
Thrombocytopenia in Virus Infections

 , , ,
, , ,
Abstract
1. Introduction
2. Search Strategy
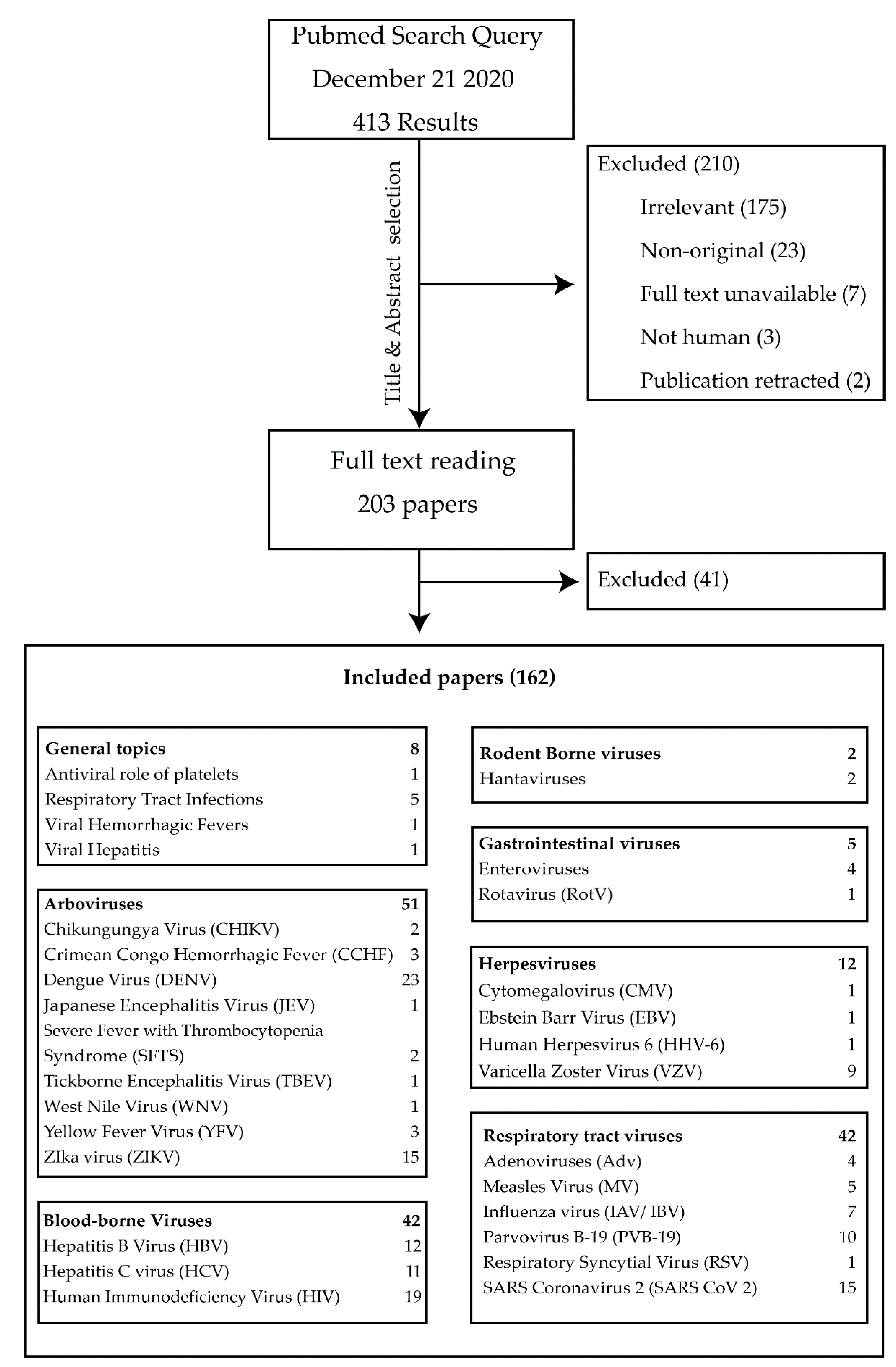
3. Results
3.1. Platelets and Viral Infections: General Principles
3.1.1. Aggregation
3.1.2. Impaired Hematopoiesis
3.1.3. Sequestration and Intravascular Destruction
3.1.4. Platelet Expression of Pattern Recognition Receptors (PRR)
3.1.5. Platelets Can Induce Inflammation and Secrete Anti-Microbial Proteins
3.1.6. Platelets Act as Antigen Presenting Cells (APCs)
3.2. Arboviruses
3.2.1. Dengue Virus
3.2.2. Chikungunya virus (CHIKV)
3.2.3. Crimean Congo Hemorrhagic Fever (CCHF)
3.2.4. Japanese Encephalitis Virus (JEV)
3.2.5. Severe Fever with Thrombocytopenia Syndrome (SFTS)
3.2.6. Tick-Borne Encephalitis Virus (TBEV)
3.2.7. Viral Hemorrhagic Fevers (VHF)
3.2.8. West Nile Virus (WNV)
3.2.9. Yellow Fever Virus (YFV)
3.2.10. Zika Virus (ZIKV)
3.3. Blood-Borne Viruses
3.3.1. Hepatitis B and C (HBV and HBC)
3.3.2. Human Immunodeficiency Virus (HIV)
3.4. Rodent-Borne Viruses
Hantavirus
3.5. Gastrointestinal Tract Viruses
3.5.1. Enteroviruses
3.5.2. Rotavirus (RotV)
3.6. Herpesviruses
3.6.1. Cytomegalovirus (CMV)
3.6.2. Epstein Barr Virus (EBV)
3.6.3. Human Herpesvirus 6 (HHV-6)
3.6.4. Varicella Zostervirus (VZV)
3.7. Respiratory Tract Infections
3.7.1. Adenoviruses (Adv)
3.7.2. Influenza Virus (IAV/IBV)
3.7.3. Measles Virus (MV)
3.7.4. Parvovirus B19 (PVB-19)
3.7.5. Respiratory Syncytial Virus
3.7.6. SARS Coronavirus 2 (SARS-CoV-2, COVID-19)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| ACE | Angiotensin Converting Enzyme |
| ADAMTS13 | A Disintegrin And Metalloproteinase with a ThromboSpondin type 1 domain |
| AdV | Adenovirus |
| APC | Antigen Presenting Cell |
| aPTT | activated Partial Thromboplastin Time |
| ARDS | Acute Respiratory Distress Syndrome |
| CAR | Coxsackie-Adeno Receptor |
| c-ART | combination Anti-Retroviral Therapy |
| CCHF | Crimean Congo Hemorrhagic Fever |
| CCL | Chemokine Ligand |
| CD | Cluster of Differentiation |
| CHIKV | Chikungungya Virus |
| CLEC | C-type Lectin |
| c-MPL | Myeloproliferative Leukemia Protein |
| CMV | Cytomegalovirus |
| COVID-19 | Coronavirus Disease 2019 |
| CoxV | Coxsackie virus |
| DC-SIGN | Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin |
| DENV | Dengue Virus |
| DIC | Disseminated Intravascular Coagulation |
| DNA | Desoxyribonucleic Acid |
| EBV | Ebstein Barr Virus |
| EDTA | Ethylenediaminetetraacetic acid |
| EM | Electron Microscopy |
| FcϒR2A | Fc Gamma Receptor 2a |
| FDP | Fibrinogen Degradation Product |
| FGF-4 | Fibroblast Growth Factor |
| GM-CSF | Granulocyte-Monocyte Colony Stimulating Factor |
| gMDSC | Granulocytic Myeloid-derived suppressor cell |
| GP | Glycoprotein |
| GVHD | graft versus host disease |
| HBV | Hepatitis B Virus |
| HCC | Hepatocellular Carcinoma |
| HCV | Hepatitis C Virus |
| HDV | Hepatitis Delta Virus |
| HFRS | Hemorrhagic Fever and Renal Syndrome |
| HHV-6 | Human Herpesvirus 6 |
| HIV | Human Immunodeficiency Virus |
| HLA | Human Leukocyte Antigen |
| HLH | Hemophagocytic Lymphohistiocytosis |
| HSP | Heparan Sulfate Proteoglycan |
| HUVEC | human vascular endothelial cells |
| IAV/IBV | Influenza A/B Virus |
| ICU | Intensive Care Unit |
| Ig | Immunoglobulin |
| IL | Interleukin |
| INR | International Normalized Ratio |
| ITP | Immune Thrombocytopenia |
| IUT | Intrauterine Transfusion |
| IVIG | Intravenous Immunoglobulin |
| JEV | Japanese Encephalitis Virus |
| LCMV | Lymphocytic Choriomeningitis Virus |
| LDH | Lactate Dehydrogenase |
| MHC | Major Histocompatibility Complex |
| MPV | Mean Platelet Volume |
| MRI | Magnetic Resonance Imaging |
| MV | Measles Virus |
| NET | Neutrophil Extracellular Trap |
| NS1 | Nonstructural Protein 1 |
| OR | Odds Ratio |
| PAF | Platelet Activating Factor |
| PAMP | Pattern Associated Molecular Pattern |
| PAR | Protease Activating Receptor |
| PCR | Polymerase Chain Reaction |
| PDW | Platelet Distribution Width |
| PF4 | Platelet Factor 4 |
| PLR | Platelet Lymphocyte Ratio |
| PMA | Platelet-Monocyte Aggregate |
| PMP | Platelet Microbicidal Peptides |
| PRR | Pattern Recognition Receptor |
| PS | phospatidylserine |
| PSGL-1 | P-Selectin Glycoprotein Ligand-1 |
| PT | Prothrombin Time |
| PUUV | Puumala virus |
| PVB-19 | Parvovirus B19 |
| RBC | Red Blood Cell |
| RNA | Ribonucleic Acid |
| RotV | Rotavirus |
| RSV | Respiratory Syncytial Virus |
| SARS-CoV-2 | SARS Coronavirus 2 |
| sCD40L | soluble CD40 ligand |
| SDF-1 | Stromal Derived Factor 1 |
| SFTS | Severe Fever with Thrombocytopenia Syndrome |
| Tat | Transactivating factor |
| TBEV | Tickborne Encephalitis Virus |
| TF | Tissue Factor |
| TLR | Toll-Like Receptor |
| TPO | Thrombopoietin |
| VHF | Viral Hemorrhagic Fever |
| VZV | Varicella Zoster Virus |
| WBC | White Blood Cell |
| WNV | West Nile Virus |
| YFV | Yellow Fever Virus |
| ZIKV | Zika Virus |
| γHV68 | murine gammaherpesvirus 68 |
References
- Sellers, S.A.; Hagan, R.S.; Hayden, F.G.; Fischer, W.A., 2nd. The hidden burden of influenza: A review of the extra-pulmonary complications of influenza infection. Influenza Other Respir. Viruses. 2017, 11, 372–393. [Google Scholar] [CrossRef]
- Wasano, K.; Ishikawa, T.; Kawasaki, T.; Yamamoto, S.; Tomisato, S.; Shinden, S.; Minami, S.; Wakabayashi, T.; Ogawa, K. Novel pre-therapeutic scoring system using patient and haematological data to predict facial palsy prognosis. Clin. Otolaryngol. 2017, 42, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, P.L.; Wendelboe, A.; Lucero-Obusan, C.A.; Ryono, R.A.; Winters, M.A.; Oda, G.; Martinez, M.; Saavedra, S.; Holodniy, M. Zika virus infection in the Veterans Health Administration (VHA), 2015–2016. PLoS Negl. Trop. Dis. 2018, 12, e0006416. [Google Scholar] [CrossRef] [PubMed]
- Pakos, I.S.; Lo, K.B.; Salacup, G.; Pelayo, J.; Bhargav, R.; Peterson, E.; Gul, F.; DeJoy, R., 3rd; Albano, J.; Patarroyo-Aponte, G.; et al. Characteristics of peripheral blood differential counts in hospitalized patients with COVID-19. Eur. J. Haematol. 2020, 105, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Martín-Rojas, R.M.; Pérez-Rus, G.; Delgado-Pinos, V.E.; Domingo-González, A.; Regalado-Artamendi, I.; Alba-Urdiales, N.; Demelo-Rodríguez, P.; Monsalvo, S.; Rodríguez-Macías, G.; Ballesteros, M.; et al. COVID-19 coagulopathy: An in-depth analysis of the coagulation system. Eur. J. Haematol. 2020, 105, 741–750. [Google Scholar] [CrossRef]
- Eren, S.H.; Zengin, S.; Büyüktuna, S.A.; Gözel, M.G. Clinical severity in forecasting platelet to lymphocyte ratio in Crimean-Congo hemorrhagic fever patients. J. Med Microbiol. 2016, 65, 1100–1104. [Google Scholar] [CrossRef]
- Kuo, Y.H.; Kee, K.M.; Hsu, N.T.; Wang, J.H.; Hsiao, C.C.; Chen, Y.; Lu, S.N. Using AST-platelet ratio index and fibrosis 4 index for detecting chronic hepatitis C in a large-scale community screening. PLoS ONE 2019, 14, e0222196. [Google Scholar] [CrossRef]
- Zhu, Y.F.; Tan, Y.F.; Xu, X.; Zheng, J.L.; Zhang, B.H.; Tang, H.R.; Yang, J.Y. Gamma-glutamyl transpeptidase-to-platelet ratio and the fibrosis-4 index in predicting hepatitis B virus-related hepatocellular carcinoma development in elderly chronic hepatitis B patients in China: A single-center retrospective study. Medicine 2019, 98, e18319. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lan, Q.; Lin, L.; Lu, J.; Ye, C.; Tao, Q.; Cui, M.; Zheng, S.; Zhang, X.; Xue, Y. Gamma-glutamyl transpeptidase-to-platelet ratio predicts the prognosis in HBV-associated acute-on-chronic liver failure. Clin. Chim. Acta 2018, 476, 92–97. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, J.; Wang, J.; Xie, T.; Zhang, Q.; Feng, S.; Deng, H.; Zhong, B. Platelet-to-lymphocyte ratio (PLR) and neutrophil-to-lymphocyte ratio (NLR) are associated with chronic hepatitis B virus (HBV) infection. Int. Immunopharmacol. 2017, 51, 1–8. [Google Scholar] [CrossRef]
- Wang, Q.; Blank, S.; Fiel, M.I.; Kadri, H.; Luan, W.; Warren, L.; Zhu, A.; Deaderick, P.A.; Sarpel, U.; Labow, D.M.; et al. The Severity of Liver Fibrosis Influences the Prognostic Value of Inflammation-Based Scores in Hepatitis B-Associated Hepatocellular Carcinoma. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S1125–S1132. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Shang, Y.; Wang, J.; Zhang, X.; Su, D.; Zhao, S.; Wang, Q.; Liu, L.; Li, Y.; et al. Ratios of neutrophil-to-lymphocyte and platelet-to-lymphocyte predict all-cause mortality in inpatients with coronavirus disease 2019 (COVID-19): A retrospective cohort study in a single medical centre. Epidemiol. Infect. 2020, 148, e211. [Google Scholar] [CrossRef]
- Thandassery, R.B.; Al Kaabi, S.; Soofi, M.E.; Mohiuddin, S.A.; John, A.K.; Al Mohannadi, M.; Al Ejji, K.; Yakoob, R.; Derbala, M.F.; Wani, H.; et al. Mean Platelet Volume, Red Cell Distribution Width to Platelet Count Ratio, Globulin Platelet Index, and 16 Other Indirect Noninvasive Fibrosis Scores: How Much Do Routine Blood Tests Tell About Liver Fibrosis in Chronic Hepatitis C? J. Clin. Gastroenterol. 2016, 50, 518–523. [Google Scholar] [CrossRef]
- Ng, K.J.; Tseng, C.W.; Chang, T.T.; Tzeng, S.J.; Hsieh, Y.H.; Hung, T.H.; Huang, H.T.; Wu, S.F.; Tseng, K.C. Aspartate aminotransferase to platelet ratio index and sustained virologic response are associated with progression from hepatitis C associated liver cirrhosis to hepatocellular carcinoma after treatment with pegylated interferon plus ribavirin. Clin. Interv. Aging 2016, 11, 1035–1041. [Google Scholar] [PubMed]
- Tseng, P.L.; Wang, J.H.; Hung, C.H.; Tung, H.D.; Chen, T.M.; Huang, W.S.; Liu, S.L.; Hu, T.H.; Lee, C.M.; Lu, S.N. Comparisons of noninvasive indices based on daily practice parameters for predicting liver cirrhosis in chronic hepatitis B and hepatitis C patients in hospital and community populations. Kaohsiung J. Med Sci. 2013, 29, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wu, J.; Xu, J.; Xu, J.; Xian, J.; Xue, S.; Ye, J. Association between Aspartate Aminotransferase-to-Platelet Ratio Index and Hepatocellular Carcinoma Risk in Patients with Chronic Hepatitis: A Meta-Analysis of Cohort Study. Dis. Markers 2019, 2019, 2046825. [Google Scholar] [CrossRef]
- Jun, B.G.; Park, E.J.; Lee, W.C.; Jang, J.Y.; Jeong, S.W.; Kim, Y.D.; Cheon, G.J.; Cho, Y.S.; Lee, S.H.; Kim, H.S.; et al. Platelet count is associated with sustained virological response rates in treatments for chronic hepatitis C. Korean J. Intern. Med. 2019, 34, 989–997. [Google Scholar] [CrossRef]
- Lee, J.; Kim, M.Y.; Kang, S.H.; Kim, J.; Uh, Y.; Yoon, K.J.; Kim, H.S. The gamma-glutamyl transferase to platelet ratio and the FIB-4 score are noninvasive markers to determine the severity of liver fibrosis in chronic hepatitis B infection. Br. J. Biomed. Sci. 2018, 75, 128–132. [Google Scholar] [CrossRef]
- Nishikawa, H.; Iguchi, E.; Koshikawa, Y.; Ako, S.; Inuzuka, T.; Takeda, H.; Nakajima, J.; Matsuda, F.; Sakamoto, A.; Henmi, S.; et al. The effect of pegylated interferon-alpha2b and ribavirin combination therapy for chronic hepatitis C infection in elderly patients. BMC Res. Notes 2012, 5, 135. [Google Scholar] [CrossRef]
- Menacho, I.; Sequeira, E.; Muns, M.; Barba, O.; Leal, L.; Clusa, T.; Fernandez, E.; Moreno, L.; Raben, D.; Lundgren, J.; et al. Comparison of two HIV testing strategies in primary care centres: Indicator-condition-guided testing vs. testing of those with non-indicator conditions. HIV Med. 2013, 14 (Suppl. 3), 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.K.; Raben, D.; Reekie, J.; Rayment, M.; Mocroft, A.; Esser, S.; Leon, A.; Begovac, J.; Brinkman, K.; Zangerle, R.; et al. Feasibility and effectiveness of indicator condition-guided testing for HIV: Results from HIDES I (HIV indicator diseases across Europe study). PLoS ONE 2013, 8, e52845. [Google Scholar] [CrossRef]
- Søgaard, O.S.; Lohse, N.; Østergaard, L.; Kronborg, G.; Røge, B.; Gerstoft, J.; Sørensen, H.T.; Obel, N. Morbidity and risk of subsequent diagnosis of HIV: A population based case control study identifying indicator diseases for HIV infection. PLoS ONE 2012, 7, e32538. [Google Scholar]
- Tominski, D.; Katchanov, J.; Driesch, D.; Daley, M.B.; Liedtke, A.; Schneider, A.; Slevogt, H.; Arastéh, K.; Stocker, H. The late-presenting HIV-infected patient 30 years after the introduction of HIV testing: Spectrum of opportunistic diseases and missed opportunities for early diagnosis. HIV Med. 2017, 18, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J. Milestones in understanding platelet production: A historical overview. Br. J. Haematol. 2014, 165, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Vinholt, P.J. The role of platelets in bleeding in patients with thrombocytopenia and hematological disease. Clin. Chem. Lab. Med. 2019, 57, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Behrens, K.; Alexander, W.S. Cytokine control of megakaryopoiesis. Growth Factors (Chur, Switzerland) 2018, 36, 89–103. [Google Scholar] [CrossRef]
- de Graaf, C.A.; Metcalf, D. Thrombopoietin and hematopoietic stem cells. Cell Cycle (Georgetown, Tex) 2011, 10, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Assinger, A. Platelets and infection—An emerging role of platelets in viral infection. Front. Immunol. 2014, 5, 649. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.W.; Schwertz, H.; Weyrich, A.S. Platelet mRNA: The meaning behind the message. Curr. Opin. Hematol. 2012, 19, 385–391. [Google Scholar] [CrossRef]
- Sutherland, M.R.; Simon, A.Y.; Serrano, K.; Schubert, P.; Acker, J.P.; Pryzdial, E.L. Dengue virus persists and replicates during storage of platelet and red blood cell units. Transfusion 2016, 56, 1129–1137. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, J.; Gao, L.; Pan, S. Spurious Thrombocytopenia in Automated Platelet Count. Lab. Med. 2018, 49, 130–133. [Google Scholar] [CrossRef]
- Stasi, R. How to approach thrombocytopenia. Hematol. Amer. Society Hematol. Educ. Prog. 2012, 2012, 191–197. [Google Scholar] [CrossRef]
- Kolb-Mäurer, A.; Goebel, W. Susceptibility of hematopoietic stem cells to pathogens: Role in virus/bacteria tropism and path-ogenesis. FEMS Microbiol. Lett. 2003, 226, 203–207. [Google Scholar] [CrossRef]
- Metcalf Pate, K.A.; Lyons, C.E.; Dorsey, J.L.; Queen, S.E.; Adams, R.J.; Morrell, C.N.; Mankowski, J.L. TGFβ-Mediated Downregulation of Throm-bopoietin Is Associated with Platelet Decline in Asymptomatic SIV Infection. J. Acquir. Immune Defic. Syndr. 2014, 65, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Isomura, H.; Yoshida, M.; Namba, H.; Fujiwara, N.; Ohuchi, R.; Uno, F.; Oda, M.; Seino, Y.; Yamada, M. Suppressive effects of human herpesvirus-6 on thrombopoietin-inducible megakaryocytic colony formation in vitro. J. Gen. Virol. 2000, 81 Pt 3, 663–673. [Google Scholar] [CrossRef]
- Chelucci, C.; Federico, M.; Guerriero, R.; Mattia, G.; Casella, I.; Pelosi, E.; Testa, U.; Mariani, G.; Hassan, H.J.; Peschle, C. Productive Human Immunodeficiency Virus-1 Infection of Purified Megakaryocytic Progenitors/Precursors and Maturing Megakaryocytes. Blood 1998, 91, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Crapnell, K.; Zanjani, E.D.; Chaudhuri, A.; Ascensao, J.L.; St Jeor, S.; Maciejewski, J.P. In vitro infection of megakaryocytes and their precursors by human cytomegalovirus. Blood 2000, 95, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jeffers, L.J.; Garon, C.; Fischer, E.R.; Scheffel, J.; Moore, B.; Reddy, K.R.; Demedina, M.; Schiff, E.R. Persistence of hepatitis C virus in a human megakaryoblastic leukaemia cell line. J. Viral Hepat. 1999, 6, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Assinger, A.; Kral, J.B.; Yaiw, K.C.; Schrottmaier, W.C.; Kurzejamska, E.; Wang, Y.; Mohammad, A.A.; Religa, P.; Rahbar, A.; Schabbauer, G.; et al. Human Cytomegalovirus–Platelet Interaction Triggers Toll-Like Receptor 2–Dependent Proinflammatory and Proangiogenic Responses. Arter. Thromb. Vasc. Biol. 2014, 34, 801–809. [Google Scholar] [CrossRef]
- Flaujac, C.; Boukour, S.; Cramer-Bordé, E. Platelets and viruses: An ambivalent relationship. Cell. Mol. Life Sci. 2009, 67, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Speth, C.; Löffler, J.; Krappmann, S.; Lass-Flörl, C.; Rambach, G. Platelets as immune cells in infectious diseases. Future Microbiol. 2013, 8, 1431–1451. [Google Scholar] [CrossRef] [PubMed]
- Seyoum, M.; Enawgaw, B.; Melku, M. Human blood platelets and viruses: Defense mechanism and role in the removal of viral pathogens. Thromb. J. 2018, 16, 16. [Google Scholar] [CrossRef]
- Maugeri, N.; Cattaneo, M.; Rovere-Querini, P.; Manfredi, A.A. Platelet clearance by circulating leukocytes: A rare event or a determinant of the “immune continuum”? Platelets 2014, 25, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef]
- Antoniak, S.; Mackman, N. Multiple roles of the coagulation protease cascade during virus infection. Blood 2014, 123, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Chacko, G.W.; Osborne, J.M.; Brandt, J.T. The Fc receptor for immunoglobulin G (Fc gamma RII) on human platelets. Semin. Thromb. Hemost. 1995, 21, 1–9. [Google Scholar] [CrossRef]
- Cox, D.; Kerrigan, S.W.; Watson, S.P. Platelets and the innate immune system: Mechanisms of bacterial-induced platelet activation. J. Thromb. Haemost. 2011, 9, 1097–1107. [Google Scholar] [CrossRef]
- Goeijenbier, M.; van Wissen, M.; van de Weg, C.; Jong, E.; Gerdes, V.E.; Meijers, J.C.; Brandjes, D.P.; van Gorp, E.C. Review: Viral infections and mechanisms of thrombosis and bleeding. J. Med Virol. 2012, 84, 1680–1696. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Hottz, E.D.; Bozza, F.A.; Bozza, P.T. Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front. Med. 2018, 5, 121. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef]
- Mohan, K.V.; Rao, S.S.; Atreya, C.D. Antiviral activity of selected antimicrobial peptides against vaccinia virus. Antivir. Res. 2010, 86, 306–311. [Google Scholar] [CrossRef]
- Zufferey, A.; Schvartz, D.; Nolli, S.; Reny, J.-L.; Sanchez, J.-C.; Fontana, P. Characterization of the platelet granule proteome: Evidence of the presence of MHC1 in alpha-granules. J. Proteom. 2014, 101, 130–140. [Google Scholar] [CrossRef]
- Colberg, L.; Cammann, C.; Greinacher, A.; Seifert, U. Structure and function of the ubiquitin-proteasome system in platelets. J. Thromb. Haemost. 2020, 18, 771–780. [Google Scholar] [CrossRef]
- Chapman, L.M.; Aggrey, A.A.; Field, D.J.; Srivastava, K.; Ture, S.; Yui, K.; Topham, D.J.; Baldwin, W.M., 3rd; Morrell, C.N. Platelets Present Antigen in the Context of MHC Class I. J. Immunol. 2012, 189, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Tomashek, K.M.; Lorenzi, O.D.; Andújar-Pérez, D.A.; Torres-Velásquez, B.C.; Hunsperger, E.A.; Munoz-Jordan, J.L.; Perez-Padilla, J.; Rivera, A.; Gonzalez-Zeno, G.E.; Sharp, T.M.; et al. Clinical and epidemiologic characteristics of dengue and other etiologic agents among patients with acute febrile illness, Puerto Rico, 2012–2015. PLoS Negl. Trop. Dis. 2017, 11, e0005859. [Google Scholar] [CrossRef] [PubMed]
- Dhanoa, A.; Rajasekaram, G.; Hassan, S.S.; Ramadas, A.; Azreen Adnan, N.A.; Lau, C.F.; Chan, T.S.; Ngim, C.F. Risk factors and clinical outcome of profound thrombocytopenia in adult patients with DENV infections. Platelets 2017, 28, 724–727. [Google Scholar] [CrossRef]
- Tramontini Gomes de Sousa Cardozo, F.; Baimukanova, G.; Lanteri, M.C.; Keating, S.M.; Moraes Ferreira, F.; Heitman, J.; Pannuti, C.S.; Pati, S.; Romano, C.M.; Cerdeira Sabino, E. Serum from dengue virus-infected patients with and without plasma leakage differentially affects endothelial cells barrier function in vitro. PLoS ONE 2017, 12, e0178820. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.C.; Agarwal, N.; Parikh, S.C. Detection of dengue NS1 antigen, alongside IgM plus IgG and concurrent platelet enu-meration during an outbreak. Asian Pac. J. Trop. Med. 2011, 4, 672. [Google Scholar] [CrossRef]
- Arya, S.C.; Agarwal, N. Thrombocytopenia progression in dengue cases during the 2010 outbreak in Indian capital metropolis. Platelets 2011, 22, 476–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Núñez-Avellaneda, D.; Mosso-Pani, M.A.; Sánchez-Torres, L.E.; Castro-Mussot, M.E.; Corona-de la Peña, N.A.; Salazar, M.I. Dengue Virus Induces the Release of sCD40L and Changes in Levels of Membranal CD42b and CD40L Molecules in Human Platelets. Viruses 2018, 10, 357. [Google Scholar] [CrossRef] [PubMed]
- Trugilho, M.R.O.; Hottz, E.D.; Brunoro, G.V.F.; Teixeira-Ferreira, A.; Carvalho, P.C.; Salazar, G.A.; Zimmerman, G.A.; Bozza, F.A.; Bozza, P.T.; Perales, J. Platelet proteome reveals novel pathways of platelet activation and platelet-mediated immunoregulation in dengue. PLoS Pathog. 2017, 13, e1006385. [Google Scholar] [CrossRef]
- de Jong, W.; Asmarawati, T.P.; Verbeek, I.; Rusli, M.; Hadi, U.; van Gorp, E.; Goeijenbier, M. Point-of-care thrombocyte function testing using multiple-electrode aggregometry in dengue patients: An explorative study. BMC Infect. Dis. 2020, 20, 580. [Google Scholar] [CrossRef]
- Chao, C.H.; Wu, W.C.; Lai, Y.C.; Tsai, P.J.; Perng, G.C.; Lin, Y.S.; Yeh, T.M. Dengue virus nonstructural protein 1 activates platelets via Toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. 2019, 15, e1007625. [Google Scholar] [CrossRef]
- Sung, P.S.; Huang, T.F.; Hsieh, S.L. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat. Commun. 2019, 10, 2402. [Google Scholar] [CrossRef]
- Hottz, E.D.; Medeiros-de-Moraes, I.M.; Vieira-de-Abreu, A.; de Assis, E.F.; Vals-de-Souza, R.; Castro-Faria-Neto, H.C.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; Bozza, P.T. Platelet Activation and Apoptosis Modulate Monocyte Inflammatory Responses in Dengue. J. Immunol. 2014, 193, 1864–1872. [Google Scholar] [CrossRef]
- Tsai, J.J.; Jen, Y.H.; Chang, J.S.; Hsiao, H.M.; Noisakran, S.; Perng, G.C. Frequency Alterations in Key Innate Immune Cell Components in the Peripheral Blood of Dengue Patients Detected by FACS Analysis. J. Innate Immun. 2011, 3, 530–540. [Google Scholar] [CrossRef]
- Ojha, A.; Bhasym, A.; Mukherjee, S.; Annarapu, G.K.; Bhakuni, T.; Akbar, I.; Seth, T.; Vikram, N.K.; Vrati, S.; Basu, A.; et al. Platelet Factor 4 Promotes Rapid Replication and Propagation of Dengue and Japanese Encephalitis Viruses. EBioMedicine 2019, 39, 332–347. [Google Scholar] [CrossRef]
- Masri, M.F.B.; Mantri, C.K.; Rathore, A.P.S.; John, A.L.S. Peripheral serotonin causes dengue virus–induced thrombocytopenia through 5HT2 receptors. Blood 2019, 133, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Wijewickrama, A.; Fernando, S.; Jeewandara, C.; Ginneliya, A.; Samarasekara, S.; Madushanka, P.; Punchihewa, C.; Paranavitane, S.; Idampitiya, D.; et al. A preliminary study on efficacy of rupatadine for the treatment of acute dengue infection. Sci. Rep. 2018, 8, 3857. [Google Scholar] [CrossRef]
- Ojha, A.; Nandi, D.; Batra, H.; Singhal, R.; Annarapu, G.K.; Bhattacharyya, S.; Seth, T.; Dar, L.; Medigeshi, G.R.; Vrati, S.; et al. Platelet activation determines the severity of thrombocytopenia in dengue infection. Sci. Rep. 2017, 7, 41697. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.W.; Yang, Y.W.; Chu, Y.T.; Lin, C.F.; Chang, C.P.; Yeh, T.M.; Anderson, R.; Lin, Y.S. Anti-dengue virus nonstructural protein 1 antibodies contribute to platelet phagocytosis by macrophages. Thromb. Haemost. 2016, 115, 646–656. [Google Scholar]
- Wang, T.T.; Sewatanon, J.; Memoli, M.J.; Wrammert, J.; Bournazos, S.; Bhaumik, S.K.; Pinsky, B.A.; Chokephaibulkit, K.; Onlamoon, N.; Pattanapanyasat, K.; et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science 2017, 355, 395–398. [Google Scholar] [CrossRef]
- Wan, S.W.; Lu, Y.T.; Huang, C.H.; Lin, C.F.; Anderson, R.; Liu, H.S.; Yeh, T.M.; Yen, Y.T.; Wu-Hsieh, B.A.; Lin, Y.S. Protection against dengue virus infection in mice by ad-ministration of antibodies against modified nonstructural protein 1. PLoS ONE 2014, 9, e92495. [Google Scholar] [CrossRef]
- Wan, S.W.; Chen, P.W.; Chen, C.Y.; Lai, Y.C.; Chu, Y.T.; Hung, C.Y.; Lee, H.; Wu, H.F.; Chuang, Y.C.; Lin, J.; et al. Therapeutic Effects of Monoclonal Antibody against Dengue Virus NS1 in a STAT1 Knockout Mouse Model of Dengue Infection. J. Immunol. 2017, 199, 2834–2844. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.Y.; Sutherland, M.R.; Pryzdial, E.L. Dengue virus binding and replication by platelets. Blood 2015, 126, 378–385. [Google Scholar] [CrossRef]
- Tomo, S.; Mohan, S.; Ramachandrappa, V.S.; Samadanam, D.M.; Suresh, S.; Pillai, A.B.; Tamilarasu, K.; Ramachandran, R.; Rajendiran, S. Dynamic modulation of DC-SIGN and FcΥR2A receptors expression on platelets in dengue. PLoS ONE 2018, 13, e0206346. [Google Scholar] [CrossRef]
- Noisakran, S.; Onlamoon, N.; Pattanapanyasat, K.; Hsiao, H.M.; Songprakhon, P.; Angkasekwinai, N.; Chokephaibulkit, K.; Villinger, F.; Ansari, A.A.; Perng, G.C. Role of CD61+ cells in thrombocytopenia of dengue patients. Int. J. Hematol. 2012, 96, 600–610. [Google Scholar] [CrossRef]
- Attatippaholkun, N.; Kosaisawe, N.; Yaowalak, U.P.; Supraditaporn, P.; Lorthongpanich, C.; Pattanapanyasat, K.; Issaragrisil, S. Selective Tropism of Dengue Virus for Human Glycoprotein Ib. Sci. Rep. 2018, 8, 2688. [Google Scholar] [CrossRef]
- Torres, J.R.; Falleiros-Arlant, L.H.; Dueñas, L.; Pleitez-Navarrete, J.; Salgado, D.M.; Castillo, J.B. Congenital and perinatal complications of chikungunya fever: A Latin American experience. Int. J. Infect. Dis. 2016, 51, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Shahid, U.; Farooqi, J.Q.; Barr, K.L.; Mahmood, S.F.; Jamil, B.; Imitaz, K.; Azizullah, Z.; Malik, F.R.; Prakoso, D.; Long, M.T.; et al. Comparison of clinical presentation and out-comes of Chikungunya and Dengue virus infections in patients with acute undifferentiated febrile illness from the Sindh region of Pa-kistan. PLoS Negl. Trop. Dis. 2020, 14, e0008086. [Google Scholar] [CrossRef] [PubMed]
- Doğan, H.O.; Büyüktuna, S.A.; Kapancik, S.; Bakir, S. Evaluation of the associations between endothelial dysfunction, inflamma-tion and coagulation in Crimean-Congo hemorrhagic fever patients. Arch. Virol. 2018, 163, 609–616. [Google Scholar] [CrossRef]
- Yilmaz, H.; Yilmaz, G.; Menteşe, A.; Kostakoğlu, U.; Karahan, S.C.; Köksal, İ. Prognostic impact of platelet distribution width in patients with Crimean-Congo hemorrhagic fever. J. Med Virol. 2016, 88, 1862–1866. [Google Scholar] [CrossRef]
- Solomon, T.; Dung, N.M.; Kneen, R.; Gainsborough, M.; Vaughn, D.W.; Khanh, V.T. Japanese encephalitis. J. Neurol. Neurosurg. Psychiatry 2000, 68, 405. [Google Scholar] [CrossRef]
- Ma'roef, C.N.; Dhenni, R.; Megawati, D.; Fadhilah, A.; Lucanus, A.; Artika, I.M.; Masyeni, S.; Lestarini, A.; Sari, K.; Suryana, K.; et al. Japanese encephalitis virus infection in non-encephalitic acute febrile illness patients. PLoS Negl. Trop. Dis. 2020, 14, e0008454. [Google Scholar] [CrossRef]
- Liu, S.; Chai, C.; Wang, C.; Amer, S.; Lv, H.; He, H.; Sun, J.; Lin, J. Systematic review of severe fever with thrombocytopenia syndrome:virology, epidemiology, and clinical characteristics. Rev. Med Virol. 2013, 24, 90–102. [Google Scholar] [CrossRef]
- Hofmann, H.; Li, X.; Zhang, X.; Liu, W.; Kühl, A.; Kaup, F.; Soldan, S.S.; González-Scarano, F.; Weber, F.; He, Y.; et al. Severe fever with thrombocytopenia virus glycoproteins are targeted by neutralizing antibodies and can use DC-SIGN as a receptor for pH-dependent entry into human and animal cell lines. J. Virol. 2013, 87, 4384–4394. [Google Scholar] [CrossRef]
- Li, X.K.; Lu, Q.B.; Chen, W.W.; Xu, W.; Liu, R.; Zhang, S.F.; Du, J.; Li, H.; Yao, K.; Zhai, D.; et al. Arginine deficiency is involved in thrombocytopenia and immuno-suppression in severe fever with thrombocytopenia syndrome. Sci. Transl. Med. 2018, 10, eaat4162. [Google Scholar] [CrossRef] [PubMed]
- Ruzek, D.; Avšič Županc, T.; Borde, J.; Chrdle, A.; Eyer, L.; Karganova, G.; Kholodilov, I.; Knap, N.; Kozlovskaya, L.; Matveev, A.; et al. Tick-borne encephalitis in Europe and Russia: Review of pathogenesis, clinical features, therapy, and vaccines. Antivir. Res. 2019, 164, 23–51. [Google Scholar] [CrossRef]
- Moniuszko, A.; Pancewicz, S.; Czupryna, P.; Grygorczuk, S.; Świerzbińska, R.; Kondrusik, M.; Penza, P.; Zajkowska, J. ssICAM-1, IL-21 and IL-23 in patients with tick borne encephalitis and neuroborreliosis. Cytokine 2012, 60, 468–472. [Google Scholar] [CrossRef]
- Ahmad, A.; Ashraf, S.; Komai, S. Are developing countries prepared to face Ebola-like outbreaks? Virol. Sin. 2015, 30, 234–237. [Google Scholar] [CrossRef]
- Loria, G.D.; Romagnoli, P.A.; Moseley, N.B.; Rucavado, A.; Altman, J.D. Platelets support a protective immune response to LCMV by preventing splenic necrosis. Blood 2013, 121, 940–950. [Google Scholar] [CrossRef]
- Paddock, C.D.; Nicholson, W.L.; Bhatnagar, J.; Goldsmith, C.S.; Greer, P.W.; Hayes, E.B.; Risko, J.A.; Henderson, C.; Blackmore, C.G.; Lanciotti, R.S.; et al. Fatal Hemorrhagic Fever Caused by West Nile Virus in the United States. Clin. Infect. Dis. 2006, 42, 1527–1535. [Google Scholar] [CrossRef]
- Hayes, C.; Stephens, L.; Fridey, J.L.; Snyder, R.E.; Groves, J.A.; Stramer, S.L.; Klapper, E. Probable transfusion transmission of West Nile virus from an apheresis platelet that screened non-reactive by individual donor-nucleic acid testing. Transfusion 2019, 60, 424–429. [Google Scholar] [CrossRef]
- Lataillade, L.G.; Vazeille, M.; Obadia, T.; Madec, Y.; Mousson, L.; Kamgang, B.; Chen, C.H.; Failloux, A.B.; Yen, P.S. Risk of yellow fever virus transmission in the Asia-Pacific region. Nat. Commun. 2020, 11, 5801. [Google Scholar] [CrossRef] [PubMed]
- Garske, T.; Van Kerkhove, M.D.; Yactayo, S.; Ronveaux, O.; Lewis, R.F.; Staples, J.E.; Perea, W.; Ferguson, N.M. Yellow Fever in Africa: Estimating the Burden of Disease and Impact of Mass Vaccination from Outbreak and Serological Data. PLoS Med. 2014, 11, e1001638. [Google Scholar] [CrossRef] [PubMed]
- Oliosi, E.; Serero Corcos, C.; Barroso, P.F.; Bleibtreu, A.; Grard, G.; De Filippis, B.A.M.; Caumes, E. Yellow fever in two unvaccinated French tourists to Brazil, January and March, 2018. Eurosurveillance 2018, 23, 1800240. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.L.; Joelsons, D.; Leite, G.F.C.; Malbouisson, L.M.S.; Song, A.T.W.; Perondi, B.; Andrade, L.C.; Pinto, L.F.; D’Albuquerque, L.A.C.; Segurado, A.A.C. Severe yellow fever in Brazil: Clinical charac-teristics and management. J. Travel Med. 2019, 26, taz040. [Google Scholar] [CrossRef] [PubMed]
- Brandão-de-Resende, C.; Cunha, L.H.M.; Oliveira, S.L.; Pereira, L.S.; Oliveira, J.G.F.; Santos, T.A.; Vasconcelos-Santos, D.V. Characterization of Retinopathy Among Patients with Yellow Fever During 2 Outbreaks in Southeastern Brazil. JAMA Ophthalmol. 2019, 137, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Ko, A.I.; Baud, D. Zika Virus Infection—After the Pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef]
- Langerak, T.; Brinkman, T.; Mumtaz, N.; Arron, G.; Hermelijn, S.; Baldewsingh, G.; Wongsokarijo, M.; Resida, L.; Rockx, B.; Koopmans, M.P.G.; et al. Zika virus seroprevalence in urban and rural areas of Suriname in 2017. J. Infect. Dis. 2019, 220, 28–31. [Google Scholar] [CrossRef]
- Saba Villarroel, P.M.; Nurtop, E.; Pastorino, B.; Roca, Y.; Drexler, J.F.; Gallian, P.; Jaenisch, T.; Leparc-Goffart, I.; Priet, S.; Ninove, L.; et al. Zika virus epidemiology in Bolivia: A sero-prevalence study in volunteer blood donors. PLoS Negl. Trop. Dis. 2018, 12, e0006239. [Google Scholar] [CrossRef]
- Netto, E.M.; Moreira-Soto, A.; Pedroso, C.; Höser, C.; Funk, S.; Kucharski, A.J.; Rockstroh, A.; Kümmerer, B.M.; Sampaio, G.S.; Luz, E.; et al. High Zika Virus Seroprevalence in Salvador, Northeastern Brazil Limits the Potential for Further Outbreaks. mBio 2017, 8. [Google Scholar] [CrossRef]
- Van Dyne, E.A.; Neaterour, P.; Rivera, A.; Bello-Pagan, M.; Adams, L.; Munoz-Jordan, J.; Baez, P.; Garcia, M.; Waterman, S.H.; Reyes, N.; et al. Incidence and Outcome of Severe and Nonsevere Thrombocytopenia Associated with Zika Virus Infection-Puerto Rico, 2016. Open Forum Infect. Dis. 2019, 6, ofy325. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cui, X.; Wu, N.; Song, R.; Yang, W.; Zhang, W.; Fan, D.; Chen, Z.; An, J. A unique case of human Zika virus infection in association with severe liver injury and coagulation disorders. Sci. Rep. 2017, 7, 11393. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.S.; Araujo, M.T.; Martins Filho, A.J.; Oliveira, C.S.; Nunes, B.T.; Cruz, A.C.; Nascimento, A.G.; Medeiros, R.C.; Caldas, C.A.; Araujo, F.C.; et al. Zika virus epidemic in Brazil. I. Fatal disease in adults: Clinical and laboratorial aspects. J. Clin. Virol. 2016, 85, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Sokal, A.; D’Ortenzio, E.; Houhou-Fidouh, N.; Brichler, S.; Dorchies, J.; Cabras, O.; Leparc-Goffart, I.; Yazdanpanah, Y.; Matheron, S. Zika virus infection: Report of the first imported cases in a Paris travel centre. J. Travel Med. 2016, 24. [Google Scholar] [CrossRef]
- Bandeira, A.C.; Gois, L.L.; Campos, G.S.; Sardi, S.; Yssel, H.; Vieillard, V.; Autran, B.; Grassi, M.F.R. Clinical and laboratory findings of acute Zika virus infection in patients from Salvador during the first Brazilian epidemic. Braz. J. Infect. Dis. 2020, 24, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.H.L.; Ho, H.J.; Chow, A.; Wong, J.; Kyaw, W.M.; Tan, A.; Chia, P.Y.; Choy, C.Y.; Tan, G.; Yeo, T.W.; et al. Correlation of clinical illness with viremia in Zika virus disease during an outbreak in Singapore. BMC Infect. Dis. 2018, 18, 301. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.M.; Muñoz-Jordán, J.; Perez-Padilla, J.; Bello-Pagán, M.I.; Rivera, A.; Pastula, D.M.; Salinas, J.L.; Martínez Mendez, J.H.; Méndez, M.; Powers, A.M.; et al. Zika Virus Infection Associated With Severe Thrombocytopenia. Clin. Infect. Dis. 2016, 63, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Chraïbi, S.; Najioullah, F.; Bourdin, C.; Pegliasco, J.; Deligny, C.; Résière, D.; Meniane, J.C. Two cases of thrombocytopenic purpura at onset of Zika virus infection. J. Clin. Virol. 2016, 83, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Boyer Chammard, T.; Schepers, K.; Breurec, S.; Messiaen, T.; Destrem, A.L.; Mahevas, M.; Soulillou, A.; Janaud, L.; Curlier, E.; Herrmann-Storck, C.; et al. Severe Thrombocytopenia after Zika Virus Infection, Guadeloupe, 2016. Emerg. Infect. Dis. 2017, 23, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Zea-Vera, A.F.; Parra, B. Zika virus (ZIKV) infection related with immune thrombocytopenic purpura (ITP) exacerbation and antinuclear antibody positivity. Lupus 2016, 26, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Roth, H.; Schneider, L.; Eberle, R.; Lausen, J.; Modlich, U.; Blümel, J.; Baylis, S.A. Zika virus infection studies with CD34 + hematopoietic and megakaryocyte-erythroid progenitors, red blood cells and platelets. Transfusion 2020, 60, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Hunsberger, S.; Ortega-Villa, A.M.; Powers, J.H., 3rd; León, H.A.R.; Sosa, S.C.; Hernández, E.R.; Nájera Cancino, J.G.; Nason, M.; Lumbard, K.; Sepulveda, J.; et al. Patterns of signs, symptoms and laboratory values associated with Zika, dengue and undefined acute illnesses in a dengue endemic region: Secondary analysis of a prospective cohort study in southern México. Int. J. Infect. Dis. 2020, 98, 241–248. [Google Scholar] [CrossRef]
- Musso, D.; Nhan, T.X.; de Pina, J.J.; Marchi, J.; Texier, G. The Use of Simple Laboratory Parameters in the Differential Diagnosis of Acute-Phase Zika and Dengue Viruses. Intervirology 2019, 62, 51–56. [Google Scholar] [CrossRef]
- Yan, G.; Pang, L.; Cook, A.R.; Ho, H.J.; Win, M.S.; Khoo, A.L.; Wong, J.G.X.; Lee, C.K.; Yan, B.; Jureen, R.; et al. Distinguishing Zika and Dengue Viruses through Simple Clinical Assessment, Singapore. Emerg. Infect. Dis. 2018, 24, 1565–1568. [Google Scholar] [CrossRef]
- Wang, R.Q.; Zhang, Q.S.; Zhao, S.X.; Niu, X.M.; Du, J.H.; Du, H.J.; Nan, Y.M. Gamma-glutamyl transpeptidase to platelet ratio index is a good noninvasive biomarker for predicting liver fibrosis in Chinese chronic hepatitis B patients. J. Int. Med Res. 2016, 44, 1302–1313. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, M.; Chen, G.; An, Y.; Lv, C.; Wang, Y.; Shang, Q. Reassessment of gamma-glutamyl transpeptidase to platelet ratio (GPR): A large-sample, dynamic study based on liver biopsy in a Chinese population with chronic hepatitis B virus (HBV) infection. Gut 2017, 67, 989–991. [Google Scholar] [CrossRef]
- Lee, H.S.; Kweon, Y.O.; Tak, W.Y.; Park, S.Y.; Kang, E.J.; Lee, Y.L.; Yang, H.M.; Park, H.W. Advanced fibrosis is not a negative pretreatment predictive factor for genotype 2 or 3 chronic hepatitis C patients. Clin. Mol. Hepatol. 2013, 19, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Ismael, M.N.; Forde, J.; Milla, E.; Khan, W.; Cabrera, R. Utility of Inflammatory Markers in Predicting Hepatocellular Carcinoma Survival after Liver Transplantation. BioMed Res. Int. 2019, 2019, 7284040. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.C.; Liu, X.L.; Zeng, F.R.; Chen, Z.; Wu, D.H. Platelet-to-lymphocyte ratio acts as an independent risk factor for patients with hepatitis B virus-related hepatocellular carcinoma who received transarterial chemoembolization. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 2302–2309. [Google Scholar] [PubMed]
- Shen, S.L.; Fu, S.J.; Chen, B.; Kuang, M.; Li, S.Q.; Hua, Y.P.; Liang, L.J.; Guo, P.; Hao, Y.; Peng, B.G. Preoperative aspartate aminotransferase to platelet ratio is an inde-pendent prognostic factor for hepatitis B-induced hepatocellular carcinoma after hepatic resection. Ann. Surg. Oncol. 2014, 21, 3802–3809. [Google Scholar] [CrossRef]
- Dou, J.; Lou, Y.; Wu, J.; Lu, Y.; Jin, Y. Thrombocytopenia in patients with hepatitis B virus-related chronic hepatitis: Evaluation of the immature platelet fraction. Platelets 2014, 25, 399–404. [Google Scholar] [CrossRef]
- Cho, S.Y.; Lee, A.; Lee, H.J.; Suh, J.T.; Park, T.S. Mean platelet volume in Korean patients with hepatic diseases. Platelets 2012, 23, 648–649. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Wang, F.; Zhang, J.; Yang, L.; Gong, X.; Fan, Y.; Xu, K.; Li, J.; Lu, Y.; Mei, L.; et al. Impact of hepatitis B virus (HBV) infection on platelet response to clopidogrel in patients undergoing coronary stent implantation. Thromb. Res. 2018, 167, 119–124. [Google Scholar] [CrossRef]
- Lima, D.S.; Murad Júnior, A.J.; Barreira, M.A.; Fernandes, G.C.; Coelho, G.R.; Garcia, J.H.P. Liver Transplantation in Hepatitis Delta: South America Experience. Arquivos de Gastroenterologia 2018, 55, 14–17. [Google Scholar] [CrossRef]
- Onan, E.; Uskudar, O.; Coşkun, Y.; Akkiz, H. Higher hepatitis C [correction of hepatis C] virus concentration in platelets than in plasma in a patient with ITP. Platelets 2012, 23, 413–414. [Google Scholar] [CrossRef][Green Version]
- Olariu, M.; Olariu, C.; Olteanu, D. Thrombocytopenia in chronic hepatitis C. J. Gastrointest. Liver Dis. 2010, 19, 381–385. [Google Scholar]
- Ariede, J.R.; Pardini, M.I.; Silva, G.F.; Grotto, R.M. Platelets can be a biological compartment for the Hepatitis C Virus. Braz. J. Microbiol. 2015, 46, 627–629. [Google Scholar] [CrossRef]
- Espírito-Santo, M.P.; Brandão-Mello, C.E.; Marques, V.A.; Lampe, E.; Almeida, A.J. Analysis of hepatitis C virus (HCV) RNA load in platelets of HCV-monoinfected patients receiving antiviral therapy. Ann. Hepatol. 2013, 12, 373–379. [Google Scholar] [CrossRef]
- Ragin, A.B.; D’Souza, G.; Reynolds, S.; Miller, E.; Sacktor, N.; Selnes, O.A.; Martin, E.; Visscher, B.R.; Becker, J.T. Platelet decline as a predictor of brain injury in HIV infection. J. NeuroVirology 2011, 17, 487–495. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pastori, D.; Esposito, A.; Carnevale, R.; Bartimoccia, S.; Novo, M.; Fantauzzi, A.; Di Campli, F.; Pignatelli, P.; Violi, F.; Mezzaroma, I. HIV-1 induces in vivo platelet activation by enhancing platelet NOX2 activity. J. Infect. 2015, 70, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Nkambule, B.B.; Davison, G.M.; Ipp, H. The evaluation of platelet indices and markers of inflammation, coagulation and disease progression in treatment-naïve, asymptomatic HIV-infected individuals. Int. J. Lab. Hematol. 2015, 37, 450–458. [Google Scholar] [CrossRef]
- Mena, Á.; Meijide, H.; Vázquez, P.; Castro, Á.; López, S.; Bello, L.; Serrano, J.; Baliñas, J.; Pedreira, J.D. HIV Increases Mean Platelet Volume During Asymptomatic HIV Infection in Treatment-Naive Patients. J. Acquir. Immune Defic. Syndr. 2011, 57, e112–e113. [Google Scholar] [CrossRef]
- Beck, Z.; Jagodzinski, L.L.; Eller, M.A.; Thelian, D.; Matyas, G.R.; Kunz, A.N.; Alving, C.R. Platelets and erythrocyte-bound platelets bind in-fectious HIV-1 in plasma of chronically infected patients. PLoS ONE 2013, 8, e81002. [Google Scholar] [CrossRef]
- Auerbach, D.J.; Lin, Y.; Miao, H.; Cimbro, R.; Difiore, M.J.; Gianolini, M.E.; Furci, L.; Biswas, P.; Fauci, A.S.; Lusso, P. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 9569–9574. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Quirino-Teixeira, A.C.; Valls-de-Souza, R.; Zimmerman, G.A.; Bozza, F.A.; Bozza, P.T. Platelet function in HIV plus dengue coinfection associates with reduced inflammation and milder dengue illness. Sci. Rep. 2019, 9, 7096. [Google Scholar] [CrossRef]
- Real, F.; Capron, C.; Sennepin, A.; Arrigucci, R.; Zhu, A.; Sannier, G.; Zheng, J.; Xu, L.; Massé, J.M.; Greffe, S.; et al. Platelets from HIV-infected individuals on antiretroviral drug therapy with poor CD4+ T cell recovery can harbor replication-competent HIV despite viral suppression. Sci. Transl. Med. 2020, 12, eaat6263. [Google Scholar] [CrossRef]
- Nkambule, B.B.; Davison, G.; Ipp, H. Platelet leukocyte aggregates and markers of platelet aggregation, immune activation and disease progression in HIV infected treatment naive asymptomatic individuals. J. Thromb. Thrombolysis 2015, 40, 458–467. [Google Scholar] [CrossRef]
- Liang, H.; Duan, Z.; Li, D.; Li, D.; Wang, Z.; Ren, L.; Shen, T.; Shao, Y. Higher levels of circulating monocyte–platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell. Mol. Immunol. 2015, 12, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.V.; Davidson, D.C.; Kiebala, M.; Maggirwar, S.B. Detection of circulating platelet–monocyte complexes in persons infected with human immunodeficiency virus type-1. J. Virol. Methods 2012, 181, 170–176. [Google Scholar] [CrossRef]
- Singh, M.V.; Davidson, D.C.; Jackson, J.W.; Singh, V.B.; Silva, J.; Ramirez, S.H.; Maggirwar, S.B. Characterization of Platelet–Monocyte Complexes in HIV-1–Infected Individuals: Possible Role in HIV-Associated Neuroinflammation. J. Immunol. 2014, 192, 4674–4684. [Google Scholar] [CrossRef]
- Davidson, D.C.; Hirschman, M.P.; Spinelli, S.L.; Morrell, C.N.; Schifitto, G.; Phipps, R.P.; Maggirwar, S.B. Antiplatelet Activity of Valproic Acid Contributes to Decreased Soluble CD40 Ligand Production in HIV Type 1-Infected Individuals. J. Immunol. 2011, 186, 584–591. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, W.; Nardi, M.A.; Li, Z. HIV-1 Tat-induced platelet activation and release of CD154 contribute to HIV-1-associated autoimmune thrombocytopenia. J. Thromb. Haemost. 2011, 9, 562–573. [Google Scholar] [CrossRef]
- Damien, P.; Cognasse, F.; Lucht, F.; Suy, F.; Pozzetto, B.; Garraud, O.; Hamzeh-Cognasse, H. Highly Active Antiretroviral Therapy Alters Inflammation Linked to Platelet Cytokines in HIV-1-Infected Patients. J. Infect. Dis. 2013, 208, 868–870. [Google Scholar] [CrossRef] [PubMed][Green Version]
- O’Halloran, J.A.; Dunne, E.; Gurwith, M.; Lambert, J.S.; Sheehan, G.J.; Feeney, E.R.; Pozniak, A.; Reiss, P.; Kenny, D.; Mallon, P. The effect of initiation of antiretroviral therapy on monocyte, endothelial and platelet function in HIV-1 infection. HIV Med. 2015, 16, 608–619. [Google Scholar] [CrossRef]
- Connolly-Andersen, A.M.; Sundberg, E.; Ahlm, C.; Hultdin, J.; Baudin, M.; Larsson, J.; Dunne, E.; Kenny, D.; Lindahl, T.L.; Ramström, S.; et al. Increased Thrombopoiesis and Platelet Activation in Hantavirus-Infected Patients. J. Infect. Dis. 2015, 212, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Laine, O.; Mäkelä, S.; Mustonen, J.; Helminen, M.; Vaheri, A.; Lassila, R.; Joutsi-Korhonen, L. Platelet ligands and ADAMTS13 during Puumala hantavirus infection and associated thrombocytopenia. Blood Coagul. Fibrinolysis 2011, 22, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Laine, O.; Joutsi-Korhonen, L.; Lassila, R.; Koski, T.; Huhtala, H.; Vaheri, A.; Mäkelä, S.; Mustonen, J. Hantavirus infection-induced thrombocytopenia triggers increased production but associates with impaired aggregation of platelets except for collagen. Thromb. Res. 2015, 136, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Lütteke, N.; Raftery, M.J.; Lalwani, P.; Lee, M.H.; Giese, T.; Voigt, S.; Bannert, N.; Schulze, H.; Krüger, D.H.; Schönrich, G. Switch to high-level virus replication and HLA class I upregulation in differentiating megakaryocytic cells after infection with pathogenic hantavirus. Virology 2010, 405, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Goeijenbier, M.; Meijers, J.C.; Anfasa, F.; Roose, J.M.; van de Weg, C.A.; Bakhtiari, K.; Henttonen, H.; Vaheri, A.; Osterhaus, A.D.; van Gorp, E.C.; et al. Effect of Puumala hantavirus infection on human umbilical vein endothelial cell hemostatic function: Platelet interactions, increased tissue factor expression and fibrinolysis regulator release. Front. Microbiol. 2015, 6, 220. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Yoshioka, S.; Gosho, H.; Miyazoe, S.; Suenaga, H.; Aoki, M.; Hashimoto, K. A neonatal case of coxsackievirus B3 vertical infection with symptoms of hemophagocytic lymphohistiocytosis. IDCases 2020, 20, e00738. [Google Scholar] [CrossRef] [PubMed]
- Kaga, A.; Katata, Y.; Suzuki, A.; Otani, K.; Watanabe, H.; Kitaoka, S.; Kumaki, S. Perinatal Coxsackievirus B3 Infection with Transient Thrombocytopenia. Tohoku J. Exp. Med. 2016, 239, 135–138. [Google Scholar] [CrossRef][Green Version]
- Hara, S.; Kawada, J.; Kawano, Y.; Yamashita, T.; Minagawa, H.; Okumura, N.; Ito, Y. Hyperferritinemia in neonatal and infantile human parechovirus-3 infection in comparison with other infectious diseases. J. Infect. Chemother. 2014, 20, 15–19. [Google Scholar] [CrossRef]
- Negrotto, S.; Jaquenod de Giusti, C.; Rivadeneyra, L.; Ure, A.E.; Mena, H.A.; Schattner, M.; Gomez, R.M. Platelets interact with Cox-sackieviruses B and have a critical role in the pathogenesis of virus-induced myocarditis. J. Thromb. Haemost. 2015, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef]
- Mete, E.; Akelma, A.Z.; Cizmeci, M.N.; Bozkaya, D.; Kanburoglu, M.K. Decreased mean platelet volume in children with acute rotavirus gastroenteritis. Platelets 2014, 25, 51–54. [Google Scholar] [CrossRef]
- Plosa, E.J.; Esbenshade, J.C.; Fuller, M.P.; Weitkamp, J.-H. Cytomegalovirus Infection. Pediatr. Rev. 2012, 33, 156. [Google Scholar] [CrossRef] [PubMed]
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious Mononucleosis. Curr. Top. Microbiol. Immunol. 2015, 390 Pt 1, 211–240. [Google Scholar]
- Freeman, M.L.; Burkum, C.E.; Lanzer, K.G.; Roberts, A.D.; Pinkevych, M.; Itakura, A.; Kummer, L.W.; Szaba, F.M.; Davenport, M.P.; McCarty, O.J.; et al. Gammaherpesvirus latency induces antibody-associated thrombocytopenia in mice. J. Autoimmun. 2013, 42, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Dulery, R.; Salleron, J.; Dewilde, A.; Rossignol, J.; Boyle, E.M.; Gay, J.; de Berranger, E.; Coiteux, V.; Jouet, J.P.; Duhamel, A.; et al. Early human herpesvirus type 6 reactivation after al-logeneic stem cell transplantation: A large-scale clinical study. Biol. Blood Marrow Transplant. 2012, 18, 1080–1089. [Google Scholar] [CrossRef]
- Scotch, A.H.; Hoss, E.; Orenstein, R.; Budavari, A.I. Disseminated Varicella-Zoster Virus After Vaccination in an Immunocompetent Patient. J. Am. Osteopat. Assoc. 2016, 116, 402–405. [Google Scholar] [CrossRef][Green Version]
- Kim, S.T.; Park, K.H.; Oh, S.C.; Seo, J.H.; Shin, S.W.; Kim, J.S.; Kim, Y.H. Varicella Zoster Virus Infection during Chemotherapy in Solid Cancer Patients. Oncology 2012, 82, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, H.; Takahashi, N.; Nanjo, H.; Kawabata, Y.; Hirokawa, M.; Sawada, K. Varicella-Zoster Virus-associated Fulminant Hepatitis Following Allogeneic Hematopoietic Stem Cell Transplantation for Multiple Myeloma. Intern. Med. 2013, 52, 1727–1730. [Google Scholar] [CrossRef]
- Zhang, W.; Ruan, Q.L.; Yan, F.; Hu, Y.K. Fatal hemorrhagic varicella in a patient with abdominal pain: A case report. BMC Infect. Dis. 2020, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Furuto, Y.; Kawamura, M.; Namikawa, A.; Takahashi, H.; Shibuya, Y. Successful management of visceral disseminated varicella zoster virus infection during treatment of membranous nephropathy: A case report. BMC Infect. Dis. 2019, 19, 625. [Google Scholar] [CrossRef]
- Bollea-Garlatti, M.L.; Bollea-Garlatti, L.A.; Vacas, A.S.; Torre, A.C.; Kowalczuk, A.M.; Galimberti, R.L.; Ferreyro, B.L. Clinical Characteristics and Outcomes in a Population With Disseminated Herpes Zoster: A Retrospective Cohort Study. Actas Dermo-Sifiliográficas 2017, 108, 145–152. [Google Scholar] [CrossRef]
- Nagel, M.A.; Traktinskiy, I.; Azarkh, Y.; Kleinschmidt-DeMasters, B.; Hedley-Whyte, T.; Russman, A.; VanEgmond, E.M.; Stenmark, K.; Frid, M.; Mahalingam, R.; et al. Varicella zoster virus vasculopathy: Analysis of virus-infected arteries. Neurology 2011, 77, 364–370. [Google Scholar] [CrossRef]
- Hoshino, T.; Toi, S.; Toda, K.; Uchiyama, Y.; Yoshizawa, H.; Iijima, M.; Shimizu, Y.; Kitagawa, K. Ischemic Stroke due to Virologically-Confirmed Varicella Zoster Virus Vasculopathy: A Case Series. J. Stroke Cerebrovasc. Dis. 2019, 28, 338–343. [Google Scholar] [CrossRef]
- Uthayakumar, A.; Harrington, D. Spontaneous splenic rupture complicating primary varicella zoster infection: A case report. BMC Res. Notes 2018, 11, 334. [Google Scholar] [CrossRef]
- Tsappa, I.; Missouris, C.; Psarellis, S. Acyclovir-induced thrombocytopaenia in a patient with SLE. BMJ Case Rep. 2018, 2018, bcr-2018. [Google Scholar] [CrossRef]
- Shibusawa, M.; Motomura, S.; Hidai, H.; Tsutsumi, H.; Fujita, A. Varicella infection complicated by marked thrombocytopenia. Jpn. J. Infect. Dis. 2014, 67, 292–294. [Google Scholar] [CrossRef]
- Yoshida, T.; Higuchi, T.; Suzuki, L.; Koyamada, R.; Okada, S. Relapse of Immune Thrombocytopenia Associated with Varicella 20 Years after Splenectomy. Intern. Med. 2014, 53, 2721–2723. [Google Scholar] [CrossRef]
- Diniz, L.M.O.; Maia, M.M.M.; Oliveira, Y.V.; Mourão, M.S.F.; Couto, A.V.; Mota, V.C.; Versiani, C.M.; Silveira, P.; Romanelli, R.M.C. Study of Complications of Varicella-Zoster Virus Infection in Hospitalized Children at a Reference Hospital for Infectious Disease Treatment. Hosp. Pediatr. 2018, 8, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Choi, S.M.; Lee, J.; Park, Y.S.; Lee, C.H.; Yim, J.J.; Yoo, C.G.; Kim, Y.W.; Han, S.K.; Lee, S.M. Respiratory virus of severe pneumonia in South Korea: Prevalence and clinical implications. PLoS ONE 2018, 13, e0198902. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Liabsuetrakul, T.; Sangsupawanich, P.; Xia, X.; He, P.; Cao, H.; McNeil, E. Efficacy and safety of Laggera pterodonta in children 3–24 months with acute bronchiolitis: A randomized controlled trial. Clin. Respir. J. 2017, 11, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Al Shibli, A.; Alkuwaiti, N.; Hamie, M.; Abukhater, D.; Noureddin, M.B.; Amri, A.; Al Kaabi, S.; Al Kaabi, A.; Harbi, M.; Narchi, H. Significance of platelet count in children admitted with bronchiolitis. World J. Clin. Pediatrics 2017, 6, 118–123. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Xiao, Q.Y.; Xie, X.H.; Deng, Y.; Ren, L.; Tian, D.Y.; Luo, Z.X.; Luo, J.; Fu, Z.; Huang, A.L.; et al. Association between secondary thrombocytosis and viral respiratory tract infections in children. Sci. Rep. 2016, 6, 22964. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Low, J.G.; Wyone, W.; Oon, L.L.; Tan, B.H. Survey of Respiratory Virus in Patients Hospitalised for Acute Exacerbations of Heart Failure—A Prospective Observational Study. Ann. Acad. Med. Singap. 2018, 47, 445–450. [Google Scholar]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Gupalo, E.; Buriachkovskaia, L.; Othman, M. Human platelets express CAR with localization at the sites of intercellular interac-tion. Virol. J. 2011, 8, 456. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.Y.; Yu, X.N.; Qu, Z.Y.; Zhang, A.A.; Xing, Y.L.; Jiang, L.X.; Shang, L.; Wang, Y.C. Adenovirus type 3 induces platelet activation in vitro. Mol. Med. Rep. 2014, 9, 370–374. [Google Scholar] [CrossRef]
- Gupalo, E.; Kuk, C.; Qadura, M.; Buriachkovskaia, L.; Othman, M. Platelet–adenovirus vs. inert particles interaction: Effect on aggregation and the role of platelet membrane receptors. Platelets 2013, 24, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Escutenaire, S.; Cerullo, V.; Diaconu, I.; Ahtiainen, L.; Hannuksela, P.; Oksanen, M.; Haavisto, E.; Karioja-Kallio, A.; Holm, S.L.; Kangasniemi, L.; et al. In vivo and in vitro distribution of type 5 and fiber-modified oncolytic adenoviruses in human blood compartments. Ann. Med. 2011, 43, 151–163. [Google Scholar] [CrossRef]
- Zhu, R.; Chen, C.; Wang, Q.; Zhang, X.; Lu, C.; Sun, Y. Routine blood parameters are helpful for early identification of influenza infection in children. BMC Infect. Dis. 2020, 20, 864. [Google Scholar] [CrossRef]
- Rose, J.J.; Voora, D.; Cyr, D.D.; Lucas, J.E.; Zaas, A.K.; Woods, C.W.; Newby, L.K.; Kraus, W.E.; Ginsburg, G.S. Gene Expression Profiles Link Respiratory Viral Infection, Platelet Response to Aspirin, and Acute Myocardial Infarction. PLoS ONE 2015, 10, e0132259. [Google Scholar] [CrossRef]
- Sugiyama, M.G.; Gamage, A.; Zyla, R.; Armstrong, S.M.; Advani, S.; Advani, A.; Wang, C.; Lee, W.L. Influenza Virus Infection Induces Plate-let-Endothelial Adhesion Which Contributes to Lung Injury. J. Virol. 2016, 90, 1812–1823. [Google Scholar] [CrossRef]
- Jansen, A.J.G.; Spaan, T.; Low, H.Z.; Di Iorio, D.; van den Brand, J.; Tieke, M.; Barendrecht, A.; Rohn, K.; van Amerongen, G.; Stittelaar, K.; et al. Influenza-induced thrombocytopenia is dependent on the subtype and sialoglycan receptor and increases with virus pathogenicity. Blood Adv. 2020, 4, 2967–2978. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1780. [Google Scholar] [CrossRef]
- Boilard, E.; Paré, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Lévesque, T.; Borgeat, P.; Flamand, L. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 2014, 123, 2854–2863. [Google Scholar] [CrossRef]
- Almohammadi, A.; Lundin, M.S.; Abro, C.; Hrinczenko, B. Epistaxis and gross haematuria with severe thrombocytopaenia asso-ciated with influenza vaccination. BMJ Case Rep. 2019, 12, e229423. [Google Scholar] [CrossRef]
- Hamiel, U.; Kventsel, I.; Youngster, I. Recurrent Immune Thrombocytopenia After Influenza Vaccination: A Case Report. Pediatrics 2016, 138, e20160124. [Google Scholar] [CrossRef]
- Laksono, B.M.; de Vries, R.D.; Verburgh, R.J.; Visser, E.G.; de Jong, A.; Fraaij, P.L.A.; Ruijs, W.L.M.; Nieuwenhuijse, D.F.; van den Ham, H.J.; Koopmans, M.P.G.; et al. Studies into the mechanism of mea-sles-associated immune suppression during a measles outbreak in the Netherlands. Nat. Commun. 2018, 9, 4944. [Google Scholar] [CrossRef] [PubMed]
- Moss, W.J. Measles. Lancet 2017, 390, 2490–2502. [Google Scholar] [CrossRef]
- Premaratna, R.; Luke, N.; Perera, H.; Gunathilake, M.; Amarasena, P.; Chandrasena, T.G. Sporadic cases of adult measles: A research article. BMC Res. Notes 2017, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Sunnetcioglu, M.; Baran, A.I.; Sunnetcioglu, A.; Mentes, O.; Karadas, S.; Aypak, A. Clinical and laboratory features of adult measles cases detected in Van, Turkey. J. Pak. Med. Assoc. 2015, 65, 273–276. [Google Scholar] [PubMed]
- Celesia, B.M.; Fontana, R.; Pinzone, M.R.; Cuccia, M.; Bellissimo, F.; Rapisarda, L.; Rinnone, S.; Rapisarda, V.; Pavone, P.; Cacopardo, B.; et al. A measles outbreak in Catania, Sicily: The importance of high vaccination coverage and early notification of cases for health and economic reasons. Le Infezioni Medicina 2014, 22, 222–226. [Google Scholar] [PubMed]
- Oncel, I.; Saltik, S.; Anlar, B. Subacute sclerosing panencephalitis and immune thrombocytopenia: More than a coincidence? Med Hypotheses 2018, 111, 70–72. [Google Scholar] [CrossRef]
- Okazaki, N.; Takeguchi, M.; Sonoda, K.; Handa, Y.; Kakiuchi, T.; Miyahara, H.; Akiyoshi, K.; Korematsu, S.; Suenobu, S.; Izumi, T. Detection of platelet-binding anti-measles and anti-rubella virus IgG antibodies in infants with vaccine-induced thrombocytopenic purpura. Vaccine 2011, 29, 4878–4880. [Google Scholar] [CrossRef]
- Agra, I.K.R.; Amorim Filho, A.G.; Lin, L.H.; Biancolin, S.E.; Francisco, R.P.V.; Brizot, M.L. Parameters Associated with Adverse Fetal Outcomes in Parvovirus B19 Congenital Infection. Revista Brasileira de Ginecologia e Obstetrícia 2017, 39, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Melamed, N.; Whittle, W.; Kelly, E.N.; Windrim, R.; Seaward, P.G.; Keunen, J.; Keating, S.; Ryan, G. Fetal thrombocytopenia in pregnancies with fetal human parvovirus-B19 infection. Am. J. Obstet. Gynecol. 2015, 212, 793.e1–793.e8. [Google Scholar] [CrossRef]
- Rajput, R.; Sehgal, A.; Jain, D.; Sen, R.; Gupta, A. Acute Parvovirus B19 Infection Leading to Severe Aplastic Anemia in a Previously Healthy Adult Female. Indian J. Hematol. Blood Transfus. 2012, 28, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Park, S.; Lee, G.W.; Koh, E.H.; Kim, H.Y. Parvovirus B19 infection presenting with neutropenia and thrombocytopenia: Three case reports. Medicine 2019, 98, e16993. [Google Scholar] [CrossRef]
- Yaguchi, D.; Marui, N.; Matsuo, M. Three Adult Cases of HPV-B19 Infection with Concomitant Leukopenia and Low Platelet Counts. Clin. Med. Insights Case Rep. 2015, 8, 19–22. [Google Scholar] [CrossRef]
- Furkan Demir, B.; Karahan Meteris, A.; Yirgin, G.; Comoglu, M.; Katipoglu, B.; Yılmaz, N.; Ates, I. Parvovırus-induced thrombocy-topenıa. Hematol. Oncol. Stem Cell Ther. 2019, 12, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Jain, P.; Prakash, S.; Kumar, A.; Khan, D.N.; Seth, A.; Gupta, S.; Kant, R. Genotype 3b of human parvovirus B19 detected from hospitalized children with solid malignancies in a North Indian tertiary care hospital. J. Med Virol. 2016, 88, 1922–1929. [Google Scholar] [CrossRef]
- Jitschin, R.; Peters, O.; Plentz, A.; Turowski, P.; Segerer, H.; Modrow, S. Impact of parvovirus B19 infection on paediatric patients with haematological and/or oncological disorders. Clin. Microbiol. Infect. 2011, 17, 1336–1342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bachelier, K.; Biehl, S.; Schwarz, V.; Kindermann, I.; Kandolf, R.; Sauter, M.; Ukena, C.; Yilmaz, A.; Sliwa, K.; Bock, C.T.; et al. Parvovirus B19-induced vascular damage in the heart is associated with elevated circulating endothelial microparticles. PLoS ONE 2017, 12, e0176311. [Google Scholar] [CrossRef]
- Wurster, T.; Pölzelbauer, C.; Schönberger, T.; Paul, A.; Seizer, P.; Stellos, K.; Schuster, A.; Botnar, R.M.; Gawaz, M.; Bigalke, B. Green Fluorescent Protein (GFP) Color Reporter Gene Visualizes Parvovirus B19 Non-Structural Segment 1 (NS1) Transfected Endothelial Modification. PLoS ONE 2012, 7, e33602. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Griffiths, C.; Drews, S.J.; Marchant, D.J. Respiratory Syncytial Virus: Infection, Detection, and New Options for Prevention and Treatment. Clin. Microbiol. Rev. 2017, 30, 277. [Google Scholar] [CrossRef]
- Kullaya, V.I.; de Mast, Q.; van der Ven, A.; elMoussaoui, H.; Kibiki, G.; Simonetti, E.; de Jonge, M.I.; Ferwerda, G. Platelets Modulate Innate Immune Response Against Human Respiratory Syncytial Virus In Vitro. Viral Immunol. 2017, 30, 576–581. [Google Scholar] [CrossRef]
- Bels, J.L.M.; van Kuijk, S.M.J.; Ghossein-Doha, C.; Tijssen, F.H.; van Gassel, R.J.J.; Tas, J.; Collaborators, M.; Schnabel, R.M.; Aries, M.J.H.; van de Poll, M.C.G.; et al. Decreased serial scores of severe organ failure assessments are associated with survival in mechanically ventilated patients; the prospective Maastricht Intensive Care COVID cohort. J. Crit. Care 2021, 62, 38–45. [Google Scholar] [CrossRef]
- Kaptein, F.H.J.; Stals, M.A.M.; Grootenboers, M.; Braken, S.J.E.; Burggraaf, J.L.I.; van Bussel, B.C.T.; Cannegieter, S.C.; ten Cate, H.; Endeman, H.; Gommers, D.A.M.P.J.; et al. Incidence of thrombotic compli-cations and overall survival in hospitalized patients with COVID-19 in the second and first wave. Thromb. Res. 2020. [Google Scholar]
- Brüggemann, R.A.G.; Spaetgens, B.; Gietema, H.A.; Brouns, S.H.A.; Stassen, P.M.; Magdelijns, F.J.; Rennenberg, R.J.; Henry, R.M.A.; Mulder, M.M.G.; van Bussel, B.C.T.; et al. The prevalence of pulmonary embolism in patients with COVID-19 and respiratory decline: A three-setting comparison. Thromb. Res. 2020, 196, 486–490. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- The COVID-19 Autopsy. The first COVID-19 autopsy in Spain performed during the early stages of the pandemic. Revista Espanola de Patologia 2020, 53, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated with Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to im-munothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. Erythrocyte, Platelet, Serum Ferritin, and P-Selectin Pathophysiology Implicated in Severe Hypercoagulation and Vascular Complications in COVID-19. Int. J. Mol. Sci. 2020, 21, 8234. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Rokni, M.; Ahmadikia, K.; Asghari, S.; Mashaei, S.; Hassanali, F. Comparison of clinical, para-clinical and laboratory findings in survived and deceased patients with COVID-19: Diagnostic role of inflammatory indications in determining the severity of illness. BMC Infect. Dis. 2020, 20, 869. [Google Scholar] [CrossRef]
- Lu, G.; Wang, J. Dynamic changes in routine blood parameters of a severe COVID-19 case. Clin. Chim. Acta 2020, 508, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Motta, A.; Strollo, M.; Banfi, G.; Locatelli, M. Routine blood tests as a potential diagnostic tool for COVID-19. Clin. Chem. Lab. Med. 2020, 58, 1095–1099. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Yuan, Y.; Huang, F.; Ma, R.; Luo, B.; Xi, Z.; Pan, T.; Liu, B.; Zhang, Y.; et al. Two waves of pro-inflammatory factors are released during the in-fluenza A virus (IAV)-driven pulmonary immunopathogenesis. PLoS Pathog. 2020, 16, e1008334. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.H.; Timmermans, S.; Nagy, M.; Visser, M.; Huckriede, J.; Aendekerk, J.P.; de Vries, F.; Potjewijd, J.; Jallah, B.; Ysermans, R.; et al. Neutrophils and Contact Activation of Co-agulation as Potential Drivers of COVID-19. Circulation 2020, 142, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and plate-let-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Nguyen, C.B.; Yeung, Z.; Sanchez, K.; Rosen, D.; Bushan, S. Evans syndrome in a patient with COVID-19. Br. J. Haematol. 2020, 190, e59–e61. [Google Scholar] [CrossRef]
- Li, H.; Wang, B.; Ning, L.; Luo, Y.; Xiang, S. Transient appearance of EDTA dependent pseudothrombocytopenia in a patient with 2019 novel coronavirus pneumonia. Platelets 2020, 31, 825–826. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G. Vaccine innovation spurred by the long wait for an Ebola virus vaccine. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Mulangu, S.; Dodd, L.E.; Davey, R.T., Jr.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- da Costa, P.S.; Ribeiro, G.M.; Junior, C.S.; da Costa Campos, L. Severe thrombotic events associated with dengue fever, Brazil. Am. J. Trop. Med. Hyg. 2012, 87, 741–742. [Google Scholar] [CrossRef]
- Liu, S.; DeLalio, L.J.; Isakson, B.E.; Wang, T.T. AXL-Mediated Productive Infection of Human Endothelial Cells by Zika Virus. Circ. Res. 2016, 119, 1183–1189. [Google Scholar] [CrossRef]
- Ramacciotti, E.; Agati, L.B.; Aguiar, V.C.R.; Wolosker, N.; Guerra, J.C.; de Almeida, R.P.; Alves, J.C.; Lopes, R.D.; Wakefield, T.W.; Comerota, A.J.; et al. Zika and Chikungunya Virus and Risk for Venous Thromboembolism. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029618821184. [Google Scholar] [CrossRef]
- Guillevin, L.; Mahr, A.; Callard, P.; Godmer, P.; Pagnoux, C.; Leray, E.; Cohen, P. Hepatitis B virus-associated polyarteritis nodosa: Clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine 2005, 84, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, O.; Feldman, D.M.; Diakow, M.; Sigal, S.H. The pathophysiology of thrombocytopenia in chronic liver disease. Hepatic Med. Évid. Res. 2016, 8, 39–50. [Google Scholar]
- Wijarnpreecha, K.; Thongprayoon, C.; Panjawatanan, P.; Ungprasert, P. Hepatitis C Virus Infection and Risk of Venous Thromboembolism: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 514–520. [Google Scholar] [CrossRef]
- Nery, F.; Chevret, S.; Condat, B.; de Raucourt, E.; Boudaoud, L.; Rautou, P.E.; Plessier, A.; Roulot, D.; Chaffaut, C.; Bourcier, V.; et al. Causes and consequences of portal vein thrombosis in 1,243 patients with cirrhosis: Results of a longitudinal study. Hepatology 2015, 61, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Wijarnpreecha, K.; Thongprayoon, C.; Panjawatanan, P.; Ungprasert, P. Hepatitis B virus infection and risk of coronary artery disease: A meta-analysis. Ann. Transl. Med. 2016, 4, 423. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.A.; Xiaoqiang, W.; Budoff, M.; Leaf, D.; Kuller, L.H.; Justice, A.C. Hepatitis C Virus Infection and the Risk of Coronary Disease. Clin. Infect. Dis. 2009, 49, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Blüm, P.; Pircher, J.; Merkle, M.; Czermak, T.; Ribeiro, A.; Mannell, H.; Krötz, F.; Hennrich, A.; Spannagl, M.; Köppel, S.; et al. Arterial thrombosis in the context of HCV-associated vascular disease can be prevented by protein C. Cell. Mol. Immunol. 2017, 14, 986–996. [Google Scholar] [CrossRef][Green Version]
- Baiocchini, A.; Del Nonno, F.; Taibi, C.; Visco-Comandini, U.; D’Offizi, G.; Piacentini, M.; Falasca, L. Liver sinusoidal endothelial cells (LSECs) modifications in patients with chronic hepatitis C. Sci. Rep. 2019, 9, 8760. [Google Scholar] [CrossRef]
- Ragab, G.; Hussein, M.A. Vasculitic syndromes in hepatitis C virus: A review. J. Adv. Res. 2017, 8, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.A.; Allain, T.J.; Mzinganjira, H.; Connor, M.D.; Smith, C.; Lucas, S.; Joekes, E.; Kampondeni, S.; Chetcuti, K.; Turnbull, I.; et al. The Role of Human Immunodeficiency Vi-rus-Associated Vasculopathy in the Etiology of Stroke. J. Infect. Dis. 2017, 216, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Morgello, S.; Klotman, M.E.; Mosoian, A.; Lento, P.A.; Berman, J.W.; Schecter, A.D. Human immunodeficiency virus (HIV) infects human arterial smooth muscle cells in vivo and in vitro: Implications for the pathogenesis of HIV-mediated vascular disease. Am. J. Pathol. 2008, 172, 1100–1111. [Google Scholar] [CrossRef]
- Nixon, C.C.; Vatakis, D.N.; Reichelderfer, S.N.; Dixit, D.; Kim, S.G.; Uittenbogaart, C.H.; Zack, J.A. HIV-1 infection of hematopoietic pro-genitor cells in vivo in humanized mice. Blood 2013, 122, 2195–2204. [Google Scholar] [CrossRef]
- Rokx, C.; Borjas Howard, J.F.; Smit, C.; Wit, F.W.; Pieterman, E.D.; Reiss, P.; Cannegieter, S.C.; Lijfering, W.M.; Meijer, K.; Bierman, W.; et al. Risk of recurrent venous thromboembolism in patients with HIV infection: A nationwide cohort study. PLoS Med. 2020, 17, e1003101. [Google Scholar] [CrossRef]
- Freiberg, M.S.; Chang, C.C.; Kuller, L.H.; Skanderson, M.; Lowy, E.; Kraemer, K.L.; Butt, A.A.; Bidwell Goetz, M.; Leaf, D.; Oursler, K.A.; et al. HIV Infection and the Risk of Acute Myocardial Infarction. JAMA Intern. Med. 2013, 173, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Van Wyk, V.; Kotzé, H.F.; Heyns, A.P. Kinetics of indium-111-labelled platelets in HIV-infected patients with and without asso-ciated thrombocytopaenia. Eur. J. Haematol. 1999, 62, 332–335. [Google Scholar] [PubMed]
- Ambler, K.L.; Vickars, L.M.; Leger, C.S.; Foltz, L.M.; Montaner, J.S.; Harris, M.; Dias Lima, V.; Leitch, H.A. Clinical Features, Treatment, and Outcome of HIV-Associated Immune Thrombocytopenia in the HAART Era. Adv. Hematol. 2012, 2012, 910954. [Google Scholar] [CrossRef] [PubMed]
- Telles, J.P.; de Andrade Perez, M.; Marcusso, R.; Correa, K.; Teixeira, R.F.A.; Tobias, W.M. Hemophagocytic syndrome in patients living with HIV: A retrospective study. Ann. Hematol. 2019, 98, 67–72. [Google Scholar] [CrossRef]


{kind=link}
| Virus | Platelet Binding (Receptors) | Platelet Activation (Receptors, Markers) | Platelet Infection (Receptors) | Platelet Replication | Vascular Endothelial Disruption | Impairs Platelet Production | Associated with Hemorrhage | Associated with Thrombo-embolism | Platelet Sequestration | PLA Formation | Autoimmunity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DENV | DC-SIGN, HSP, FcϒR2A, GPIb, CLEC2, CLEC5A [66,67,81] | Receptors: CLEC2, CLEC5A, TLR 2/4, 5HT2 [66,67,71] Markers: P-Selectin, Integrin α2b, PF4, CCL5, PS, CD40L, CD63, GP1b, GPIIb/IIIa, microparticle release, aggregation [63,64,65,66,67,70,73] | DC-SIGN, HSP, FcϒR2A (Ig bound virus) [30] | Yes [30] | Plasma leakage [59,67] | Infects megakaryocyte like cells [80,81] | With endothelial and platelet dysfunction [56,57,58] | Rare [235] | Phagocytosis [73,76] | PMA, PNA, inducing NETosis [68,69] | Immune complexes [75] |
| ZIKV | U | U | No [116] | No [116] | Infects vascular endothelial cells in vitro [236] | U | In severe cases [107,108] | Rare [237] | U | U | ITP [112,113,114] |
| YFV | U | U | U | U | Fundoscopic abnormalities [101] | U | Hepatic failure and deficiency of plasma coagulation factors. [99,100] Cirrhosis associated varices (HBV/HCV) | Microvascular thrombosis [101] | U | U | U |
| HBV | U | Markers: Morphological changes [127] resistance to antiplatelet agents [128] | U | U | Polyarteritis nodosa (rare) [238] | Impaired hepatic TPO production [131,239] | Increased risk of VTE [240] Portal vein thrombosis in cirrhosis [241]. Risk of ischemic cardiovascular disease elevated for HCV only [242,243] | Portal hypertension and hypersplenism [239] | U | U | |
| HCV | Likely, Mechanisms unknown [130,132,133] | U | Greater stability of HCV in platelets, [130,132] persistence in platelets during treatment [133] | No [132] | Endothelial activation [244], capillarization of liver sinusoidal endothelial cells [245], (non)cryoglobulinemic vasculitis [246] | U | ITP [130] | ||||
| HIV | DC-SIGN, [138] GPIIIa, CCL-3 [146] | Receptors: CXCL4, CCR3, GPIIIa. Markers: PF-4, CCL5, P-selectin, sCD40L, CCL5, Conflicting reports on morphological changes, oxidative stress.(135–137, 139) | Yes, via megakaryocyte precursors [141] | No | HIV-associated vasculopathy [247], Infects arterial smooth muscle cells [248] | Infection and impairment of hematopoietic progenitor cells [249] | No | Increased risk of VTE [250], myocardial infarction [251] and cerebrovascular disease [247] | Hyper-splenism [252] | PMA, increasing monocyte TF expression [144,145] | ITP [253], HLH [254] |
| IAV/IBV | Likely NA binding of glycans [191] | Receptors: Glycans, TLR7, [44] Immune complexes via Fc-ϒIIA receptor [193] Markers: P-selectin, [191] CD40L, C3, CD63[192] Resistance to antiplatelet agents.[189] | Phago-cytosis [191] | U | Infection of pulmonary vascular endothelial cells [190] | U | No | VTE, myocardial infarction, ischemic cerebrovascular accidents [1] | U | PNA, inducing NETosis [192] | ITP [194,195] |
| SARS CoV 2 | ACE2, conflicting evidence on platelet expression [223,224] | Receptors: ACE2, TLR4, CLEC2, CXCR-4. Markers: P-selectin, GPIb, GP IIa GPIIIb, GPIIb/IIIa, CD40L, CD63, morphological changes, aggregation, degranulation [220,222,223,224] | U | U | U | Decreased cGMP expression [224] | No | Microvascular thrombosis and VTE [216,217,218,219] | U | PMA increasing monocyte TF expression, PNA inducing NETosis [221,230] | ITP, Evans Syndrome [231] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raadsen, M.; Du Toit, J.; Langerak, T.; van Bussel, B.; van Gorp, E.; Goeijenbier, M. Thrombocytopenia in Virus Infections. J. Clin. Med. 2021, 10, 877. https://doi.org/10.3390/jcm10040877
Raadsen M, Du Toit J, Langerak T, van Bussel B, van Gorp E, Goeijenbier M. Thrombocytopenia in Virus Infections. Journal of Clinical Medicine. 2021; 10(4):877. https://doi.org/10.3390/jcm10040877
Chicago/Turabian StyleRaadsen, Matthijs, Justin Du Toit, Thomas Langerak, Bas van Bussel, Eric van Gorp, and Marco Goeijenbier. 2021. "Thrombocytopenia in Virus Infections" Journal of Clinical Medicine 10, no. 4: 877. https://doi.org/10.3390/jcm10040877
APA StyleRaadsen, M., Du Toit, J., Langerak, T., van Bussel, B., van Gorp, E., & Goeijenbier, M. (2021). Thrombocytopenia in Virus Infections. Journal of Clinical Medicine, 10(4), 877. https://doi.org/10.3390/jcm10040877