
Emerging Therapies in Immune Thrombocytopenia


Abstract
1. Introduction
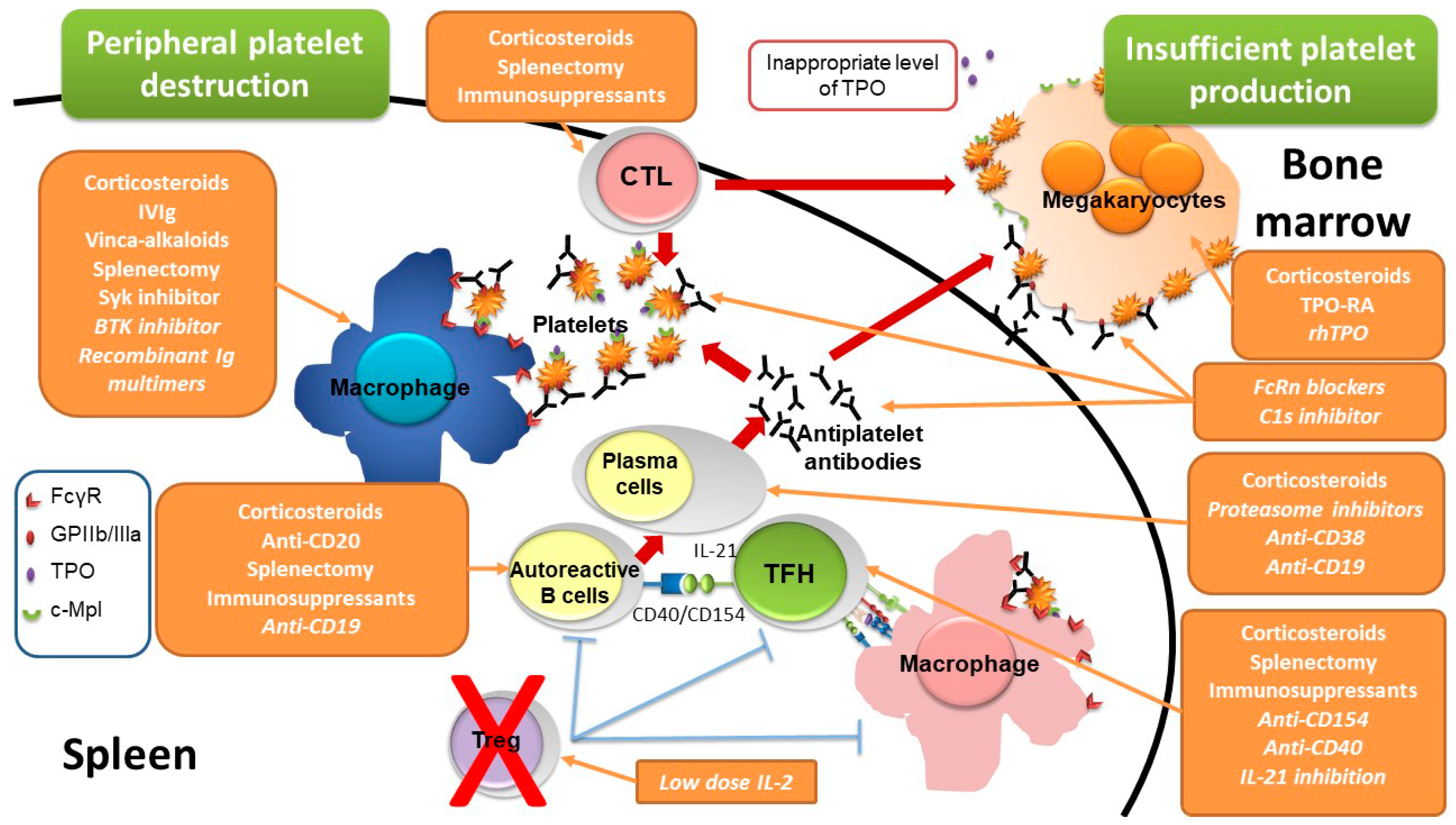
2. ITP Pathogenesis
3. Current Management of ITP
4. New Therapeutic Perspectives
4.1. New Thrombopoietin Receptor Agonists
4.2. Inhibition of the FcγR Transduction Signal
4.2.1. Spleen Tyrosine Kinase (Syk) Inhibitor
4.2.2. Bruton Tyrosine Kinase (BTK) Inhibitors
4.3. Neonatal Fc Receptor (FcRn) Inhibitors
4.4. Replacement of IVIg by Recombinant Molecules
4.5. Inhibition of the Classical Complement Pathway
4.6. Other B Cell Depleting Therapy Strategies
4.6.1. Combination Therapies
4.6.2. Anti-CD20 Targeting Therapies
4.6.3. Plasma Cell Targeting Therapies
4.7. T Cell Targeting Therapies
4.7.1. CD40/CD154 Blockade
4.7.2. IL-21 Inhibition
4.7.3. IL-2 Signaling Modulation
4.7.4. Epigenetic Modulation
4.8. Inhibition of Platelet Desialylation
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moulis, G.; Comont, T.; Adoue, D. New insights into the epidemiology of immune thrombocytopenia in adult patients: Impact for clinical practice. Rev. Med. Interne 2021, 42, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Moulis, G.; Palmaro, A.; Montastruc, J.L.; Godeau, B.; Lapeyre-Mestre, M.; Sailler, L. Epidemiology of incident immune thrombocytopenia: A nationwide population-based study in France. Blood 2014, 124, 3308–3315. [Google Scholar] [CrossRef]
- Provan, D.; Arnold, D.M.; Bussel, J.B.; Chong, B.H.; Cooper, N.; Gernsheimer, T.; Ghanima, W.; Godeau, B.; Gonzalez-Lopez, T.J.; Grainger, J.; et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019, 3, 3780–3817. [Google Scholar] [CrossRef]
- Khellaf, M.; Michel, M.; Schaeffer, A.; Bierling, P.; Godeau, B. Assessment of a therapeutic strategy for adults with severe autoimmune thrombocytopenic purpura based on a bleeding score rather than platelet count. Haematologica 2005, 90, 829–832. [Google Scholar]
- Khellaf, M.; Chabrol, A.; Mahevas, M.; Roudot-Thoraval, F.; Limal, N.; Languille, L.; Bierling, P.; Michel, M.; Godeau, B. Hydroxychloroquine is a good second-line treatment for adults with immune thrombocytopenia and positive antinuclear antibodies. Am. J. Hematol 2014, 89, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Mahevas, M.; Samson, M.; Godeau, B.; Bonnotte, B. Pathogenesis of immune thrombocytopenia. Autoimmun. Rev. 2017, 16, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Mahevas, M.; Bonnotte, B. Immune thrombocytopenia: From pathogenesis to treatment. Rev. Med. Interne 2021, 42, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.A.A.; Segel, G.B.; Burack, W.R.; Sachs, U.J.; Lissenberg-Thunnissen, S.N.; Vidarsson, G.; Bayat, B.; Cserti-Gazdewich, C.M.; Callum, J.; Lin, Y.; et al. FcgammaRI and FcgammaRIII on splenic macrophages mediate phagocytosis of anti-glycoprotein IIb/IIIa autoantibody-opsonized platelets in immune thrombocytopenia. Haematologica 2021, 106, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Okazaki, Y.; Ikeda, Y. Splenic macrophages maintain the anti-platelet autoimmune response via uptake of opsonized platelets in patients with immune thrombocytopenic purpura. J. Thromb. Haemost. 2009, 7, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; van der Wal, D.E.; Zhu, G.; Xu, M.; Yougbare, I.; Ma, L.; Vadasz, B.; Carrim, N.; Grozovsky, R.; Ruan, M.; et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat. Commun. 2015, 6, 7737. [Google Scholar] [CrossRef]
- Audia, S.; Rossato, M.; Santegoets, K.; Spijkers, S.; Wichers, C.; Bekker, C.; Bloem, A.; Boon, L.; Flinsenberg, T.; Compeer, E.; et al. Splenic TFH expansion participates in B-cell differentiation and antiplatelet-antibody production during immune thrombocytopenia. Blood 2014, 124, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Cooper, N.; Del Poeta, G.; Stipa, E.; Laura Evangelista, M.; Abruzzese, E.; Amadori, S. Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood 2008, 112, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Samson, M.; Guy, J.; Janikashvili, N.; Fraszczak, J.; Trad, M.; Ciudad, M.; Leguy, V.; Berthier, S.; Petrella, T.; et al. Immunologic effects of rituximab on the human spleen in immune thrombocytopenia. Blood 2011, 118, 4394–4400. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Wang, L.; Tomer, A.; Nichol, J.; Pistillo, J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004, 103, 1364–1369. [Google Scholar] [CrossRef]
- Kosugi, S.; Kurata, Y.; Tomiyama, Y.; Tahara, T.; Kato, T.; Tadokoro, S.; Shiraga, M.; Honda, S.; Kanakura, Y.; Matsuzawa, Y. Circulating thrombopoietin level in chronic immune thrombocytopenic purpura. Br. J. Haematol. 1996, 93, 704–706. [Google Scholar] [CrossRef]
- Wei, Y.; Ji, X.B.; Wang, Y.W.; Wang, J.X.; Yang, E.Q.; Wang, Z.C.; Sang, Y.Q.; Bi, Z.M.; Ren, C.A.; Zhou, F.; et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: A prospective multicenter randomized trial. Blood 2016, 127, 296–302. [Google Scholar] [CrossRef]
- Schwab, I.; Nimmerjahn, F. Intravenous immunoglobulin therapy: How does IgG modulate the immune system? Nat. Rev. Immunol. 2013, 13, 176–189. [Google Scholar] [CrossRef]
- Shimomura, M.; Hasegawa, S.; Seki, Y.; Fukano, R.; Hotta, N.; Ichiyama, T. Intravenous immunoglobulin does not increase FcgammaRIIB expression levels on monocytes in children with immune thrombocytopenia. Clin. Exp. Immunol. 2012, 169, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Audia, S.; Santegoets, K.; Laarhoven, A.G.; Vidarsson, G.; Facy, O.; Ortega-Deballon, P.; Samson, M.; Janikashvili, N.; Saas, P.; Bonnotte, B.; et al. Fcgamma receptor expression on splenic macrophages in adult immune thrombocytopenia. Clin. Exp. Immunol. 2017, 188, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Godeau, B.; Chevret, S.; Varet, B.; Lefrere, F.; Zini, J.M.; Bassompierre, F.; Cheze, S.; Legouffe, E.; Hulin, C.; Grange, M.J.; et al. Intravenous immunoglobulin or high-dose methylprednisolone, with or without oral prednisone, for adults with untreated severe autoimmune thrombocytopenic purpura: A randomised, multicentre trial. Lancet 2002, 359, 23–29. [Google Scholar] [CrossRef]
- Li, J.; van der Wal, D.E.; Zhu, L.; Vadasz, B.; Simpson, E.K.; Li, C.; Webster, M.L.; Zhu, G.; Lang, S.; Chen, P.; et al. Fc-independent phagocytosis: Implications for IVIG and other therapies in immune-mediated thrombocytopenia. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 50–58. [Google Scholar] [CrossRef]
- Peng, J.; Ma, S.H.; Liu, J.; Hou, Y.; Liu, X.M.; Niu, T.; Xu, R.R.; Guo, C.S.; Wang, X.M.; Cheng, Y.F.; et al. Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: A multicenter cohort study. J. Thromb. Haemost. 2014, 12, 497–504. [Google Scholar] [CrossRef]
- Rogier, T.; Samson, M.; Mourey, G.; Falvo, N.; Magy-Bertrand, N.; Ouandji, S.; Picque, J.B.; Greigert, H.; Mausservey, C.; Imbach, A.; et al. Antiplatelet Antibodies Do Not Predict the Response to Intravenous Immunoglobulins during Immune Thrombocytopenia. J. Clin. Med. 2020, 9, 1998. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.; Aslam, R.; Speck, E.R.; Kim, M.; Cridland, N.; Webster, M.L.; Chen, P.; Sahib, K.; Ni, H.; Lazarus, A.H.; et al. A murine model of severe immune thrombocytopenia is induced by antibody- and CD8+ T cell-mediated responses that are differentially sensitive to therapy. Blood 2010, 115, 1247–1253. [Google Scholar] [CrossRef]
- Deshayes, S.; Khellaf, M.; Zarour, A.; Layese, R.; Fain, O.; Terriou, L.; Viallard, J.F.; Cheze, S.; Graveleau, J.; Slama, B.; et al. Long-term safety and efficacy of rituximab in 248 adults with immune thrombocytopenia: Results at 5 years from the French prospective registry ITP-ritux. Am. J. Hematol. 2019, 94, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.M.; Vrbensky, J.R.; Karim, N.; Smith, J.W.; Liu, Y.; Ivetic, N.; Kelton, J.G.; Nazy, I. The effect of rituximab on anti-platelet autoantibody levels in patients with immune thrombocytopenia. Br. J. Haematol. 2017, 178, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Lee, C.S.; Seery, C.; Imahiyerobo, A.A.; Thompson, M.V.; Catellier, D.; Turenne, I.G.; Patel, V.L.; Basciano, P.A.; Elstrom, R.L.; et al. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica 2014, 99, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Mahevas, M.; Patin, P.; Huetz, F.; Descatoire, M.; Cagnard, N.; Bole-Feysot, C.; Le Gallou, S.; Khellaf, M.; Fain, O.; Boutboul, D.; et al. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J. Clinc. Investig. 2013, 123, 432–442. [Google Scholar] [CrossRef]
- Thai, L.H.; Le Gallou, S.; Robbins, A.; Crickx, E.; Fadeev, T.; Zhou, Z.; Cagnard, N.; Megret, J.; Bole, C.; Weill, J.C.; et al. BAFF and CD4(+) T cells are major survival factors for long-lived splenic plasma cells in a B-cell-depletion context. Blood 2018, 131, 1545–1555. [Google Scholar] [CrossRef]
- Audia, S.; Samson, M.; Mahevas, M.; Ferrand, C.; Trad, M.; Ciudad, M.; Gautheron, A.; Seaphanh, F.; Leguy, V.; Berthier, S.; et al. Preferential splenic CD8(+) T-cell activation in rituximab-nonresponder patients with immune thrombocytopenia. Blood 2013, 122, 2477–2486. [Google Scholar] [CrossRef]
- Feng, R.; Liu, X.; Zhao, Y.; Zhu, Y.; Peng, J.; Hou, M.; Chen, C. GPIIb/IIIa autoantibody predicts better rituximab response in ITP. Br. J. Haematol 2017, 182, 305–307. [Google Scholar] [CrossRef]
- Kuter, D.J.; Bussel, J.B.; Newland, A.; Baker, R.I.; Lyons, R.M.; Wasser, J.; Viallard, J.F.; Macik, G.; Rummel, M.; Nie, K.; et al. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: Safety and efficacy. Br. J. Haematol. 2013, 161, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Rummel, M.; Boccia, R.; Macik, B.G.; Pabinger, I.; Selleslag, D.; Rodeghiero, F.; Chong, B.H.; Wang, X.; Berger, D.P. Romiplostim or standard of care in patients with immune thrombocytopenia. N. Engl. J. Med. 2010, 363, 1889–1899. [Google Scholar] [CrossRef]
- Kuter, D.J.; Bussel, J.B.; Lyons, R.M.; Pullarkat, V.; Gernsheimer, T.B.; Senecal, F.M.; Aledort, L.M.; George, J.N.; Kessler, C.M.; Sanz, M.A.; et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: A double-blind randomised controlled trial. Lancet 2008, 371, 395–403. [Google Scholar] [CrossRef]
- Bussel, J.B.; Kuter, D.J.; Pullarkat, V.; Lyons, R.M.; Guo, M.; Nichol, J.L. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009, 113, 2161–2171. [Google Scholar] [CrossRef]
- Wong, R.S.M.; Saleh, M.N.; Khelif, A.; Salama, A.; Portella, M.S.O.; Burgess, P.; Bussel, J.B. Safety and efficacy of long-term treatment of chronic/persistent ITP with eltrombopag: Final results of the EXTEND study. Blood 2017, 130, 2527–2536. [Google Scholar] [CrossRef]
- Ebbo, M.; Riviere, E.; Godeau, B. Adult immune thrombocytopenia and thrombopoietin receptor agonist: Ten years later. Rev. Med. Interne 2021, 42, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Mahevas, M.; Fain, O.; Ebbo, M.; Roudot-Thoraval, F.; Limal, N.; Khellaf, M.; Schleinitz, N.; Bierling, P.; Languille, L.; Godeau, B.; et al. The temporary use of thrombopoietin-receptor agonists may induce a prolonged remission in adult chronic immune thrombocytopenia. Results of a French observational study. Br. J. Haematol. 2014, 165, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lopez, T.J.; Pascual, C.; Alvarez-Roman, M.T.; Fernandez-Fuertes, F.; Sanchez-Gonzalez, B.; Caparros, I.; Jarque, I.; Mingot-Castellano, M.E.; Hernandez-Rivas, J.A.; Martin-Salces, M.; et al. Successful discontinuation of eltrombopag after complete remission in patients with primary immune thrombocytopenia. Am. J. Hematol. 2015, 90, E40–E43. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Bussel, J.B.; Heck, S.; He, W.; Karpoff, M.; Boulad, N.; Yazdanbakhsh, K. Improved regulatory T-cell activity in patients with chronic immune thrombocytopenia treated with thrombopoietic agents. Blood 2010, 116, 4639–4645. [Google Scholar] [CrossRef] [PubMed]
- Pell, J.; Greenwood, R.; Ingram, J.; Wale, K.; Thomas, I.; Kandiyali, R.; Mumford, A.; Dick, A.; Bagot, C.; Cooper, N.; et al. Trial protocol: A multicentre randomised trial of first-line treatment pathways for newly diagnosed immune thrombocytopenia: Standard steroid treatment versus combined steroid and mycophenolate. The FLIGHT trial. BMJ Open 2018, 8, e024427. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Arnold, D.M.; McCrae, K.R. Splenectomy for immune thrombocytopenia: Down but not out. Blood 2018, 131, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.W.; Schoonen, W.M.; Farkas, D.K.; Riis, A.; Jacobsen, J.; Fryzek, J.P.; Sorensen, H.T. Risk for hospital contact with infection in patients with splenectomy: A population-based cohort study. Ann. Intern. Med. 2009, 151, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Thai, L.H.; Mahevas, M.; Roudot-Thoraval, F.; Limal, N.; Languille, L.; Dumas, G.; Khellaf, M.; Bierling, P.; Michel, M.; Godeau, B. Long-term complications of splenectomy in adult immune thrombocytopenia. Medicine 2016, 95, e5098. [Google Scholar] [CrossRef] [PubMed]
- Rorholt, M.; Ghanima, W.; Farkas, D.K.; Norgaard, M. Risk of cardiovascular events and pulmonary hypertension following splenectomy—A Danish population-based cohort study from 1996–2012. Haematologica 2017, 102, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Najean, Y.; Rain, J.D.; Billotey, C. The site of destruction of autologous 111In-labelled platelets and the efficiency of splenectomy in children and adults with idiopathic thrombocytopenic purpura: A study of 578 patients with 268 splenectomies. Br. J. Haematol. 1997, 97, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Iki, S.; Urabe, A. The role of autoantibody-producing plasma cells in immune thrombocytopenic purpura refractory to rituximab. Am. J. Hematol. 2007, 82, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Mahevas, M.; Gerfaud-Valentin, M.; Moulis, G.; Terriou, L.; Audia, S.; Guenin, S.; Le Guenno, G.; Salles, G.; Lambotte, O.; Limal, N.; et al. Characteristics, outcome, and response to therapy of multirefractory chronic immune thrombocytopenia. Blood 2016, 128, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, W.; Chojnowski, K.; Mayer, J.; Krawczyk, K.; Jamieson, B.D.; Tian, W.; Allen, L.F. Phase 3 randomised study of avatrombopag, a novel thrombopoietin receptor agonist for the treatment of chronic immune thrombocytopenia. Br. J. Haematol. 2018, 183, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Cheloff, A.Z.; Al-Samkari, H. Avatrombopag for the treatment of immune thrombocytopenia and thrombocytopenia of chronic liver disease. J. Blood Med. 2019, 10, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Lusutrombopag: First Global Approval. Drugs 2016, 76, 155–158. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, L.; Zhang, F.; Lu, H.; Chen, X.; Wen, A.; Luo, J.; Hu, Y.; Wang, Y.; Niu, T.; et al. First-in-patient study of hetrombopag in patients with chronic idiopathic thrombocytopenic purpura. J. Thromb Haemost 2020, 18, 3053–3060. [Google Scholar] [CrossRef]
- Yang, G.; Huang, R.; Yang, S.; Zhang, X.; Yang, X.; Chen, H.; Huang, Z.; Guo, C.; Pei, Q.; Tai, Y.; et al. Effect of postdose fasting duration on hetrombopag olamine pharmacokinetics and pharmacodynamics in healthy volunteers. Br. J. Clin. Pharmacol. 2020, 86, 1528–1536. [Google Scholar] [CrossRef]
- Wang, S.; Yang, R.; Zou, P.; Hou, M.; Wu, D.; Shen, Z.; Lu, X.; Li, Y.; Chen, X.; Niu, T.; et al. A multicenter randomized controlled trial of recombinant human thrombopoietin treatment in patients with primary immune thrombocytopenia. Int. J. Hematol. 2012, 96, 222–228. [Google Scholar] [CrossRef]
- Kong, Z.; Qin, P.; Xiao, S.; Zhou, H.; Li, H.; Yang, R.; Liu, X.; Luo, J.; Li, Z.; Ji, G.; et al. A novel recombinant human thrombopoietin therapy for the management of immune thrombocytopenia in pregnancy. Blood 2017, 130, 1097–1103. [Google Scholar] [CrossRef]
- Michel, M.; Ruggeri, M.; Gonzalez-Lopez, T.J.; Alkindi, S.S.; Cheze, S.; Ghanima, W.; Tvedt, T.H.A.; Ebbo, M.; Terriou, L.; Bussel, J.B.; et al. Use of thrombopoietin receptor agonists for immune thrombocytopenia in pregnancy: Results from a multicenter study. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Fcgamma Receptor Heterogeneity in Leukocyte Functional Responses. Front. Immunol 2017, 8, 280. [Google Scholar] [CrossRef]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Trelinski, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.A.; et al. Fostamatinib for the Treatment of Adult Persistent and Chronic Immune Thrombocytopenia: Results of Two Phase 3, Randomized, Placebo-Controlled Trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Connell, N.T.; Berliner, N. Fostamatinib for the treatment of chronic immune thrombocytopenia. Blood 2019, 133, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Boccia, R.; Cooper, N.; Ghanima, W.; Boxer, M.A.; Hill, Q.A.; Sholzberg, M.; Tarantino, M.D.; Todd, L.K.; Tong, S.; Bussel, J.B. Fostamatinib is an effective second-line therapy in patients with immune thrombocytopenia. Br. J. Haematol. 2020, 190, 933–938. [Google Scholar] [CrossRef]
- Kazerounian, S.; Duquette, M.; Reyes, M.A.; Lawler, J.T.; Song, K.; Perruzzi, C.; Primo, L.; Khosravi-Far, R.; Bussolino, F.; Rabinovitz, I.; et al. Priming of the vascular endothelial growth factor signaling pathway by thrombospondin-1, CD36, and spleen tyrosine kinase. Blood 2011, 117, 4658–4666. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Izak, M.; Bussel, J.B. Long-term sustained response to fostamatinib in two patients with chronic refractory immune thrombocytopenia (ITP). Br. J. Haematol 2020, 189, 379–382. [Google Scholar] [CrossRef]
- Bussel, J.B.; Arnold, D.M.; Boxer, M.A.; Cooper, N.; Mayer, J.; Zayed, H.; Tong, S.; Duliege, A.M. Long-Term Fostamatinib Treatment of Adults with Immune Thrombocytopenia (ITP) during the Phase 3 Clinical Trial Program. Am. J. Hematol. 2019, 94, 546–553. [Google Scholar] [CrossRef]
- Hampel, P.J.; Larson, M.C.; Kabat, B.; Call, T.G.; Ding, W.; Kenderian, S.S.; Bowen, D.; Boysen, J.; Schwager, S.M.; Leis, J.F.; et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br. J. Haematol. 2018, 183, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Funk, J.O.; Gourlay, S.G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 2017, 83, 2367–2376. [Google Scholar] [CrossRef]
- Langrish, C.; Bradshaw, J.; Owens, T.; Campbell, R.; Francesco, M.; Karr, D.; Murray, S.; Quesenberry, R.; Smith, P.; Taylor, M.; et al. PRN1008, a Reversible Covalent BTK Inhibitor in Clinical Development for Immune Thrombocytopenic Purpura. In Proceedings of the 59th American Society of Hematology Annual Meeting and Exposition, Atlanta, GA, USA, 9–12 December 2017. [Google Scholar]
- Kuter, J.; Efraim, M.; Mayer, J.; McDonald, V.; Bird, R.; Regenbogen, T.; Garg, M.; Kaplan, Z.; Bandman, O.; Burns, R.; et al. Oral Rilzabrutinib, a Bruton Tyrosine Kinase Inhibitor, Showed Clinically Active and Durable Platelet Responses and Was Well-Tolerated in Patients with Heavily Pretreated Immune Thrombocytopenia. In Proceedings of the 62nd American Society of Hematology Annual Meeting and Exposition, Online Event, 2–10 December 2020. [Google Scholar]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, P.; Lledo-Garcia, R.; Watanabe, S.; Langdon, G.; Tran, D.; Bari, M.; Christodoulou, L.; Jones, E.; Price, G.; Smith, B.; et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci. Transl. Med. 2017, 9, eaan1208. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.E.; Hillson, J.L.; Tiessen, R.G.; Bosje, T.; van Iersel, M.P.; Nix, D.J.; Markowitz, L.; Cilfone, N.A.; Duffner, J.; Streisand, J.B.; et al. M281, an Anti-FcRn Antibody: Pharmacodynamics, Pharmacokinetics, and Safety Across the Full Range of IgG Reduction in a First-in-Human Study. Clin. Pharmacol. Ther. 2019, 105, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clinc. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef]
- Zuercher, A.W.; Spirig, R.; Baz Morelli, A.; Rowe, T.; Kasermann, F. Next-generation Fc receptor-targeting biologics for autoimmune diseases. Autoimmun. Rev. 2019, 18, 102366. [Google Scholar] [CrossRef]
- Robak, T.; Kazmierczak, M.; Jarque, I.; Musteata, V.; Trelinski, J.; Cooper, N.; Kiessling, P.; Massow, U.; Woltering, F.; Snipes, R.; et al. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Adv. 2020, 4, 4136–4146. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.C.; Sanchez-Gonzalez, B.; Rejto, L.; Egyed, M.; Romanyuk, N.; Godar, M.; Verschueren, K.; Gandini, D.; Ulrichts, P.; Beauchamp, J.; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am. J. Hematol. 2020, 95, 178–187. [Google Scholar] [CrossRef]
- Blumberg, L.J.; Humphries, J.E.; Jones, S.D.; Pearce, L.B.; Holgate, R.; Hearn, A.; Cheung, J.; Mahmood, A.; Del Tito, B.; Graydon, J.S.; et al. Blocking FcRn in humans reduces circulating IgG levels and inhibits IgG immune complex-mediated immune responses. Sci. Adv. 2019, 5, eaax9586. [Google Scholar] [CrossRef]
- Zhang, X.; Owens, J.; Olsen, H.S.; So, E.; Burch, E.; McCroskey, M.C.; Li, X.; Weber, G.L.; Bennett, D.; Rybin, D.; et al. A recombinant human IgG1 Fc multimer designed to mimic the active fraction of IVIG in autoimmunity. JCI Insight 2019, 4, e121905. [Google Scholar] [CrossRef]
- Zhou, H.; Olsen, H.; So, E.; Merigeon, E.; Rybin, D.; Owens, J.; LaRosa, G.; Block, D.S.; Strome, S.E.; Zhang, X. A fully recombinant human IgG1 Fc multimer (GL-2045) inhibits complement-mediated cytotoxicity and induces iC3b. Blood Adv. 2017, 1, 504–515. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Rowley, T.F.; Junker, F.; Peters, S.J.; Crilly, S.; Compson, J.; Eddleston, A.; Bjorkelund, H.; Greenslade, K.; Parkinson, M.; et al. Multivalent Fcgamma-receptor engagement by a hexameric Fc-fusion protein triggers Fcgamma-receptor internalisation and modulation of Fcgamma-receptor functions. Sci. Rep. 2017, 7, 17049. [Google Scholar] [CrossRef]
- Majewska, N.I.; Tejada, M.L.; Betenbaugh, M.J.; Agarwal, N. N-Glycosylation of IgG and IgG-Like Recombinant Therapeutic Proteins: Why Is It Important and How Can We Control It? Annu Rev. Chem Biomol. Eng. 2020, 11, 311–338. [Google Scholar] [CrossRef] [PubMed]
- Najaoui, A.; Bakchoul, T.; Stoy, J.; Bein, G.; Rummel, M.J.; Santoso, S.; Sachs, U.J. Autoantibody-mediated complement activation on platelets is a common finding in patients with immune thrombocytopenic purpura (ITP). Eur. J. Haematol. 2012, 88, 167–174. [Google Scholar] [CrossRef]
- Peerschke, E.I.; Andemariam, B.; Yin, W.; Bussel, J.B. Complement activation on platelets correlates with a decrease in circulating immature platelets in patients with immune thrombocytopenic purpura. Br. J. Haematol. 2010, 148, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Cheloff, A.Z.; Kuter, D.J.; Al-Samkari, H. Serum complement levels in immune thrombocytopenia: Characterization and relation to clinical features. Res. Pract. Thromb. Haemost. 2020, 4, 807–812. [Google Scholar] [CrossRef]
- Castelli, R.; Lambertenghi Delilliers, G.; Gidaro, A.; Cicardi, M.; Bergamaschini, L. Complement activation in patients with immune thrombocytopenic purpura according to phases of disease course. Clin. Exp. Immunol. 2020, 201, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.; Panicker, S.; Bussel, J. Classical complement pathway activation in immune thrombocytopenia purpura: Inhibition by a novel C1s inhibitor. Br. J. Haematol. 2016, 173, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Broome, C.; Röth, A.; Kuter, D.; Scully, M.; Smith, R.; Wang, J.; Jiang, X.; Reuter, C.; Daak, A.; Hobbs, W. Long-Term Safety and Efficacy of Sutimlimab in Patients with Chronic Immune Thrombocytopenia. In Proceedings of the 62nd American Society of Hematology Annual Meeting and Exposition, Online Event, 2–10 December 2020. [Google Scholar]
- Mahévas, M.; Azzaoui, I.; Crickx, E.; Canoui-Poitrine, F.; Gobert, D.; Languille, L.; Limal, N.; Guillaud, C.; Croisille, L.; Jeljeli, M.; et al. Efficacy, safety and immunological profile of combining rituximab with belimumab for adults with persistent or chronic immune thrombocytopenia: Results from a prospective phase 2b trial. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Khellaf, M.; Charles-Nelson, A.; Fain, O.; Terriou, L.; Viallard, J.F.; Cheze, S.; Graveleau, J.; Slama, B.; Audia, S.; Ebbo, M.; et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: Results from a prospective registry including 248 patients. Blood 2014, 214, 3228–3236. [Google Scholar] [CrossRef]
- Godeau, B.; Porcher, R.; Fain, O.; Lefrere, F.; Fenaux, P.; Cheze, S.; Vekhoff, A.; Chauveheid, M.P.; Stirnemann, J.; Galicier, L.; et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura—Results of a prospective multicenter phase 2 study. Blood 2008, 112, 999–1004. [Google Scholar] [CrossRef]
- Liebman, H.A.; Saleh, M.N.; Bussel, J.B.; Negrea, O.G.; Horne, H.; Wegener, W.A.; Goldenberg, D.M. Comparison of two dosing schedules for subcutaneous injections of low-dose anti-CD20 veltuzumab in relapsed immune thrombocytopenia. Haematologica 2016, 101, 1327–1332. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herishanu, Y.; Levi, S.; Kamdjou, T.; Bornstein, Y.; Ram, R.; Benyamini, N.; Varon, D.; Avivi, I.; Perry, C. Obinutuzumab in the treatment of autoimmune haemolytic anaemia and immune thrombocytopenia in patients with chronic lymphocytic leukaemia/small lymphocytic lymphoma. Br. J. Haematol. 2021, 192, e1–e4. [Google Scholar] [CrossRef]
- Li, G.; Wang, S.; Li, N.; Liu, Y.; Feng, Q.; Zuo, X.; Li, X.; Hou, Y.; Shao, L.; Ma, C.; et al. Proteasome Inhibition with Bortezomib Induces Apoptosis of Long-Lived Plasma Cells in Steroid-Resistant or Relapsed Immune Thrombocytopaenia. Thromb. Haemost. 2018, 118, 1752–1764. [Google Scholar] [CrossRef]
- Beckman, J.D.; Rollins-Raval, M.A.; Raval, J.S.; Park, Y.A.; Mazepa, M.; Ma, A. Bortezomib for Refractory Immune-Mediated Thrombocytopenia Purpura. Am. J. Ther. 2018, 25, e270–e272. [Google Scholar] [CrossRef]
- Fadlallah, J.; Michel, M.; Crickx, E.; Limal, N.; Costedoat, N.; Malphettes, M.; Fieschi, C.; Galicier, L.; Oksenhendler, E.; Godeau, B.; et al. Bortezomib and dexamethasone, an original approach for treating multi-refractory warm autoimmune haemolytic anaemia. Br. J. Haematol. 2019, 187, 124–128. [Google Scholar] [CrossRef]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2 S,3 R)- N-(( S)-3-(Cyclopent-1-en-1-yl)-1-(( R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-(( S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Migdady, Y.; Ediriwickrema, A.; Jackson, R.P.; Kadi, W.; Gupta, R.; Socola, F.; Arai, S.; Martin, B.A. Successful treatment of thrombocytopenia with daratumumab after allogeneic transplant: A case report and literature review. Blood Adv. 2020, 4, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Fedyk, E.R.; Zhao, L.; Koch, A.; Smithson, G.; Estevam, J.; Chen, G.; Lahu, G.; Roepcke, S.; Lin, J.; McLean, L. Safety, tolerability, pharmacokinetics and pharmacodynamics of the anti-CD38 cytolytic antibody TAK-079 in healthy subjects. Br. J. Clin. Pharmacol. 2020, 86, 1314–1325. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.Y.; Yeter, K.; Kotha, R.; Pong, E.; Miranda, Y.; Phung, S.; Chen, H.; Lee, S.H.; Leung, I.; Bonzon, C.; et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcgamma receptor IIb inhibitory receptor. Arthritis Rheumatol 2014, 66, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Solanilla, A.; Pasquet, J.M.; Viallard, J.F.; Contin, C.; Grosset, C.; Dechanet-Merville, J.; Dupouy, M.; Landry, M.; Belloc, F.; Nurden, P.; et al. Platelet-associated CD154 in immune thrombocytopenic purpura. Blood 2005, 105, 215–218. [Google Scholar] [CrossRef]
- Patel, V.L.; Schwartz, J.; Bussel, J.B. The effect of anti-CD40 ligand in immune thrombocytopenic purpura. Br. J. Haematol. 2008, 141, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Koyama, I.; Kawai, T.; Andrews, D.; Boskovic, S.; Nadazdin, O.; Wee, S.L.; Sogawa, H.; Wu, D.L.; Smith, R.N.; Colvin, R.B.; et al. Thrombophilia associated with anti-CD154 monoclonal antibody treatment and its prophylaxis in nonhuman primates. Transplantation 2004, 77, 460–462. [Google Scholar] [CrossRef]
- Boumpas, D.T.; Furie, R.; Manzi, S.; Illei, G.G.; Wallace, D.J.; Balow, J.E.; Vaishnaw, A. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003, 48, 719–727. [Google Scholar] [CrossRef]
- Nagahama, M.; Nomura, S.; Kanazawa, S.; Ozaki, Y.; Kagawa, H.; Fukuhara, S. Significance of chemokines and soluble CD40 ligand in patients with autoimmune thrombocytopenic purpura. Eur. J. Haematol. 2002, 69, 303–308. [Google Scholar] [CrossRef]
- Robles-Carrillo, L.; Meyer, T.; Hatfield, M.; Desai, H.; Davila, M.; Langer, F.; Amaya, M.; Garber, E.; Francis, J.L.; Hsu, Y.M.; et al. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J. Immunol. 2010, 185, 1577–1583. [Google Scholar] [CrossRef]
- Karnell, J.L.; Albulescu, M.; Drabic, S.; Wang, L.; Moate, R.; Baca, M.; Oganesyan, V.; Gunsior, M.; Thisted, T.; Yan, L.; et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 2019, 11, eaar6584. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Wakwe, W.; Higginbotham, L.B.; Mathews, D.V.; Breeden, C.P.; Stephenson, A.C.; Jenkins, J.; Strobert, E.; Price, K.; Price, L.; et al. Fc-Silent Anti-CD154 Domain Antibody Effectively Prevents Nonhuman Primate Renal Allograft Rejection. Am. J. Transplant. 2017, 17, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- de Vos, A.F.; Melief, M.J.; van Riel, D.; Boon, L.; van Eijk, M.; de Boer, M.; Laman, J.D. Antagonist anti-human CD40 antibody inhibits germinal center formation in cynomolgus monkeys. Eur. J. Immunol 2004, 34, 3446–3455. [Google Scholar] [CrossRef] [PubMed]
- Espie, P.; He, Y.; Koo, P.; Sickert, D.; Dupuy, C.; Chokote, E.; Schuler, R.; Mergentaler, H.; Ristov, J.; Milojevic, J.; et al. First-in-human clinical trial to assess pharmacokinetics, pharmacodynamics, safety, and tolerability of iscalimab, an anti-CD40 monoclonal antibody. Am. J. Transplant. 2020, 20, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, C.; Rosenstock, B.; Doan, T.; Hamilton, P.; Dunbar, P.R.; Eleftheraki, A.G.; Joseph, D.; Hilbert, J.; Schoelch, C.; Padula, S.J.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of Multiple Rising Doses of BI 655064, an Antagonistic Anti-CD40 Antibody, in Healthy Subjects: A Potential Novel Treatment for Autoimmune Diseases. J. Clin. Pharmacol. 2018, 58, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, S.; Daniluk, S.; Ptaszynski, R.; Muller-Ladner, U.; Ramanujam, M.; Rosenstock, B.; Eleftheraki, A.G.; Vinisko, R.; Petrikova, A.; Kellner, H.; et al. Effects of BI 655064, an antagonistic anti-CD40 antibody, on clinical and biomarker variables in patients with active rheumatoid arthritis: A randomised, double-blind, placebo-controlled, phase IIa study. Ann. Rheum. Dis. 2019, 78, 754–760. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, G.; Chan, B.; Pearson, J.T.; Rathanaswami, P.; Delaney, J.; Ching Lim, A.; Babcook, J.; Hsu, H.; Gavin, M.A. Interleukin-21 receptor blockade inhibits secondary humoral responses and halts the progression of preestablished disease in the (NZB x NZW)F1 systemic lupus erythematosus model. Arthritis Rheumatol. 2015, 67, 2723–2731. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef]
- Fogarty, P.F.; Seggewiss, R.; McCloskey, D.J.; Boss, C.A.; Dunbar, C.E.; Rick, M.E. Anti-interleukin-2 receptor antibody (daclizumab) treatment of corticosteroid-refractory autoimmune thrombocytopenic purpura. Haematologica 2006, 91, 277–278. [Google Scholar]
- Rosenzwajg, M.; Lorenzon, R.; Cacoub, P.; Pham, H.P.; Pitoiset, F.; El-Soufi, K.; RIbet, C.; Bernard, C.; Aractingi, S.; Banneville, B.; et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann. Rheum. Dis. 2019, 78, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.Y.; Ma, Y.H.; Li, D.Q.; Sun, T.; Li, L.Z.; Li, P.; Liu, X.G.; Zhou, H.; Hou, Y.; Liu, Y.; et al. Low-dose chidamide restores immune tolerance in ITP in mice and humans. Blood 2019, 133, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Qin, P.; Liu, Q.; Yuan, C.; Hao, Y.; Zhang, H.; Wang, Z.; Ran, X.; Chu, X.; Yu, W.; et al. A prospective, multicenter study of low dose decitabine in adult patients with refractory immune thrombocytopenia. Am. J. Hematol. 2019, 94, 1374–1381. [Google Scholar] [CrossRef]
- Grozovsky, R.; Begonja, A.J.; Liu, K.; Visner, G.; Hartwig, J.H.; Falet, H.; Hoffmeister, K.M. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat. Med. 2015, 21, 47–54. [Google Scholar] [CrossRef]
- Revilla, N.; Corral, J.; Minano, A.; Mingot-Castellano, M.E.; Campos, R.M.; Velasco, F.; Gonzalez, N.; Galvez, E.; Berrueco, R.; Fuentes, I.; et al. Multirefractory primary immune thrombocytopenia; targeting the decreased sialic acid content. Platelets 2019, 30, 743–751. [Google Scholar] [CrossRef]
- Marini, I.; Zlamal, J.; Faul, C.; Holzer, U.; Hammer, S.; Pelzl, L.; Bethge, W.; Althaus, K.; Bakchoul, T. Autoantibody-mediated desialylation impairs human thrombopoiesis and platelet life span. Haematologica 2021, 106, 196–207. [Google Scholar] [CrossRef]
- Shaim, H.; McCaffrey, P.; Trieu, J.A.; DeAnda, A.; Yates, S.G. Evaluating the effects of oseltamivir phosphate on platelet counts: A retrospective review. Platelets 2020, 31, 1080–1084. [Google Scholar] [CrossRef]


{kind=link}
| Therapy | Targets/Mechanisms of Action | Drugs/Molecules | Development Stage |
|---|---|---|---|
| Steroids | Broad action on immune cells (macrophages, T and B cells) limiting platelet destruction | Prednisone | Licensed |
| Dexamethasone | |||
| IVIg |
| Licensed | |
| |||
| TPO-RAs | Increase in platelet production from megakaryocytes | Eltrombopag |
|
| Romiplostim |
| ||
| Avatrombopag |
| ||
| Lusutrombopag |
| ||
| Hetrombopag |
| ||
| rhTPO |
| ||
| Immunosuppressants | Inhibition of T and B cells | Azathioprine |
|
| Mycophenolate mofetil |
| ||
| Cyclophosphamide |
| ||
| Syk inhibitors | Decrease in platelet destruction by inhibition of macrophage phagocytosis | Fostamatinib | Licensed (US) |
| BTK inhibitors | Decrease in platelet destruction by inhibition of macrophage phagocytosis | - Rilzabrutinib | Phase 3 (NCT04562766) |
| FcRn blockers | Increase in antiplatelet antibody clearance, thus decreasing peripheral platelet destruction and immune response against megakaryocytes |
|
|
|
| ||
|
| ||
|
| ||
|
| ||
| Recombinant immunoglobulin multimers |
|
|
|
|
|
| |
|
| ||
| Complement inhibitors | Decrease in complement dependent cytotoxicity | Sutimlimab (C1s inhibitor) | Phase 2 (NCT04669600) |
| B cell depleting therapies |
| Anti-CD20: | |
|
|
| |
|
| ||
|
| ||
| Plasma cell targeting therapies | Decrease in short and long-lived plasma cells, thus decreasing antiplatelet antibodies | Proteasome inhibitors: | |
|
| ||
|
| ||
| Anti-CD38: | |||
|
| ||
|
| ||
| B cell and plasma cell targeting therapies | Decrease in short and long-lived plasma cells, thus decreasing antiplatelet antibodies | Anti-CD19: | |
|
| ||
|
| ||
| T cell targeting therapies |
| Anti-CD154: | |
|
|
| |
|
|
| |
|
| ||
|
| ||
| Anti-CD40: | |||
|
| ||
|
| ||
| IL-21 inhibition: | |||
|
| ||
|
| ||
| Neuraminidase inhibitors | Inhibiting platelet desialylation | Oseltamivir | Phase 1/2 (NCT03520049) |
| Epigenetic modulation |
|
| |
|
| ||
| Splenectomy |
| ||
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Audia, S.; Bonnotte, B. Emerging Therapies in Immune Thrombocytopenia. J. Clin. Med. 2021, 10, 1004. https://doi.org/10.3390/jcm10051004
Audia S, Bonnotte B. Emerging Therapies in Immune Thrombocytopenia. Journal of Clinical Medicine. 2021; 10(5):1004. https://doi.org/10.3390/jcm10051004
Chicago/Turabian StyleAudia, Sylvain, and Bernard Bonnotte. 2021. "Emerging Therapies in Immune Thrombocytopenia" Journal of Clinical Medicine 10, no. 5: 1004. https://doi.org/10.3390/jcm10051004
APA StyleAudia, S., & Bonnotte, B. (2021). Emerging Therapies in Immune Thrombocytopenia. Journal of Clinical Medicine, 10(5), 1004. https://doi.org/10.3390/jcm10051004