spike S1 glycoprotein is a TLR4 agonist, upregulates ACE2 expression and induces pro-inflammatory M1 macrophage polarisation. bioRxiv Prepr. Serv. Biol. 2021, 8, 455921. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L. The Membrane Protein of Severe Acute Respiratory Syndrome Coronavirus Functions as a Novel Cytosolic Pathogen-Associated Molecular Pattern To Promote Beta Interferon Induction via a Toll-Like-Receptor-Related TRAF3-Independent Mechanism. mBio 2016, 7, e01872-15. [Google Scholar] [CrossRef]
- Cyprian, F.; Sohail, M.U.; Abdelhafez, I.; Salman, S.; Attique, Z.; Kamareddine, L.; Al-Asmakh, M. SARS-CoV-2 and immune-microbiome interactions: Lessons from respiratory viral infections. Int. J. Infect. Dis. 2021, 105, 540–550. [Google Scholar] [CrossRef]
- Weaver, L.K.; Minichino, D.; Biswas, C.; Chu, N.; Lee, J.J.; Bittinger, K.; Albeituni, S.; Nichols, K.E.; Behrens, E.M. Microbiota-dependent signals are required to sustain TLR-mediated immune responses. JCI Insight 2019, 4, e124370. [Google Scholar] [CrossRef]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef]
- Duan, Y.; Zeng, L.; Zheng, C.; Song, B.; Li, F.; Kong, X.; Xu, K. Inflammatory Links Between High Fat Diets and Diseases. Front. Immunol. 2018, 9, 2649. [Google Scholar] [CrossRef]
- Poulain-Godefroy, O.; Le Bacquer, O.; Plancq, P.; Lecoeur, C.; Pattou, F.; Fruhbeck, G.; Froguel, P. Inflammatory role of Toll-like receptors in human and murine adipose tissue. Mediat. Inflamm. 2010, 2010, 823486. [Google Scholar] [CrossRef]
- Cai, D.; Liu, T. Hypothalamic inflammation: A double-edged sword to nutritional diseases. Ann. N. Y. Acad. Sci. 2011, 1243, E1–E39. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef]
- Dickie, L.J.; Church, L.D.; Coulthard, L.R.; Mathews, R.J.; Emery, P.; McDermott, M.F. Vitamin D3 down-regulates intracellular Toll-like receptor 9 expression and Toll-like receptor 9-induced IL-6 production in human monocytes. Rheumatology 2010, 49, 1466–1471. [Google Scholar] [CrossRef]
- Martinez-Moreno, J.; Hernandez, J.C.; Urcuqui-Inchima, S. Effect of high doses of vitamin D supplementation on dengue virus replication, Toll-like receptor expression, and cytokine profiles on dendritic cells. Mol. Cell Biochem. 2020, 464, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Shen, L.; Ye, X.; Zhou, D.; He, Y.; Zhang, H. Mechanism of immunosuppression in zebrafish (Danio rerio) spleen induced by environmentally relevant concentrations of perfluorooctanoic acid. Chemosphere 2020, 249, 126200. [Google Scholar] [CrossRef] [PubMed]
- Piuri, G.; Soriano, J.; Speciani, M.C.; Speciani, A.F. B cell activating factor (BAFF) and platelet activating factor (PAF) could both be markers of non-IgE-mediated reactions. Clin. Transl. Allergy 2013, 3, O5. [Google Scholar] [CrossRef]
- Zhang, H.; Fang, W.; Wang, D.; Gao, N.; Ding, Y.; Chen, C. The role of interleukin family in perfluorooctanoic acid (PFOA)-induced immunotoxicity. J. Hazard. Mater. 2014, 280, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martinez-Colon, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. J. Infect. Dis. 2020, 221, 1762–1769. [Google Scholar] [CrossRef]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020, 11, 3434. [Google Scholar] [CrossRef]
- Mehta, P.; Fajgenbaum, D.C. Is severe COVID-19 a cytokine storm syndrome: A hyperinflammatory debate. Curr. Opin. Rheumatol. 2021, 33, 419–430. [Google Scholar] [CrossRef]
- KE1868. Available online: https://aopwiki.org/events/1868 (accessed on 16 March 2022).
- Bergamaschi, L.; Mescia, F.; Turner, L.; Hanson, A.; Kotagiri, P.; Dunmore, B.J.; Ruffieux, H.; De Sa, A.; Huhn, O.; Morgan, M.D.; et al. Delayed bystander CD8 T cell activation, early immune pathology and persistent dysregulation characterise severe COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Bonafe, M.; Prattichizzo, F.; Giuliani, A.; Storci, G.; Sabbatinelli, J.; Olivieri, F. Inflamm-aging: Why older men are the most susceptible to SARS-CoV-2 complicated outcomes. Cytokine Growth Factor Rev. 2020, 53, 33–37. [Google Scholar] [CrossRef]
- Kovacs, E.J.; Boe, D.M.; Boule, L.A.; Curtis, B.J. Inflammaging and the Lung. Clin. Geriatr. Med. 2017, 33, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.R. Age-related alterations in immune responses to West Nile virus infection. Clin. Exp. Immunol. 2017, 187, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Zust, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, K.; van Dijken, H.; Wijsman, L.; Gomersbach, A.; Schouten, T.; Kool, J.; Lenz, S.; Roholl, P.; Meijer, A.; van Kasteren, P.B.; et al. Pathology and Immunity After SARS-CoV-2 Infection in Male Ferrets Is Affected by Age and Inoculation Route. Front. Immunol. 2021, 12, 750229. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; de Lang, A.; van den Brand, J.M.; Leijten, L.M.; van Ijcken, W.F.; Eijkemans, M.J.; van Amerongen, G.; Kuiken, T.; Andeweg, A.C.; Osterhaus, A.D.; et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010, 6, e1000756. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, V.; Gadi, N.; Spihlman, A.P.; Wu, S.C.; Choi, C.H.; Moulton, V.R. Aging, Immunity, and COVID-19: How Age Influences the Host Immune Response to Coronavirus Infections? Front. Physiol. 2020, 11, 571416. [Google Scholar] [CrossRef]
- Takahashi, T.; Ellingson, M.K.; Wong, P.; Israelow, B.; Lucas, C.; Klein, J.; Silva, J.; Mao, T.; Oh, J.E.; Tokuyama, M.; et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 2020, 588, 315–320. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Scully, E.P.; Haverfield, J.; Ursin, R.L.; Tannenbaum, C.; Klein, S.L. Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat. Rev. Immunol. 2020, 20, 442–447. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Bairey Merz, N.; Barnes, P.J.; Brinton, R.D.; Carrero, J.-J.; DeMeo, D.L.; De Vries, G.J.; Epperson, C.N.; Govindan, R.; Klein, S.L.; et al. Sex and gender: Modifiers of health, disease, and medicine. Lancet 2020, 396, 565–582. [Google Scholar] [CrossRef]
- Goncalves, J.; Juliano, A.M.; Charepe, N.; Alenquer, M.; Athayde, D.; Ferreira, F.; Archer, M.; Amorim, M.J.; Serrano, F.; Soares, H. Secretory IgA and T cells targeting SARS-CoV-2 spike protein are transferred to the breastmilk upon mRNA vaccination. Cell Rep. Med. 2021, 2, 100468. [Google Scholar] [CrossRef]
- Agrawal, S.; Salazar, J.; Tran, T.M.; Agrawal, A. Sex-Related Differences in Innate and Adaptive Immune Responses to SARS-CoV-2. Front. Immunol. 2021, 12, 739757. [Google Scholar] [CrossRef]
- Singh, D.; Wasan, H.; Reeta, K.H. Heme oxygenase-1 modulation: A potential therapeutic target for COVID-19 and associated complications. Free Radic Biol. Med. 2020, 161, 263–271. [Google Scholar] [CrossRef]
- Rowley, A.H. Understanding SARS-CoV-2-related multisystem inflammatory syndrome in children. Nat. Rev. Immunol. 2020, 20, 453–454. [Google Scholar] [CrossRef]
- Caterino, M.; Gelzo, M.; Sol, S.; Fedele, R.; Annunziata, A.; Calabrese, C.; Fiorentino, G.; D’Abbraccio, M.; Dell’Isola, C.; Fusco, F.M.; et al. Dysregulation of lipid metabolism and pathological inflammation in patients with COVID-19. Sci. Rep. 2021, 11, 2941. [Google Scholar] [CrossRef]
- Hubler, M.J.; Kennedy, A.J. Role of lipids in the metabolism and activation of immune cells. J. Nutr. Biochem. 2016, 34, 1–7. [Google Scholar] [CrossRef]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Im, S.S.; Yousef, L.; Blaschitz, C.; Liu, J.Z.; Edwards, R.A.; Young, S.G.; Raffatellu, M.; Osborne, T.F. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011, 13, 540–549. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Pugliese, G.; Barrea, L.; Savastano, S.; Colao, A. Commentary: Obesity: The “Achilles heel” for COVID-19? Metabolism 2020, 108, 154251. [Google Scholar] [CrossRef]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes, E.S.A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef]
- Yang, L.; Nilsson-Payant, B.E.; Han, Y.; Jaffre, F.; Zhu, J.; Wang, P.; Zhang, T.; Redmond, D.; Houghton, S.; Moller, R.; et al. Cardiomyocytes recruit monocytes upon SARS-CoV-2 infection by secreting CCL2. Stem Cell Rep. 2021, 16, 2274–2288. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Jaffre, F.; Nilsson-Payant, B.E.; Bram, Y.; Wang, P.; Zhu, J.; Zhang, T.; Redmond, D.; Houghton, S.; et al. An Immuno-Cardiac Model for Macrophage-Mediated Inflammation in COVID-19 Hearts. Circ. Res. 2021, 129, 33–46. [Google Scholar] [CrossRef]
- Kawai, T.; Elliott, K.J.; Scalia, R.; Eguchi, S. Contribution of ADAM17 and related ADAMs in cardiovascular diseases. Cell Mol. Life Sci. 2021, 78, 4161–4187. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Iwasaka, J.; Nakamura, M.; Akatsu, T.; Shimoda, Y.; Hiramori, K. Increased expression of tumor necrosis factor-alpha converting enzyme and tumor necrosis factor-alpha in peripheral blood mononuclear cells in patients with advanced congestive heart failure. Eur. J. Heart Fail 2004, 6, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Nakamura, M.; Saitoh, H.; Satoh, H.; Maesawa, C.; Segawa, I.; Tashiro, A.; Hiramori, K. Tumor necrosis factor-alpha-converting enzyme and tumor necrosis factor-alpha in human dilated cardiomyopathy. Circulation 1999, 99, 3260–3265. [Google Scholar] [CrossRef] [PubMed]
- Palacios, Y.; Ruiz, A.; Ramon-Luing, L.A.; Ocana-Guzman, R.; Barreto-Rodriguez, O.; Sanchez-Moncivais, A.; Tecuatzi-Cadena, B.; Regalado-Garcia, A.G.; Pineda-Gudino, R.D.; Garcia-Martinez, A.; et al. Severe COVID-19 Patients Show an Increase in Soluble TNFR1 and ADAM17, with a Relationship to Mortality. Int. J. Mol. Sci. 2021, 22, 8423. [Google Scholar] [CrossRef]
- Ben Moftah, M.; Eswayah, A. Intricate relationship between SARS-CoV-2-induced shedding and cytokine storm generation: A signaling inflammatory pathway augmenting COVID-19. Health Sci. Rev. 2022, 2, 100011. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e117. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L., Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef]
- Gou, W.; Fu, Y.; Yue, L.; Chen, G.D.; Cai, X.; Shuai, M.; Xu, F.; Yi, X.; Chen, H.; Zhu, Y.; et al. Gut microbiota, inflammation, and molecular signatures of host response to infection. J. Genet. Genom. 2021, 48, 792–802. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef]
- Armengaud, J.; Grenga, L.; Pible, O.; Miotello, G.; Culotta, K.; Ruat, S.; Roncato, M.-A.; Gas, F.; Bellanger, L.; Claret, P.-G.; et al. Taxonomical and functional changes in COVID-19 faecal microbiome are related to SARS-CoV-2 faecal load. Res. Sq. Prepr. 2021. [Google Scholar] [CrossRef]
- Cristofori, F.; Dargenio, V.N.; Dargenio, C.; Miniello, V.L.; Barone, M.; Francavilla, R. Anti-Inflammatory and Immunomodulatory Effects of Probiotics in Gut Inflammation: A Door to the Body. Front. Immunol. 2021, 12, 578386. [Google Scholar] [CrossRef]
- Larsen, J.M. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; Czarlewski, W.; Zuberbier, T.; Mullol, J.; Blain, H.; Cristol, J.P.; De La Torre, R.; Le Moing, V.; Pizarro Lozano, N.; Bedbrook, A.; et al. Spices to Control COVID-19 Symptoms: Yes, but Not Only. Int. Arch. Allergy Immunol. 2021, 182, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; Cristol, J.P.; Czarlewski, W.; Anto, J.M.; Martineau, A.; Haahtela, T.; Fonseca, S.C.; Iaccarino, G.; Blain, H.; Fiocchi, A.; et al. Nrf2-interacting nutrients and COVID-19: Time for research to develop adaptation strategies. Clin. Transl. Allergy 2020, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zheng, W.; Cheng, L.; Li, M.; Huang, J.; Bao, S.; Xu, Q.; Ma, Z. Citrus fruits are rich in flavonoids for immunoregulation and potential targeting ACE2. Nat. Prod. Bioprospect 2022, 12, 4. [Google Scholar] [CrossRef]
- Chuchawankul, S.; Khorana, N.; Poovorawan, Y. Piperine inhibits cytokine production by human peripheral blood mononuclear cells. Genet. Mol. Res. 2012, 11, 617–627. [Google Scholar] [CrossRef]
- Mittal, R.; Gupta, R.L. In vitro antioxidant activity of piperine. Methods Find Exp. Clin. Pharmacol. 2000, 22, 271–274. [Google Scholar] [CrossRef]
- Kim, J.; Jayaprakasha, G.K.; Muthuchamy, M.; Patil, B.S. Structure-function relationships of citrus limonoids on p38 MAP kinase activity in human aortic smooth muscle cells. Eur. J. Pharmacol. 2011, 670, 44–49. [Google Scholar] [CrossRef]
- Kole, L.; Giri, B.; Manna, S.K.; Pal, B.; Ghosh, S. Biochanin-A, an isoflavon, showed anti-proliferative and anti-inflammatory activities through the inhibition of iNOS expression, p38-MAPK and ATF-2 phosphorylation and blocking NFkappaB nuclear translocation. Eur. J. Pharmacol. 2011, 653, 8–15. [Google Scholar] [CrossRef]
- North, C.J.; Venter, C.S.; Jerling, J.C. The effects of dietary fibre on C-reactive protein, an inflammation marker predicting cardiovascular disease. Eur. J. Clin. Nutr. 2009, 63, 921–933. [Google Scholar] [CrossRef]
- Magni, P.; Ruscica, M.; Dozio, E.; Rizzi, E.; Beretta, G.; Maffei Facino, R. Parthenolide inhibits the LPS-induced secretion of IL-6 and TNF-alpha and NF-kappaB nuclear translocation in BV-2 microglia. Phytother. Res. 2012, 26, 1405–1409. [Google Scholar] [CrossRef]
- Bahrami, M.; Kamalinejad, M.; Latifi, S.A.; Seif, F.; Dadmehr, M. Cytokine storm in COVID-19 and parthenolide: Preclinical evidence. Phytother. Res. 2020, 34, 2429–2430. [Google Scholar] [CrossRef]
- Laponogov, I.; Gonzalez, G.; Shepherd, M.; Qureshi, A.; Veselkov, D.; Charkoftaki, G.; Vasiliou, V.; Youssef, J.; Mirnezami, R.; Bronstein, M.; et al. Network machine learning maps phytochemically rich “Hyperfoods” to fight COVID-19. Hum. Genom. 2021, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.H.; Elsadek, A.A.M.; Mahmoud, M.A.; Elsadek, B.E.M. Vitamin D Receptor Gene Polymorphisms and Risk of Knee Osteoarthritis: Possible Correlations with TNF-alpha, Macrophage Migration Inhibitory Factor, and 25-Hydroxycholecalciferol Status. Biochem. Genet. 2022, 60, 611–628. [Google Scholar] [CrossRef]
- Ksiazek, A.; Zagrodna, A.; Bohdanowicz-Pawlak, A.; Lwow, F.; Slowinska-Lisowska, M. Relationships between Vitamin D and Selected Cytokines and Hemogram Parameters in Professional Football Players-Pilot Study. Int. J. Environ. Res. Public Health 2021, 18, 7124. [Google Scholar] [CrossRef] [PubMed]
- Helming, L.; Bose, J.; Ehrchen, J.; Schiebe, S.; Frahm, T.; Geffers, R.; Probst-Kepper, M.; Balling, R.; Lengeling, A. 1alpha,25-Dihydroxyvitamin D3 is a potent suppressor of interferon gamma-mediated macrophage activation. Blood 2005, 106, 4351–4358. [Google Scholar] [CrossRef]
- Munshi, R.; Hussein, M.H.; Toraih, E.A.; Elshazli, R.M.; Jardak, C.; Sultana, N.; Youssef, M.R.; Omar, M.; Attia, A.S.; Fawzy, M.S.; et al. Vitamin D insufficiency as a potential culprit in critical COVID-19 patients. J. Med. Virol. 2021, 93, 733–740. [Google Scholar] [CrossRef]
- Kurt, O.K.; Zhang, J.; Pinkerton, K.E. Pulmonary health effects of air pollution. Curr. Opin. Pulm. Med. 2016, 22, 138–143. [Google Scholar] [CrossRef]
- Zhao, J.; Gao, Z.; Tian, Z.; Xie, Y.; Xin, F.; Jiang, R.; Kan, H.; Song, W. The biological effects of individual-level PM(2.5) exposure on systemic immunity and inflammatory response in traffic policemen. Occup. Environ. Med. 2013, 70, 426–431. [Google Scholar] [CrossRef]
- Jia, H.; Liu, Y.; Guo, D.; He, W.; Zhao, L.; Xia, S. PM2.5-induced pulmonary inflammation via activating of the NLRP3/caspase-1 signaling pathway. Environ. Toxicol. 2021, 36, 298–307. [Google Scholar] [CrossRef]
- Tsai, D.H.; Amyai, N.; Marques-Vidal, P.; Wang, J.L.; Riediker, M.; Mooser, V.; Paccaud, F.; Waeber, G.; Vollenweider, P.; Bochud, M. Effects of particulate matter on inflammatory markers in the general adult population. Part Fibre Toxicol. 2012, 9, 24. [Google Scholar] [CrossRef]
- Ljungman, P.; Bellander, T.; Schneider, A.; Breitner, S.; Forastiere, F.; Hampel, R.; Illig, T.; Jacquemin, B.; Katsouyanni, K.; von Klot, S.; et al. Modification of the interleukin-6 response to air pollution by interleukin-6 and fibrinogen polymorphisms. Environ. Health Perspect. 2009, 117, 1373–1379. [Google Scholar] [CrossRef]
- Kido, T.; Tamagawa, E.; Bai, N.; Suda, K.; Yang, H.H.; Li, Y.; Chiang, G.; Yatera, K.; Mukae, H.; Sin, D.D.; et al. Particulate matter induces translocation of IL-6 from the lung to the systemic circulation. Am. J. Respir. Cell Mol. Biol. 2011, 44, 197–204. [Google Scholar] [CrossRef]
- Winquist, A.; Schauer, J.J.; Turner, J.R.; Klein, M.; Sarnat, S.E. Impact of ambient fine particulate matter carbon measurement methods on observed associations with acute cardiorespiratory morbidity. J. Expo Sci. Environ. Epidemiol. 2015, 25, 215–221. [Google Scholar] [CrossRef]
- Rydman, E.M.; Ilves, M.; Koivisto, A.J.; Kinaret, P.A.; Fortino, V.; Savinko, T.S.; Lehto, M.T.; Pulkkinen, V.; Vippola, M.; Hameri, K.J.; et al. Inhalation of rod-like carbon nanotubes causes unconventional allergic airway inflammation. Part Fibre Toxicol. 2014, 11, 48. [Google Scholar] [CrossRef]
- Kinaret, P.; Ilves, M.; Fortino, V.; Rydman, E.; Karisola, P.; Lahde, A.; Koivisto, J.; Jokiniemi, J.; Wolff, H.; Savolainen, K.; et al. Inhalation and Oropharyngeal Aspiration Exposure to Rod-Like Carbon Nanotubes Induce Similar Airway Inflammation and Biological Responses in Mouse Lungs. ACS Nano 2017, 11, 291–303. [Google Scholar] [CrossRef]
- Labib, S.; Williams, A.; Yauk, C.L.; Nikota, J.K.; Wallin, H.; Vogel, U.; Halappanavar, S. Nano-risk Science: Application of toxicogenomics in an adverse outcome pathway framework for risk assessment of multi-walled carbon nanotubes. Part Fibre Toxicol. 2016, 13, 15. [Google Scholar] [CrossRef]
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 720109. [Google Scholar] [CrossRef]
- Glencross, D.A.; Ho, T.R.; Camina, N.; Hawrylowicz, C.M.; Pfeffer, P.E. Air pollution and its effects on the immune system. Free Radic Biol. Med. 2020, 151, 56–68. [Google Scholar] [CrossRef]
- Yamasaki, K.; Whalen, B.; Van Eeden, S. Impact of particulate matter (PM) exposure on lung macrophage phenotype and phagocytic activity. Eur. Respir. J. 2020, 56, 1968. [Google Scholar] [CrossRef]
- Signorini, C.; Pignatti, P.; Coccini, T. How Do Inflammatory Mediators, Immune Response and Air Pollution Contribute to COVID-19 Disease Severity? A Lesson to Learn. Life 2021, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Al-Kindi, S.G.; Brook, R.D.; Biswal, S.; Rajagopalan, S. Environmental determinants of cardiovascular disease: Lessons learned from air pollution. Nat. Rev. Cardiol. 2020, 17, 656–672. [Google Scholar] [CrossRef]
- Forbes, L.J.; Patel, M.D.; Rudnicka, A.R.; Cook, D.G.; Bush, T.; Stedman, J.R.; Whincup, P.H.; Strachan, D.P.; Anderson, R.H. Chronic exposure to outdoor air pollution and markers of systemic inflammation. Epidemiology 2009, 20, 245–253. [Google Scholar] [CrossRef]
- Ostro, B.; Malig, B.; Broadwin, R.; Basu, R.; Gold, E.B.; Bromberger, J.T.; Derby, C.; Feinstein, S.; Greendale, G.A.; Jackson, E.A.; et al. Chronic PM2.5 exposure and inflammation: Determining sensitive subgroups in mid-life women. Environ. Res. 2014, 132, 168–175. [Google Scholar] [CrossRef]
- Mescoli, A.; Maffei, G.; Pillo, G.; Bortone, G.; Marchesi, S.; Morandi, E.; Ranzi, A.; Rotondo, F.; Serra, S.; Vaccari, M.; et al. The Secretive Liaison of Particulate Matter and SARS-CoV-2. A Hypothesis and Theory Investigation. Front. Genet. 2020, 11, 579964. [Google Scholar] [CrossRef]
- Jankowska-Kieltyka, M.; Roman, A.; Nalepa, I. The Air We Breathe: Air Pollution as a Prevalent Proinflammatory Stimulus Contributing to Neurodegeneration. Front. Cell Neurosci. 2021, 15, 647643. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, M.A.; Rivest, S. The HPA—Immune Axis and the Immunomodulatory Actions of Glucocorticoids in the Brain. Front. Immunol. 2014, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Deek, S.A. Chronic exposure to air pollution implications on COVID-19 severity. Med. Hypotheses 2020, 145, 110303. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Hong, Y.; Li, Z.; Yang, Z.; Lei, B.; Liu, J.; Cai, Z. Immunometabolism-modulation and immunotoxicity evaluation of perfluorooctanoic acid in macrophage. Ecotoxicol. Environ. Saf. 2021, 215, 112128. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Guo, C.; Lin, X.; Chan, T.F.; Lai, K.P.; Chen, J. Integrative omics analyses uncover the mechanism underlying the immunotoxicity of perfluorooctanesulfonate in human lymphocytes. Chemosphere 2020, 256, 127062. [Google Scholar] [CrossRef] [PubMed]
- Sorli, J.B.; Lag, M.; Ekeren, L.; Perez-Gil, J.; Haug, L.S.; Da Silva, E.; Matrod, M.N.; Gutzkow, K.B.; Lindeman, B. Per- and polyfluoroalkyl substances (PFASs) modify lung surfactant function and pro-inflammatory responses in human bronchial epithelial cells. Toxicol. In Vitro 2020, 62, 104656. [Google Scholar] [CrossRef]
- Corsini, E.; Sangiovanni, E.; Avogadro, A.; Galbiati, V.; Viviani, B.; Marinovich, M.; Galli, C.L.; Dell’Agli, M.; Germolec, D.R. In vitro characterization of the immunotoxic potential of several perfluorinated compounds (PFCs). Toxicol. Appl. Pharmacol. 2012, 258, 248–255. [Google Scholar] [CrossRef]
- Han, R.; Hu, M.; Zhong, Q.; Wan, C.; Liu, L.; Li, F.; Zhang, F.; Ding, W. Perfluorooctane sulphonate induces oxidative hepatic damage via mitochondria-dependent and NF-kappaB/TNF-alpha-mediated pathway. Chemosphere 2018, 191, 1056–1064. [Google Scholar] [CrossRef]
- Pennings, J.L.; Jennen, D.G.; Nygaard, U.C.; Namork, E.; Haug, L.S.; van Loveren, H.; Granum, B. Cord blood gene expression supports that prenatal exposure to perfluoroalkyl substances causes depressed immune functionality in early childhood. J. Immunotoxicol. 2016, 13, 173–180. [Google Scholar] [CrossRef]
- Abraham, K.; Mielke, H.; Fromme, H.; Volkel, W.; Menzel, J.; Peiser, M.; Zepp, F.; Willich, S.N.; Weikert, C. Internal exposure to perfluoroalkyl substances (PFASs) and biological markers in 101 healthy 1-year-old children: Associations between levels of perfluorooctanoic acid (PFOA) and vaccine response. Arch. Toxicol. 2020, 94, 2131–2147. [Google Scholar] [CrossRef]
- Therapeutics and COVID-19: Living Guideline; WHO/2019-NCoV/Therapeutics/2021.1. 2021. Available online: https://apps.who.int/iris/bitstream/handle/10665/342368/WHO-2019-nCoV-therapeutics-2021.2-eng.pdf (accessed on 11 May 2022).
- (EMA), E.M.A. RoActemra. 2021. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/roactemra (accessed on 11 May 2022).
- Jorgensen, S.C.J.; Tse, C.L.Y.; Burry, L.; Dresser, L.D. Baricitinib: A Review of Pharmacology, Safety, and Emerging Clinical Experience in COVID-19. Pharmacotherapy 2020, 40, 843–856. [Google Scholar] [CrossRef]
- Bekerman, E.; Neveu, G.; Shulla, A.; Brannan, J.; Pu, S.Y.; Wang, S.; Xiao, F.; Barouch-Bentov, R.; Bakken, R.R.; Mateo, R.; et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J. Clin. Investig. 2017, 127, 1338–1352. [Google Scholar] [CrossRef]
- Neveu, G.; Ziv-Av, A.; Barouch-Bentov, R.; Berkerman, E.; Mulholland, J.; Einav, S. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J. Virol. 2015, 89, 4387–4404. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef]
- Buijsers, B.; Yanginlar, C.; Maciej-Hulme, M.L.; de Mast, Q.; van der Vlag, J. Beneficial non-anticoagulant mechanisms underlying heparin treatment of COVID-19 patients. EBioMedicine 2020, 59, 102969. [Google Scholar] [CrossRef]
- KE1931. Available online: https://aopwiki.org/events/1931 (accessed on 16 March 2022).
- Rhee, S.H. Lipopolysaccharide: Basic biochemistry, intracellular signaling, and physiological impacts in the gut. Intest. Res. 2014, 12, 90–95. [Google Scholar] [CrossRef]
- Openshaw, P.J. Crossing barriers: Infections of the lung and the gut. Mucosal. Immunol. 2009, 2, 100–102. [Google Scholar] [CrossRef]
- Cardinale, V.; Capurso, G.; Ianiro, G.; Gasbarrini, A.; Arcidiacono, P.G.; Alvaro, D. Intestinal permeability changes with bacterial translocation as key events modulating systemic host immune response to SARS-CoV-2: A working hypothesis. Dig. Liver Dis. 2020, 52, 1383–1389. [Google Scholar] [CrossRef]
- Giron, L.B.; Dweep, H.; Yin, X.; Wang, H.; Damra, M.; Goldman, A.R.; Gorman, N.; Palmer, C.S.; Tang, H.Y.; Shaikh, M.W.; et al. Plasma Markers of Disrupted Gut Permeability in Severe COVID-19 Patients. Front. Immunol. 2021, 12, 686240. [Google Scholar] [CrossRef]
- Prasad, R.; Patton, M.J.; Floyd, J.L.; Vieira, C.P.; Fortmann, S.; DuPont, M.; Harbour, A.; Jeremy, C.S.; Wright, J.; Lamendella, R.; et al. Plasma microbiome in COVID-19 subjects: An indicator of gut barrier defects and dysbiosis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tran, L.; Greenwood-Van Meerveld, B. Age-associated remodeling of the intestinal epithelial barrier. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1045–1056. [Google Scholar] [CrossRef]
- Wilms, E.; Troost, F.J.; Elizalde, M.; Winkens, B.; de Vos, P.; Mujagic, Z.; Jonkers, D.; Masclee, A.A.M. Intestinal barrier function is maintained with aging—A comprehensive study in healthy subjects and irritable bowel syndrome patients. Sci. Rep. 2020, 10, 475. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhu, Y.; Huang, X.; Zhang, W.; Han, Z.; Liu, S. Association between TLR2 and TLR4 Gene Polymorphisms and the Susceptibility to Inflammatory Bowel Disease: A Meta-Analysis. PLoS ONE 2015, 10, e0126803. [Google Scholar] [CrossRef]
- Torok, H.P.; Bellon, V.; Konrad, A.; Lacher, M.; Tonenchi, L.; Siebeck, M.; Brand, S.; De Toni, E.N. Functional Toll-Like Receptor (TLR)2 polymorphisms in the susceptibility to inflammatory bowel disease. PLoS ONE 2017, 12, e0175180. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, S.; Zhang, J.; Lei, S.; Zhou, J. Correlations between TLR polymorphisms and inflammatory bowel disease: A meta-analysis of 49 case-control studies. Immunol. Res. 2019, 67, 142–150. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Massier, L.; Bluher, M.; Kovacs, P.; Chakaroun, R.M. Impaired Intestinal Barrier and Tissue Bacteria: Pathomechanisms for Metabolic Diseases. Front. Endocrinol. 2021, 12, 616506. [Google Scholar] [CrossRef] [PubMed]
- Iacob, S.; Iacob, D.G. Infectious Threats, the Intestinal Barrier, and Its Trojan Horse: Dysbiosis. Front. Microbiol. 2019, 10, 1676. [Google Scholar] [CrossRef] [PubMed]
- Salvi, P.S.; Cowles, R.A. Butyrate and the Intestinal Epithelium: Modulation of Proliferation and Inflammation in Homeostasis and Disease. Cells 2021, 10, 1775. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Scaldaferri, F.; Petito, V.; Gasbarrini, A. Commensal Clostridia: Leading players in the maintenance of gut homeostasis. Gut Pathog. 2013, 5, 23. [Google Scholar] [CrossRef]
- Dean, P.; Kenny, B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol. Microbiol. 2004, 54, 665–675. [Google Scholar] [CrossRef]
- Viswanathan, V.K.; Koutsouris, A.; Lukic, S.; Pilkinton, M.; Simonovic, I.; Simonovic, M.; Hecht, G. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect. Immun. 2004, 72, 3218–3227. [Google Scholar] [CrossRef]
- Tafazoli, F.; Magnusson, K.E.; Zheng, L. Disruption of epithelial barrier integrity by Salmonella enterica serovar typhimurium requires geranylgeranylated proteins. Infect. Immun. 2003, 71, 872–881. [Google Scholar] [CrossRef]
- Fasano, A.; Baudry, B.; Pumplin, D.W.; Wasserman, S.S.; Tall, B.D.; Ketley, J.M.; Kaper, J.B. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. USA 1991, 88, 5242–5246. [Google Scholar] [CrossRef]
- Wu, C.; Xu, Q.; Cao, Z.; Pan, D.; Zhu, Y.; Wang, S.; Liu, D.; Song, Z.; Jiang, W.; Ruan, Y.; et al. The volatile and heterogeneous gut microbiota shifts of COVID-19 patients over the course of a probiotics-assisted therapy. Clin. Transl. Med. 2021, 11, e643. [Google Scholar] [CrossRef]
- Sun, Z.; Song, Z.G.; Liu, C.; Tan, S.; Lin, S.; Zhu, J.; Dai, F.H.; Gao, J.; She, J.L.; Mei, Z.; et al. Gut microbiome alterations and gut barrier dysfunction are associated with host immune homeostasis in COVID-19 patients. BMC Med. 2022, 20, 24. [Google Scholar] [CrossRef]
- Leech, B.; McIntyre, E.; Steel, A.; Sibbritt, D. Risk factors associated with intestinal permeability in an adult population: A systematic review. Int. J. Clin. Pract. 2019, 73, e13385. [Google Scholar] [CrossRef]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef]
- Ferro, Y.; Pujia, R.; Maurotti, S.; Boragina, G.; Mirarchi, A.; Gnagnarella, P.; Mazza, E. Mediterranean Diet a Potential Strategy against SARS-CoV-2 Infection: A Narrative Review. Medicina 2021, 57, 1389. [Google Scholar] [CrossRef]
- Assa, A.; Vong, L.; Pinnell, L.J.; Avitzur, N.; Johnson-Henry, K.C.; Sherman, P.M. Vitamin D deficiency promotes epithelial barrier dysfunction and intestinal inflammation. J. Infect. Dis. 2014, 210, 1296–1305. [Google Scholar] [CrossRef]
- Du, J.; Chen, Y.; Shi, Y.; Liu, T.; Cao, Y.; Tang, Y.; Ge, X.; Nie, H.; Zheng, C.; Li, Y.C. 1,25-Dihydroxyvitamin D Protects Intestinal Epithelial Barrier by Regulating the Myosin Light Chain Kinase Signaling Pathway. Inflamm. Bowel Dis. 2015, 21, 2495–2506. [Google Scholar] [CrossRef]
- Eslamian, G.; Ardehali, S.H.; Hajimohammadebrahim-Ketabforoush, M.; Vahdat Shariatpanahi, Z. Association of intestinal permeability with admission vitamin D deficiency in patients who are critically ill. J. Investig. Med. 2020, 68, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.A.; Engen, P.A.; Soberanes, S.; Urich, D.; Forsyth, C.B.; Nigdelioglu, R.; Chiarella, S.E.; Radigan, K.A.; Gonzalez, A.; Jakate, S.; et al. Particulate matter air pollution causes oxidant-mediated increase in gut permeability in mice. Part Fibre Toxicol. 2011, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Rashid, F.; Ahmad, S.; Irudayaraj, J.M.K. Effect of Perfluorooctanoic Acid on the Epigenetic and Tight Junction Genes of the Mouse Intestine. Toxics 2020, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sun, S.; Wu, X.; Yang, S.; Wu, Y.; Zhao, J.; Zhang, H.; Chen, W. Intestinal environmental disorders associate with the tissue damages induced by perfluorooctane sulfonate exposure. Ecotoxicol. Environ. Saf. 2020, 197, 110590. [Google Scholar] [CrossRef]
- Zhang, L.; Rimal, B.; Nichols, R.G.; Tian, Y.; Smith, P.B.; Hatzakis, E.; Chang, S.C.; Butenhoff, J.L.; Peters, J.M.; Patterson, A.D. Perfluorooctane sulfonate alters gut microbiota-host metabolic homeostasis in mice. Toxicology 2020, 431, 152365. [Google Scholar] [CrossRef]
- Huang, J.; Wang, Q.; Liu, S.; Lai, H.; Tu, W. Comparative chronic toxicities of PFOS and its novel alternatives on the immune system associated with intestinal microbiota dysbiosis in adult zebrafish. J. Hazard. Mater. 2022, 425, 127950. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Pawelec, G.; Khalil, A.; Cohen, A.A.; Hirokawa, K.; Witkowski, J.M.; Franceschi, C. Immunology of Aging: The Birth of Inflammaging. Clin. Rev. Allergy Immunol. 2021, 1–14. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Domingo, P.; Mur, I.; Pomar, V.; Corominas, H.; Casademont, J.; de Benito, N. The four horsemen of a viral Apocalypse: The pathogenesis of SARS-CoV-2 infection (COVID-19). EBioMedicine 2020, 58, 102887. [Google Scholar] [CrossRef]
- Hampton, T. Insight on Sex-Based Immunity Differences, With COVID-19 Implications. JAMA 2020, 324, 1274. [Google Scholar] [CrossRef]
- Viveiros, A.; Rasmuson, J.; Vu, J.; Mulvagh, S.L.; Yip, C.Y.Y.; Norris, C.M.; Oudit, G.Y. Sex differences in COVID-19: Candidate pathways, genetics of ACE2, and sex hormones. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H296–H304. [Google Scholar] [CrossRef]
- Schurz, H.; Salie, M.; Tromp, G.; Hoal, E.G.; Kinnear, C.J.; Moller, M. The X chromosome and sex-specific effects in infectious disease susceptibility. Hum. Genom. 2019, 13, 2. [Google Scholar] [CrossRef]
- Hawkes, S.; Pantazis, A.; Purdie, A.; Gautam, A.; Kiwuwa-Muyingo, S.; Buse, K.; Tanaka, S.; Borkotoky, K.; Sharma, S.; Verma, R. Sex-disaggregated data matters: Tracking the impact of COVID-19 on the health of women and men. Econ. Politica 2022, 39, 55–73. [Google Scholar] [CrossRef]
- The Sex, Gender and COVID-19 Project. Available online: https://globalhealth5050.org/the-sex-gender-and-covid-19-project/ (accessed on 16 March 2022).
- Brady, E.; Nielsen, M.W.; Andersen, J.P.; Oertelt-Prigione, S. Lack of consideration of sex and gender in clinical trials for COVID-19. medRxiv 2020, 09, 20193680. [Google Scholar] [CrossRef]
- Choi, S.W.; Mak, T.S.; O’Reilly, P.F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232. [Google Scholar] [CrossRef]
- Goldstein, M.R.; Poland, G.A.; Graeber, A.C.W. Does apolipoprotein E genotype predict COVID-19 severity? QJM 2020, 113, 529–530. [Google Scholar] [CrossRef]
- Hubacek, J.A.; Dusek, L.; Majek, O.; Adamek, V.; Cervinkova, T.; Dlouha, D.; Pavel, J.; Adamkova, V. CCR5Delta32 deletion as a protective factor in Czech first-wave COVID-19 subjects. Physiol. Res. 2021, 70, 111–115. [Google Scholar] [CrossRef]
- Valenti, L.; Griffini, S.; Lamorte, G.; Grovetti, E.; Uceda Renteria, S.C.; Malvestiti, F.; Scudeller, L.; Bandera, A.; Peyvandi, F.; Prati, D.; et al. Chromosome 3 cluster rs11385942 variant links complement activation with severe COVID-19. J. Autoimmun. 2021, 117, 102595. [Google Scholar] [CrossRef]
- Zeberg, H.; Paabo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef]
- Zeberg, H.; Paabo, S. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc. Natl. Acad. Sci. USA 2021, 118, e2026309118. [Google Scholar] [CrossRef]
- Zhou, S.; Butler-Laporte, G.; Nakanishi, T.; Morrison, D.R.; Afilalo, J.; Afilalo, M.; Laurent, L.; Pietzner, M.; Kerrison, N.; Zhao, K.; et al. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med. 2021, 27, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chi, J.; Lv, W.; Wang, Y. Obesity and diabetes as high-risk factors for severe coronavirus disease 2019 (Covid-19). Diabetes Metab. Res. Rev. 2021, 37, e3377. [Google Scholar] [CrossRef] [PubMed]
- Atmosudigdo, I.S.; Pranata, R.; Lim, M.A.; Henrina, J.; Yonas, E.; Vania, R.; Radi, B. Dyslipidemia Increases the Risk of Severe COVID-19: A Systematic Review, Meta-analysis, and Meta-regression. J. Clin. Exp. Hepatol. 2021, 9, 324–333. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, Y.; Yin, Y.; Chen, W.; Li, X. Association of dyslipidemia with the severity and mortality of coronavirus disease 2019 (COVID-19): A meta-analysis. Virol. J. 2021, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Cantrell, A.C.; Zeng, H.; Zhu, S.H.; Chen, J.X. Emerging Role of Pericytes and Their Secretome in the Heart. Cells 2021, 10, 548. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Chen, C.Y.; Knox, N.C.; Marrie, R.A.; El-Gabalawy, H.; de Kievit, T.; Alfa, M.; Bernstein, C.N.; Van Domselaar, G. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome 2018, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. The hunt for a healthy microbiome. Nature 2020, 577, S6–S8. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Scientific Opinion on the substantiation of a health claim related to vitamin D and contribution to the normal function of the immune system pursuant to Article 14 of Regulation (EC) No 1924/2006. EFSA J. 2015, 13, 4096. [CrossRef][Green Version]
- Liu, R.H. Health benefits of fruit and vegetables are from additive and synergistic combinations of phytochemicals. Am. J. Clin. Nutr. 2003, 78, 517S–520S. [Google Scholar] [CrossRef]
- Tran, B.M.; Deliyannis, G.; Hachani, A.; Earnest, L.; Torresi, J.; Vincan, E. Organoid Models of SARS-CoV-2 Infection: What Have We Learned about COVID-19? Organoids 2022, 1, 2–27. [Google Scholar] [CrossRef]
- Pizzorno, A.; Padey, B.; Julien, T.; Trouillet-Assant, S.; Traversier, A.; Errazuriz-Cerda, E.; Fouret, J.; Dubois, J.; Gaymard, A.; Lescure, F.X.; et al. Characterization and Treatment of SARS-CoV-2 in Nasal and Bronchial Human Airway Epithelia. Cell Rep. Med. 2020, 1, 100059. [Google Scholar] [CrossRef]
- Ciencewicki, J.; Jaspers, I. Air pollution and respiratory viral infection. Inhal. Toxicol. 2007, 19, 1135–1146. [Google Scholar] [CrossRef]
- Frontera, A.; Cianfanelli, L.; Vlachos, K.; Landoni, G.; Cremona, G. Severe air pollution links to higher mortality in COVID-19 patients: The “double-hit” hypothesis. J. Infect. 2020, 81, 255–259. [Google Scholar] [CrossRef]
- Comunian, S.; Dongo, D.; Milani, C.; Palestini, P. Air Pollution and COVID-19: The Role of Particulate Matter in the Spread and Increase of COVID-19’s Morbidity and Mortality. Int J. Environ. Res. Public Health 2020, 17, 4487. [Google Scholar] [CrossRef]
- Setti, L.; Passarini, F.; De Gennaro, G.; Barbieri, P.; Perrone, M.G.; Borelli, M.; Palmisani, J.; Di Gilio, A.; Torboli, V.; Fontana, F.; et al. SARS-Cov-2RNA found on particulate matter of Bergamo in Northern Italy: First evidence. Environ. Res. 2020, 188, 109754. [Google Scholar] [CrossRef]
- Anand, U.; Adelodun, B.; Pivato, A.; Suresh, S.; Indari, O.; Jakhmola, S.; Jha, H.C.; Jha, P.K.; Tripathi, V.; Di Maria, F. A review of the presence of SARS-CoV-2 RNA in wastewater and airborne particulates and its use for virus spreading surveillance. Environ. Res. 2021, 196, 110929. [Google Scholar] [CrossRef]
- Barakat, T.; Muylkens, B.; Su, B.L. Is Particulate Matter of Air Pollution a Vector of Covid-19 Pandemic? Matter 2020, 3, 977–980. [Google Scholar] [CrossRef]
- Belosi, F.; Conte, M.; Gianelle, V.; Santachiara, G.; Contini, D. On the concentration of SARS-CoV-2 in outdoor air and the interaction with pre-existing atmospheric particles. Environ. Res. 2021, 193, 110603. [Google Scholar] [CrossRef]
- Tung, N.T.; Cheng, P.C.; Chi, K.H.; Hsiao, T.C.; Jones, T.; BeruBe, K.; Ho, K.F.; Chuang, H.C. Particulate matter and SARS-CoV-2: A possible model of COVID-19 transmission. Sci. Total Environ. 2021, 750, 141532. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Trottier, B.A.; Bulka, C.M.; Cormier, S.A.; Fobil, J.; Fry, R.C.; Kim, K.W.; Kleeberger, S.; Kumar, P.; Landrigan, P.J.; et al. The interplay between environmental exposures and COVID-19 risks in the health of children. Environ. Health 2021, 20, 34. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Hundreds of COVID trials could provide a deluge of new drugs. Nature 2022, 603, 25–27. [Google Scholar] [CrossRef] [PubMed]
- International Clinical Trials Registry Platform (ICTRP). Available online: https://www.who.int/clinical-trials-registry-platform (accessed on 16 March 2022).
- Coronavirus Treatment Acceleration Program (CTAP). Available online: https://www.fda.gov/drugs/coronavirus-covid-19-drugs/coronavirus-treatment-acceleration-program-ctap (accessed on 16 March 2022).
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- WHO. Biomarkers and Risk Assessment: Concepts and Principles; WHO: Geneva, Switzerland, 1993; Available online: https://apps.who.int/iris/handle/10665/39037 (accessed on 16 March 2022).
- Zare Jeddi, M.; Hopf, N.B.; Viegas, S.; Price, A.B.; Paini, A.; van Thriel, C.; Benfenati, E.; Ndaw, S.; Bessems, J.; Behnisch, P.A.; et al. Towards a systematic use of effect biomarkers in population and occupational biomonitoring. Environ. Int. 2021, 146, 106257. [Google Scholar] [CrossRef]
- Fuellen, G.; Liesenfeld, O.; Kowald, A.; Barrantes, I.; Bastian, M.; Simm, A.; Jansen, L.; Tietz-Latza, A.; Quandt, D.; Franceschi, C.; et al. The preventive strategy for pandemics in the elderly is to collect in advance samples & data to counteract chronic inflammation (inflammaging). Ageing Res. Rev. 2020, 62, 101091. [Google Scholar] [CrossRef]
- Storci, G.; De Carolis, S.; Olivieri, F.; Bonafe, M. Changes in the biochemical taste of cytoplasmic and cell-free DNA are major fuels for inflamm-aging. Semin. Immunol. 2018, 40, 6–16. [Google Scholar] [CrossRef]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Samprathi, M.; Jayashree, M. Biomarkers in COVID-19: An Up-To-Date Review. Front. Pediatr. 2020, 8, 607647. [Google Scholar] [CrossRef]
- Gogate, N.; Lyman, D.; Bell, A.; Cauley, E.; Crandall, K.A.; Joseph, A.; Kahsay, R.; Natale, D.A.; Schriml, L.M.; Sen, S.; et al. COVID-19 biomarkers and their overlap with comorbidities in a disease biomarker data model. Brief Bioinform. 2021, 22, bbab191. [Google Scholar] [CrossRef]
- Turgay Yildirim, O.; Kaya, S. The atherogenic index of plasma as a predictor of mortality in patients with COVID-19. Heart Lung 2021, 50, 329–333. [Google Scholar] [CrossRef]
- An, W.; Kang, J.S.; Wang, Q.; Kim, T.E. Cardiac biomarkers and COVID-19: A systematic review and meta-analysis. J. Infect. Public Health 2021, 14, 1191–1197. [Google Scholar] [CrossRef]
- Januzzi, J.L., Jr.; Filippatos, G.; Nieminen, M.; Gheorghiade, M. Troponin elevation in patients with heart failure: On behalf of the third Universal Definition of Myocardial Infarction Global Task Force: Heart Failure Section. Eur. Heart J. 2012, 33, 2265–2271. [Google Scholar] [CrossRef]
- Al Abbasi, B.; Torres, P.; Ramos-Tuarez, F.; Dewaswala, N.; Abdallah, A.; Chen, K.; Abdul Qader, M.; Job, R.; Aboulenain, S.; Dziadkowiec, K.; et al. Cardiac Troponin-I and COVID-19: A Prognostic Tool for In-Hospital Mortality. Cardiol. Res. 2020, 11, 398–404. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef]
- Gao, L.; Jiang, D.; Wen, X.S.; Cheng, X.C.; Sun, M.; He, B.; You, L.N.; Lei, P.; Tan, X.W.; Qin, S.; et al. Prognostic value of NT-proBNP in patients with severe COVID-19. Respir. Res. 2020, 21, 83. [Google Scholar] [CrossRef]
- Dragsted, L.O.; Gao, Q.; Scalbert, A.; Vergeres, G.; Kolehmainen, M.; Manach, C.; Brennan, L.; Afman, L.A.; Wishart, D.S.; Andres Lacueva, C.; et al. Validation of biomarkers of food intake-critical assessment of candidate biomarkers. Genes Nutr. 2018, 13, 14. [Google Scholar] [CrossRef]
- Phenol Explorer-Database on Polyphenol Content in Foods. Available online: http://phenol-explorer.eu (accessed on 16 March 2022).
- Fact Sheet For Healthcare Providers: Emergency Use-Authorization For Lagevrio™ (Molnupiravir) Capsules. Available online: https://www.merck.com/eua/molnupiravir-hcp-fact-sheet.pdf (accessed on 11 May 2022).
- Therapeutics and COVID-19: Living Guideline; WHO/2019-nCoV/therapeutics/2022.4. 2022. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2022.4 (accessed on 11 May 2022).
- Franconi, F.; Campesi, I.; Colombo, D.; Antonini, P. Sex-Gender Variable: Methodological Recommendations for Increasing Scientific Value of Clinical Studies. Cells 2019, 8, 476. [Google Scholar] [CrossRef]
- Franconi, F.; Campesi, I. Pharmacogenomics, pharmacokinetics and pharmacodynamics: Interaction with biological differences between men and women. Br. J. Pharmacol. 2014, 171, 580–594. [Google Scholar] [CrossRef]
- Zucker, I.; Prendergast, B.J. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol. Sex Differ. 2020, 11, 32. [Google Scholar] [CrossRef]
- Kumar, D.; Trivedi, N. Disease-drug and drug-drug interaction in COVID-19: Risk and assessment. Biomed. Pharmacother. 2021, 139, 111642. [Google Scholar] [CrossRef]
- Gomez-Moreno, G. Remdesivir-COVID-19: Drug interactions in dentistry. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9739–9743. [Google Scholar] [CrossRef]
- Foisy, M.M.; Yakiwchuk, E.M.; Hughes, C.A. Induction effects of ritonavir: Implications for drug interactions. Ann. Pharmacother. 2008, 42, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Rock, B.M.; Hengel, S.M.; Rock, D.A.; Wienkers, L.C.; Kunze, K.L. Characterization of ritonavir-mediated inactivation of cytochrome P450 3A4. Mol. Pharmacol. 2014, 86, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hafner, V.; Jager, M.; Matthee, A.K.; Ding, R.; Burhenne, J.; Haefeli, W.E.; Mikus, G. Effect of simultaneous induction and inhibition of CYP3A by St John’s Wort and ritonavir on CYP3A activity. Clin. Pharmacol. Ther. 2010, 87, 191–196. [Google Scholar] [CrossRef] [PubMed]
- FDA. Kevzara - Highlights of Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761037s000lbl.pdf (accessed on 11 May 2022).
- FDA. Actemra - Highlights of Prescribing Information. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125276s120s121lbl.pdf (accessed on 11 May 2022).
- Jousilahti, P.; Vartiainen, E.; Tuomilehto, J.; Puska, P. Sex, age, cardiovascular risk factors, and coronary heart disease: A prospective follow-up study of 14 786 middle-aged men and women in Finland. Circulation 1999, 99, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an independent risk factor for cardiovascular disease: A 26-year follow-up of participants in the Framingham Heart Study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.V.; Langrish, J.P.; Nair, H.; McAllister, D.A.; Hunter, A.L.; Donaldson, K.; Newby, D.E.; Mills, N.L. Global association of air pollution and heart failure: A systematic review and meta-analysis. Lancet 2013, 382, 1039–1048. [Google Scholar] [CrossRef]
- Huang, M.; Jiao, J.; Zhuang, P.; Chen, X.; Wang, J.; Zhang, Y. Serum polyfluoroalkyl chemicals are associated with risk of cardiovascular diseases in national US population. Environ. Int. 2018, 119, 37–46. [Google Scholar] [CrossRef]
- Madan, S.; Mehra, M.R. Gut dysbiosis and heart failure: Navigating the universe within. Eur. J. Heart Fail 2020, 22, 629–637. [Google Scholar] [CrossRef]
- Forkosh, E.; Ilan, Y. The heart-gut axis: New target for atherosclerosis and congestive heart failure therapy. Open Heart 2019, 6, e000993. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef]
- Lai, K.P.; Ng, A.H.; Wan, H.T.; Wong, A.Y.; Leung, C.C.; Li, R.; Wong, C.K. Dietary Exposure to the Environmental Chemical, PFOS on the Diversity of Gut Microbiota, Associated With the Development of Metabolic Syndrome. Front. Microbiol. 2018, 9, 2552. [Google Scholar] [CrossRef]
- Bouillon, R.; Marcocci, C.; Carmeliet, G.; Bikle, D.; White, J.H.; Dawson-Hughes, B.; Lips, P.; Munns, C.F.; Lazaretti-Castro, M.; Giustina, A.; et al. Skeletal and Extraskeletal Actions of Vitamin D: Current Evidence and Outstanding Questions. Endocr. Rev. 2019, 40, 1109–1151. [Google Scholar] [CrossRef]
- Benskin, L.L. A Basic Review of the Preliminary Evidence That COVID-19 Risk and Severity Is Increased in Vitamin D Deficiency. Front. Public Health 2020, 8, 513. [Google Scholar] [CrossRef]
- Fletcher, T.; Galloway, T.S.; Melzer, D.; Holcroft, P.; Cipelli, R.; Pilling, L.C.; Mondal, D.; Luster, M.; Harries, L.W. Associations between PFOA, PFOS and changes in the expression of genes involved in cholesterol metabolism in humans. Environ. Int. 2013, 57–58, 2–10. [Google Scholar] [CrossRef]
- Lauritzen, H.B.; Larose, T.L.; Oien, T.; Sandanger, T.M.; Odland, J.O.; van de Bor, M.; Jacobsen, G.W. Prenatal exposure to persistent organic pollutants and child overweight/obesity at 5-year follow-up: A prospective cohort study. Environ. Health 2018, 17, 9. [Google Scholar] [CrossRef]
- Di Nisio, A.; Rocca, M.S.; De Toni, L.; Sabovic, I.; Guidolin, D.; Dall’Acqua, S.; Acquasaliente, L.; De Filippis, V.; Plebani, M.; Foresta, C. Endocrine disruption of vitamin D activity by perfluoro-octanoic acid (PFOA). Sci. Rep. 2020, 10, 16789. [Google Scholar] [CrossRef]
- Zhao, S.; Feng, P.; Meng, W.; Jin, W.; Li, X.; Li, X. Modulated Gut Microbiota for Potential COVID-19 Prevention and Treatment. Front. Med. 2022, 9, 811176. [Google Scholar] [CrossRef]
- Kurian, S.J.; Unnikrishnan, M.K.; Miraj, S.S.; Bagchi, D.; Banerjee, M.; Reddy, B.S.; Rodrigues, G.S.; Manu, M.K.; Saravu, K.; Mukhopadhyay, C.; et al. Probiotics in Prevention and Treatment of COVID-19: Current Perspective and Future Prospects. Arch. Med. Res. 2021, 52, 582–594. [Google Scholar] [CrossRef]
- Mak, J.W.Y.; Chan, F.K.L.; Ng, S.C. Probiotics and COVID-19: One size does not fit all. Lancet Gastroenterol. Hepatol. 2020, 5, 644–645. [Google Scholar] [CrossRef]
- Calder, P.C. Nutrition, immunity and COVID-19. BMJ Nutr. Prev. Health 2020, 3, 74–92. [Google Scholar] [CrossRef]






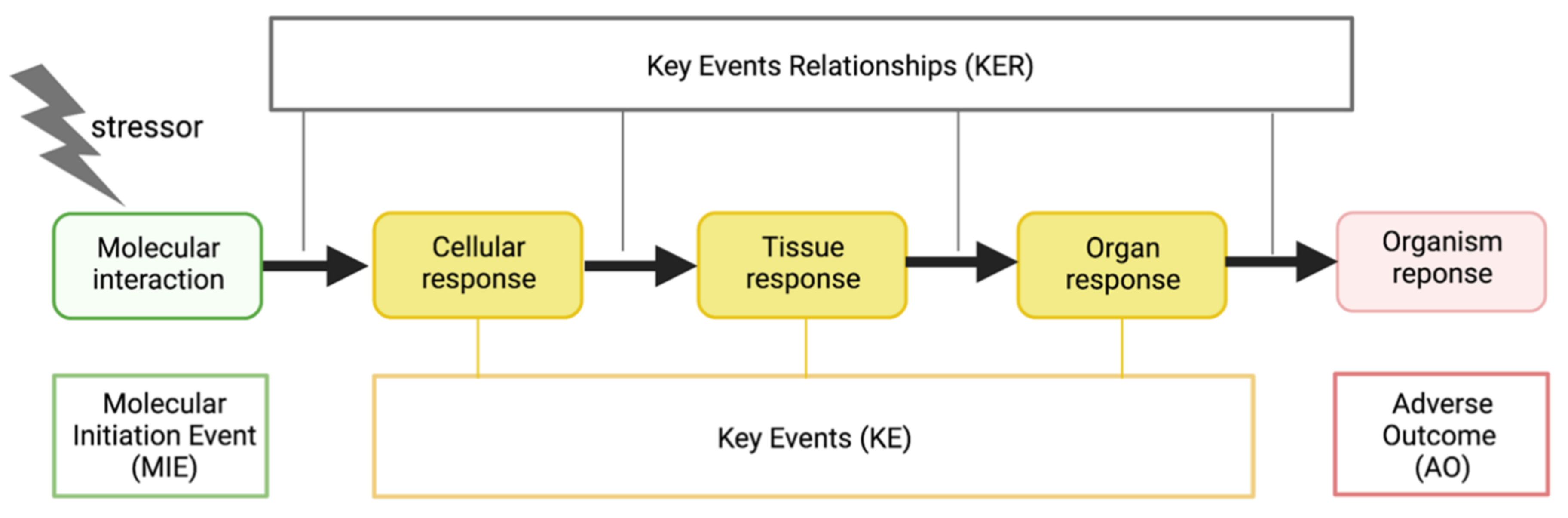{kind=link}
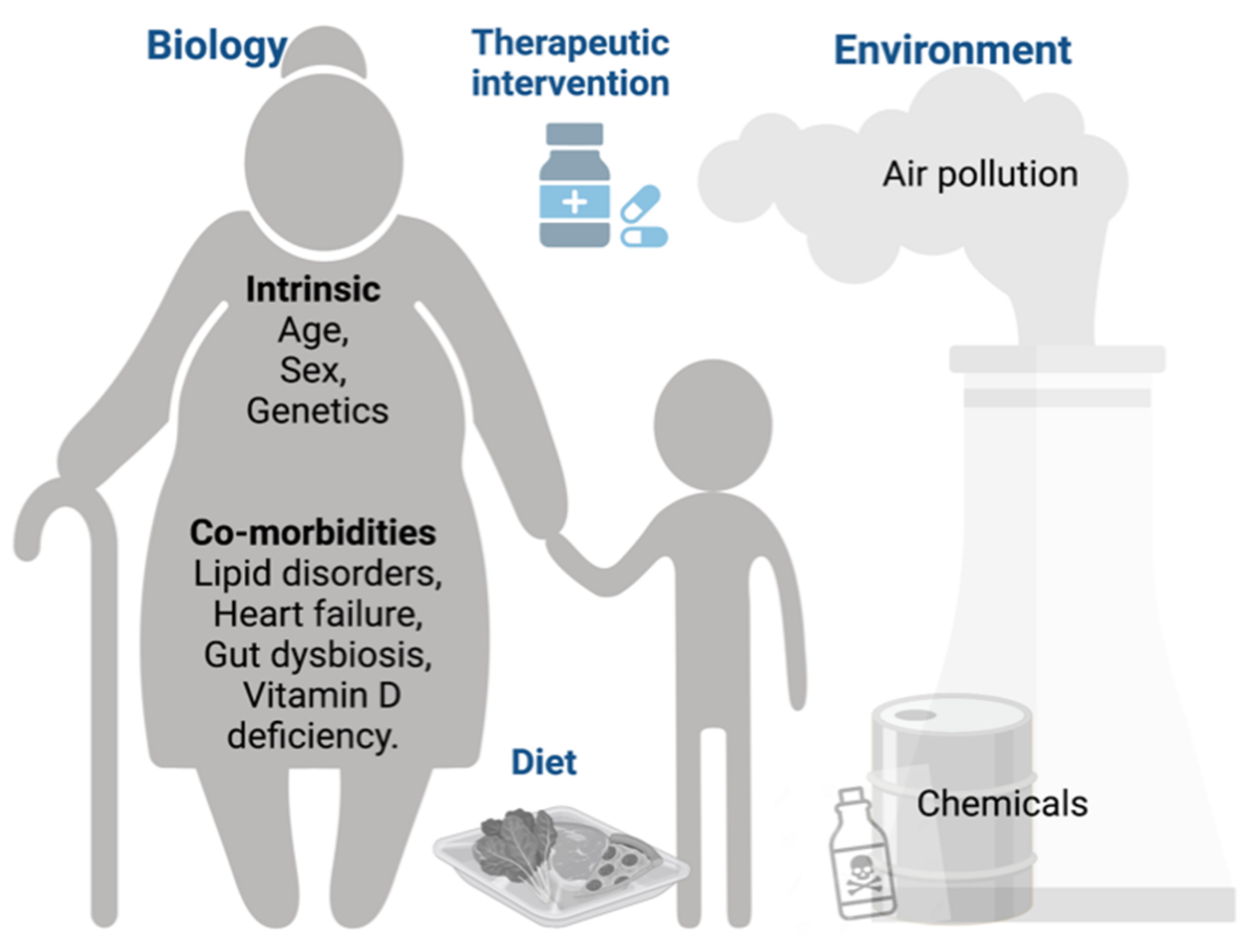{kind=link}
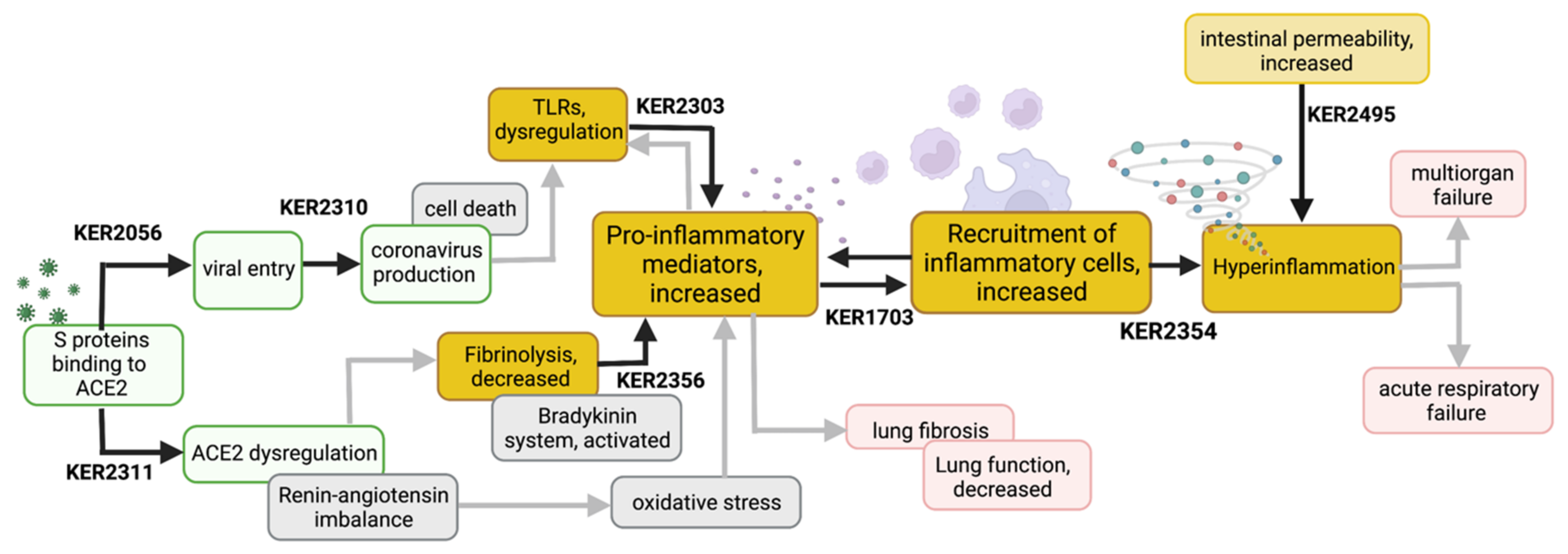{kind=link}
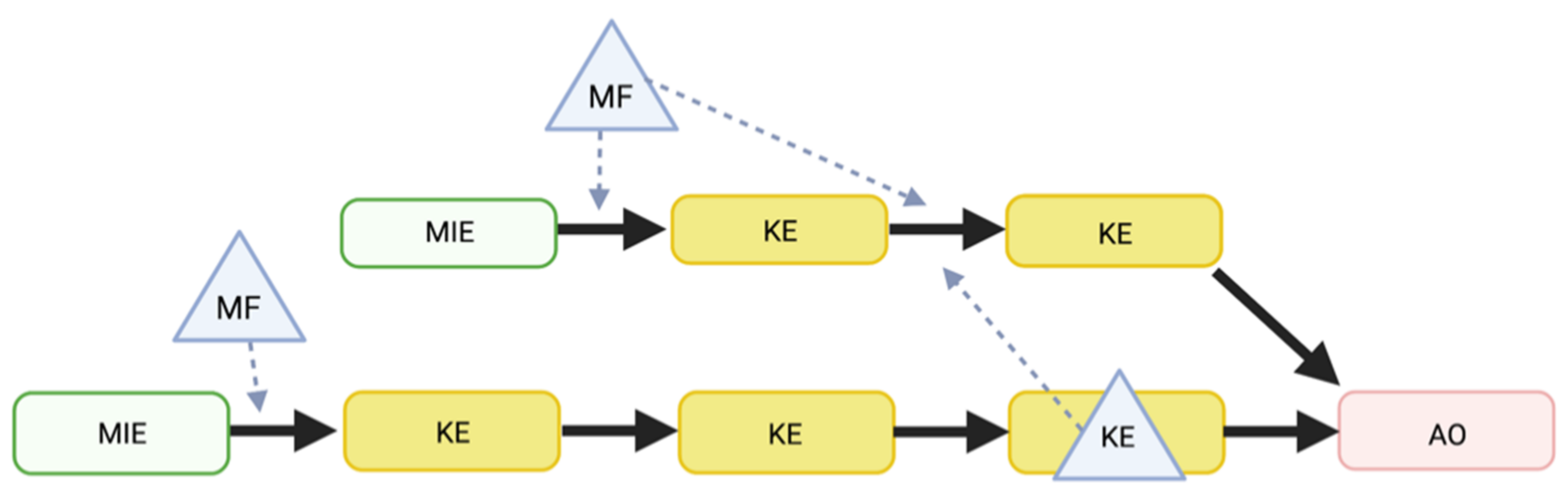{kind=link}
{kind=link}
| Clinical Trials | ||||
|---|---|---|---|---|
| Therapeutic Category | Drug Generic Name (Trade Name) | Clinical Trial | Outcomes | Reference |
| IL-6 receptor inhibitors (monoclonal antibody) | TOCILIZUMAB (Actemra/RoActemra) | RECOVERY (NCT04381936) | 621 patients (31%) who received Tocilizumab died within 28 days compared with 729 patients (35%) who received standard of care. | [106] |
| EMPACTA (NCT04372186) | Tocilizumab reduced the need for mechanical ventilation in patients with COVID-19-associated pneumonia. 12% of patients receiving Tocilizumab received mechanical ventilation compared with 19.3% of patients in the placebo group (p = 0.04); however, did not improve rates of survival. | [107] | ||
| REMAP-CAP (NCT02735707) | In critically ill patients with COVID-19 receiving organ support in ICUs, treatment with the IL-6 receptor antagonist tocilizumab improved outcomes, including survival. | [108,109] | ||
| IL-6 receptor inhibitors (monoclonal antibody) | SARILUMAB (Kevzara) | REMAP-CAP (NCT02735707) | In critically ill hospitalized adults receiving organ support in ICU mortality at Day 21 was 22.2% (10/45) for sarilumab, and 35.8% (142/397) for control. Results showed a median adjusted odds ratios in-hospital survival of 2.01 (95% credible interval, 1.18–4.71) compared with the control group. | [108] |
| Janus kinase (JAK) inhibitor | BARICITINIB (Olumiant) | The Phase 3 Adaptive COVID-19 Treatment Trial 2 (ACTT-2) (NCT04401579) | Olumiant plus Veklury (Remdesivir) significantly shortened median time to recovery from 8 days to 7 days compared with Veklury alone. Patients receiving Olumiant plus Veklury also had a significantly increased likelihood of better clinical status at 15 days and significantly fewer patients progressing to mechanical ventilation. The 28-day mortality was 5.1% in the combination group and 7.8% in the control group. | [110] |
| The Phase 3 COV-BARRIER trial (NCT04421027) | Significantly fewer patients in the Olumiant group reached 60-day all-cause mortality compared with patients in the placebo group | [111] | ||
| The Phase 3 COV-BARRIER trial (NCT04421027) | Baricitinib was the first immunomodulatory treatment to reduce COVID-19 mortality in a placebo-controlled trial | [112] | ||
| Anticoagulant | Heparin drugs: (UFH and LMWH) | REMAP-CAP, (NCT02735707) ACTIV-4a, (NCT04505774) ATTACC trial (NCT04372589) | Non-critically ill COVID-19 patients in the REMAP-CAP, ACTIV-4a, and ATTACC trials who were hospitalized for COVID-19 but not critically ill who received heparin were more likely to survive until being discharged or not need the use of supporting care compared with those who did not receive heparin. | [113] |
| A Phase 3 trial, HEP-COVID, (NCT04401293) | A therapeutic dose of LMW heparin applied prophylactically reduced the risk of blood clots among patients hospitalized with COVID-19 with “very elevated D-dimer levels” compared with standard of care for thromboprophylaxis. | [114] | ||
| Monoclonal antibodies cocktail | Casirivimab/imdevimab (REGN-COV) | RECOVERY (NCT04381936) | For patients who had not mounted an antibody response on their own (seronegative), REGN-COV significantly reduced 28-day mortality compared with usual care, but there was no significant difference between patients who had already mounted an antibody response (seropositive) and usual care. | [115,116] |
| A Phase 1/2/3 trial (NCT04425629) | The treatment resolved symptoms and reduced the SARS-CoV-2 viral load more rapidly than placebo and reduced the risk for any-cause hospitalization or mortality compared with a placebo group | [117,118] | ||
| Phase 3 trial (NCT04452318) | Treatment with subcutaneous casirivimab and imdevimab significantly reduced the incidence of symptomatic COVID-19 among recently exposed, asymptomatic individuals. | [119] | ||
| Monoclonal antibody | SOTROVIMAB (Xevudy) *known as VIR-7831 and GSK4182136 | The Phase 2/3 COMET-ICE trial (NCT04545060) | 583 patients (291 in the sotrovimab group and 292 in the placebo group). Three patients (1%) who received sotrovimab progressed to severe disease that led to being hospitalized or dying compared with 21 patients (7%) who received a placebo. | [120] |
| Antiviral | REMDESIVIR (Veklury) | ACTT-1 trial (NCT04280705) | Remdesivir, compared to placebo, improved the time to recovery (from 15 days to a median of 11 days) in adults who were hospitalized with COVID-19 and had evidence of lower respiratory tract infection. | [121] |
| CATCO trial (NCT04330690) | Hospitalized patients with COVID-19 treated with remdesivir had lower death rates (by about 4%) and 60-day mortality was 24.8% and 28.2%, respectively. Also had reduced need for oxygen and mechanical ventilation compared to people receiving standard-of-care treatments. | [122] | ||
| A phase 3, randomized, open-label study and a real-world, retrospective, longitudinal cohort study (NCT04292899 and EUPAS34303) | Remdesivir was associated with significantly greater recovery and reduced odds of death compared with standard of care in patients with severe COVID-19. The recovery rate at Day 14 was higher in patients who received remdesivir (n = 312) compared with those who received standard of care (n = 818) (74.4% vs. 59%; p < 0.001). The mortality rate at Day 14 was also lower in the remdesivir group. | [123] | ||
| PINETREE (NCT04501952) | In non-hospitalized patients who were at high risk for COVID-19 progression, a 3-day course of remdesivir demonstrated a statistically significant 87% reduction in risk for the composite primary endpoint of COVID-19 related hospitalization or all-cause death by day 28. | [124] | ||
| Antiviral (Ribonucleoside analogue) | MOLNUPIRAVIR (Lagevrio) | Phase 3 MOVe-OUT trial (NCT04575597) | Early treatment with molnupiravir reduced the risk of hospitalization or death by relative risk reduction 30% (6.8%, molnupiravir vs. 9.7%, placebo) (an absolute risk reduction of 3%) for non-hospitalized patients with mild or moderate COVID-19. | [125,126] |
| Antiviral therapeutic combination (SARS-CoV-2 protease inhibitor) | NIRMATRELVIR (former PF-07321332) + RITONAVIR (Paxlovid™) | Phase 2/3 high risk EPIC-HR trial (NCT04960202) | Patients who received Paxlovid showed an 89% reduction in hospitalization or death compared with placebo if the patient was treated within 3 days of developing symptoms and 88% if treated within five days of symptom onset. No deaths compared to placebo in non-hospitalized, high-risk adults with COVID-19. | [127,128,129] |
| Phase 2/3 EPIC-SR (NCT05011513) | Showed 0.6% of patients were hospitalized compared with 2.4% in the placebo group, a 70% reduction in hospitalization and no deaths in the treated population. | [129,130] | ||
| Number | KER Name | Links to the AOP-Wiki |
|---|---|---|
| Key Event Relationships related to initial viral infection | ||
| KER2056 | Binding to ACE2 leads to increased viral entry | https://aopwiki.org/relationships/2056 |
| KER2310 | Increased viral entry leads to Increased SARS-CoV-2 production | https://aopwiki.org/relationships/2310 |
| KER2311 | Binding to ACE2 leads to ACE2 dysregulation | https://aopwiki.org/relationships/2311 |
| Key Event Relationships related to central inflammatory processes | ||
| KER2356 | Hypofibrinolysis leads to increased proinflammatory mediators | https://aopwiki.org/relationships/2356 |
| KER2303 | TLR Dysregulation leads to increased proinflammatory mediators | https://aopwiki.org/relationships/2303 |
| KER1703 | Increased proinflammatory mediators lead to recruitment of inflammatory cells | https://aopwiki.org/relationships/1703 |
| KER2354 | Recruitment of inflammatory cells leads to hyper-inflammation | https://aopwiki.org/relationships/2354 |
| KER2495 | Intestinal hyperpermeability contributes to hyper-inflammation | https://aopwiki.org/relationships/2495 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clerbaux, L.-A.; Albertini, M.C.; Amigó, N.; Beronius, A.; Bezemer, G.F.G.; Coecke, S.; Daskalopoulos, E.P.; del Giudice, G.; Greco, D.; Grenga, L.; et al. Factors Modulating COVID-19: A Mechanistic Understanding Based on the Adverse Outcome Pathway Framework. J. Clin. Med. 2022, 11, 4464. https://doi.org/10.3390/jcm11154464
Clerbaux L-A, Albertini MC, Amigó N, Beronius A, Bezemer GFG, Coecke S, Daskalopoulos EP, del Giudice G, Greco D, Grenga L, et al. Factors Modulating COVID-19: A Mechanistic Understanding Based on the Adverse Outcome Pathway Framework. Journal of Clinical Medicine. 2022; 11(15):4464. https://doi.org/10.3390/jcm11154464
Chicago/Turabian StyleClerbaux, Laure-Alix, Maria Cristina Albertini, Núria Amigó, Anna Beronius, Gillina F. G. Bezemer, Sandra Coecke, Evangelos P. Daskalopoulos, Giusy del Giudice, Dario Greco, Lucia Grenga, and et al. 2022. "Factors Modulating COVID-19: A Mechanistic Understanding Based on the Adverse Outcome Pathway Framework" Journal of Clinical Medicine 11, no. 15: 4464. https://doi.org/10.3390/jcm11154464
APA StyleClerbaux, L.-A., Albertini, M. C., Amigó, N., Beronius, A., Bezemer, G. F. G., Coecke, S., Daskalopoulos, E. P., del Giudice, G., Greco, D., Grenga, L., Mantovani, A., Muñoz, A., Omeragic, E., Parissis, N., Petrillo, M., Saarimäki, L. A., Soares, H., Sullivan, K., & Landesmann, B. (2022). Factors Modulating COVID-19: A Mechanistic Understanding Based on the Adverse Outcome Pathway Framework. Journal of Clinical Medicine, 11(15), 4464. https://doi.org/10.3390/jcm11154464