
Roles of Ceramides in Non-Alcoholic Fatty Liver Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction on NAFLD
1.1. Steatosis and Non-Alcoholic Steatohepatitis
1.2. NAFLD, Obesity and Diabetes
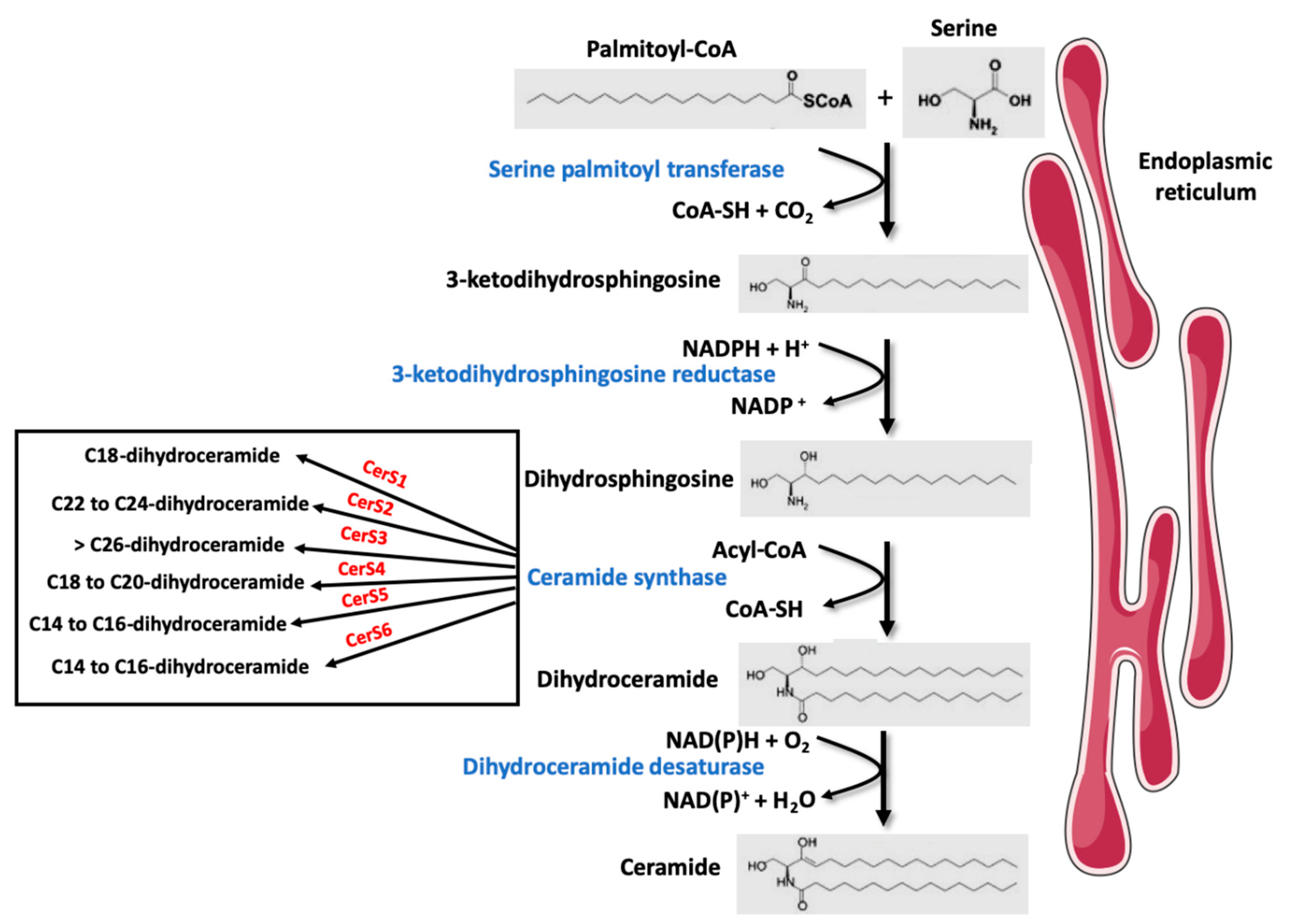
2. Overview of Sphingolipid and Ceramide Metabolism
3. Role of Sphingolipids in Hepatic Steatosis
3.1. Liver Sphingolipids Concentrations are Altered in NAFLD
3.2. Liver Ceramide Contribution to NAFLD
3.2.1. Sphingolipids and Hepatic Insulin Resistance
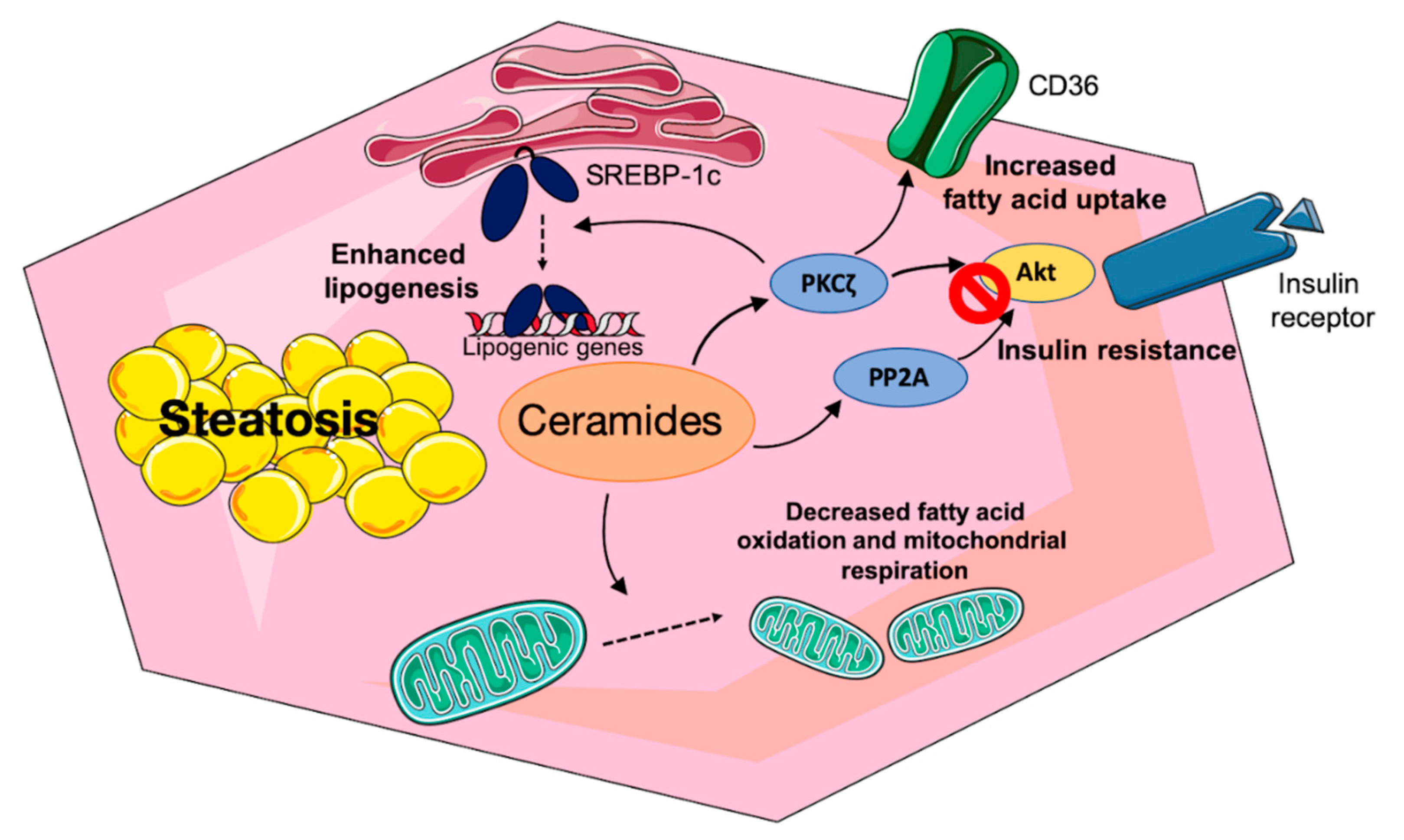
3.2.2. Mechanisms of Ceramide-Induced Hepatic Insulin Resistance
3.2.3. Direct Effect of Sphingolipid Species on Hepatic Lipid Metabolism
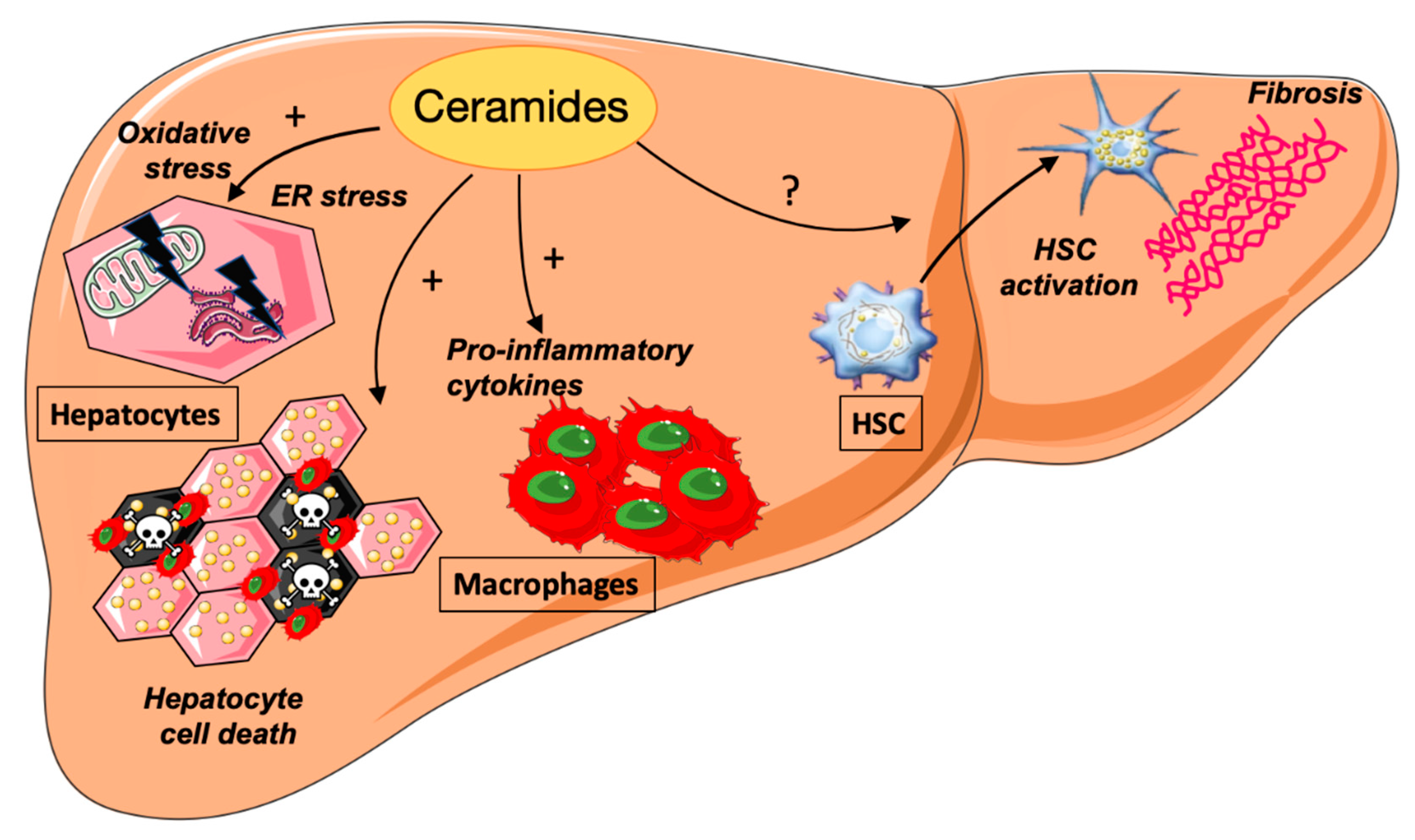
4. Ceramides and NASH
4.1. Ceramides and ER Stress
4.2. Ceramides and Hepatocyte Cell Death
4.3. Ceramides and Inflammation
4.4. Ceramides and Hepatic Fibrosis
5. Ceramides as a Therapeutic Target
Author Contributions
Funding
Conflicts of Interest
References
- Eslam, M.; Sanyal, A.J.; George, J. International Consensus Panel MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Phan, F.; Bourron, O.; Ferré, P.; Foufelle, F. Steatosis and NASH in Type 2 Diabetes. Biochimie 2017, 143, 37–41. [Google Scholar] [CrossRef]
- Unger, R.H.; Orci, L. Lipotoxic Diseases of Nonadipose Tissues in Obesity. Int. J. Obes. 2000, 24 (Suppl. 4), S28–S32. [Google Scholar] [CrossRef]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of Insulin Signaling Pathway in Rat Liver: MTORC1 Required for Stimulation of Lipogenesis, but Not Inhibition of Gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.-C.; Kim, S.H. Interrelationship between Fatty Liver and Insulin Resistance in the Development of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Thudichum, J.L.B. A Treatise on the Chemical Constituents of the Brain; Bailliere, Tindall and Cox: London, UK, 1854. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Hage Hassan, R.; Bourron, O.; Hajduch, E. Defect of Insulin Signal in Peripheral Tissues: Important Role of Ceramide. World J. Diabetes 2014, 5, 244–257. [Google Scholar] [CrossRef]
- Summers, S.A.; Chaurasia, B.; Holland, W.L. Metabolic Messengers: Ceramides. Nat. Metab. 2019, 1, 1051–1058. [Google Scholar] [CrossRef]
- Kojta, I.; Chacińska, M.; Błachnio-Zabielska, A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020, 12, 1305. [Google Scholar] [CrossRef]
- Iessi, E.; Marconi, M.; Manganelli, V.; Sorice, M.; Malorni, W.; Garofalo, T.; Matarrese, P. On the Role of Sphingolipids in Cell Survival and Death. Int. Rev. Cell Mol. Biol. 2020, 351, 149–195. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide Synthases at the Centre of Sphingolipid Metabolism and Biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef]
- Bandet, C.L.; Mahfouz, R.; Veret, J.; Sotiropoulos, A.; Poirier, M.; Giussani, P.; Campana, M.; Philippe, E.; Blachnio-Zabielska, A.; Ballaire, R.; et al. Ceramide Transporter CERT Is Involved in Muscle Insulin Signaling Defects Under Lipotoxic Conditions. Diabetes 2018, 67, 1258–1271. [Google Scholar] [CrossRef]
- Kumagai, K.; Hanada, K. Structure, Functions and Regulation of CERT, a Lipid-Transfer Protein for the Delivery of Ceramide at the ER-Golgi Membrane Contact Sites. FEBS Lett. 2019, 593, 2366–2377. [Google Scholar] [CrossRef]
- Inokuchi, J.-I.; Inamori, K.-I.; Kabayama, K.; Nagafuku, M.; Uemura, S.; Go, S.; Suzuki, A.; Ohno, I.; Kanoh, H.; Shishido, F. Biology of GM3 Ganglioside. Prog. Mol. Biol. Transl. Sci. 2018, 156, 151–195. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The Sphingolipid Salvage Pathway in Ceramide Metabolism and Signaling. Cell Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Raichur, S.; Wang, S.T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Ohman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 Haploinsufficiency Inhibits Beta-Oxidation and Confers Susceptibility to Diet-Induced Steatohepatitis and Insulin Resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef]
- Deevska, G.M.; Rozenova, K.A.; Giltiay, N.V.; Chambers, M.A.; White, J.; Boyanovsky, B.B.; Wei, J.; Daugherty, A.; Smart, E.J.; Reid, M.B.; et al. Acid Sphingomyelinase Deficiency Prevents Diet-Induced Hepatic Triacylglycerol Accumulation and Hyperglycemia in Mice. J. Biol. Chem. 2009, 284, 8359–8368. [Google Scholar] [CrossRef] [PubMed]
- Cinar, R.; Godlewski, G.; Liu, J.; Tam, J.; Jourdan, T.; Mukhopadhyay, B.; Harvey-White, J.; Kunos, G. Hepatic Cannabinoid-1 Receptors Mediate Diet-Induced Insulin Resistance by Increasing de Novo Synthesis of Long-Chain Ceramides. Hepatology 2013, 59, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, T.; Li, L.; Li, M.; Gulshan, K.; Kirwan, J.P.; Liu, X.; Previs, S.; Willard, B.; Smith, J.D.; McCullough, A. Ceramide as a Mediator of Non-Alcoholic Fatty Liver Disease and Associated Atherosclerosis. PLoS ONE 2015, 10, e0126910. [Google Scholar] [CrossRef]
- Yetukuri, L.; Katajamaa, M.; Medina-Gomez, G.; Seppanen-Laakso, T.; Vidal-Puig, A.; Oresic, M. Bioinformatics Strategies for Lipidomics Analysis: Characterization of Obesity Related Hepatic Steatosis. BMC Syst. Biol. 2007, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Longato, L.; Tong, M.; Wands, J.R.; de la Monte, S.M. High Fat Diet Induced Hepatic Steatosis and Insulin Resistance: Role of Dysregulated Ceramide Metabolism. Hepatol. Res. 2012, 42, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Raichur, S.; Brunner, B.; Bielohuby, M.; Hansen, G.; Pfenninger, A.; Wang, B.; Bruning, J.C.; Larsen, P.J.; Tennagels, N. The Role of C16:0 Ceramide in the Development of Obesity and Type 2 Diabetes: CerS6 Inhibition as a Novel Therapeutic Approach. Mol. Metab. 2019, 21, 36–50. [Google Scholar] [CrossRef]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic Diacylglycerol Content Is Associated with Hepatic Insulin Resistance in Obese Subjects. Gastroenterology 2012, 142, 1444–1446. [Google Scholar] [CrossRef]
- Kotronen, A.; Seppanen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepaa, A.L.; Oresic, M.; Yki-Jarvinen, H. Hepatic Stearoyl-CoA Desaturase (SCD)-1 Activity and Diacylglycerol but Not Ceramide Concentrations Are Increased in the Nonalcoholic Human Fatty Liver. Diabetes 2009, 58, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular Mechanism of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Barnett, A.C.; Bruce, C.R.; Schenk, S.; Horowitz, J.F.; Hoy, A.J. Regulation of Plasma Ceramide Levels with Fatty Acid Oversupply: Evidence That the Liver Detects and Secretes de Novo Synthesised Ceramide. Diabetologia 2012, 55, 2741–2746. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef]
- Lyn-Cook, L.E.; Lawton, M.; Tong, M.; Silbermann, E.; Longato, L.; Jiao, P.; Mark, P.; Wands, J.R.; Xu, H.; de la Monte, S.M. Hepatic Ceramide May Mediate Brain Insulin Resistance and Neurodegeneration in Type 2 Diabetes and Non-Alcoholic Steatohepatitis. J. Alzheimers Dis. JAD 2009, 16, 715–729. [Google Scholar] [CrossRef]
- Park, J.W.; Park, W.J.; Futerman, A.H. Ceramide Synthases as Potential Targets for Therapeutic Intervention in Human Diseases. Biochim. Biophys. Acta 2013, 1841, 671–681. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Mato, J.M.; Vance, D.; Kaplowitz, N.; Fernández-Checa, J.C. Acid Sphingomyelinase-Ceramide System in Steatohepatitis: A Novel Target Regulating Multiple Pathways. J. Hepatol. 2015, 62, 219–233. [Google Scholar] [CrossRef]
- Simha, V.; Garg, A. Lipodystrophy: Lessons in Lipid and Energy Metabolism. Curr. Opin. Lipidol. 2006, 17, 162–169. [Google Scholar] [CrossRef]
- Sankella, S.; Garg, A.; Agarwal, A.K. Activation of Sphingolipid Pathway in the Livers of Lipodystrophic Agpat2−/− Mice. J. Endocr. Soc. 2017, 1, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in Steatohepatitis Occurs despite an Increase in Tricarboxylic Acid Cycle Activity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Pacana, T. A Lipidomic Readout of Disease Progression in A Diet-Induced Mouse Model of Nonalcoholic Fatty Liver Disease. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 271–288. [Google Scholar] [PubMed]
- Montandon, S.A.; Somm, E.; Loizides-Mangold, U.; de Vito, C.; Dibner, C.; Jornayvaz, F.R. Multi-Technique Comparison of Atherogenic and MCD NASH Models Highlights Changes in Sphingolipid Metabolism. Sci. Rep. 2019, 9, 16810. [Google Scholar] [CrossRef] [PubMed]
- Haberl, E.M.; Pohl, R.; Rein-Fischboeck, L.; Höring, M.; Krautbauer, S.; Liebisch, G.; Buechler, C. Hepatic Lipid Profile in Mice Fed a Choline-Deficient, Low-Methionine Diet Resembles Human Non-Alcoholic Fatty Liver Disease. Lipids Health Dis. 2020, 19, 250. [Google Scholar] [CrossRef]
- Jiang, M.; Li, C.; Liu, Q.; Wang, A.; Lei, M. Inhibiting Ceramide Synthesis Attenuates Hepatic Steatosis and Fibrosis in Rats With Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD Progression: A Lipidomics Approach to an Epidemic. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic Ceramides Dissociate Steatosis and Insulin Resistance in Patients with Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis Model Assessment: Insulin Resistance and Beta-Cell Function from Fasting Plasma Glucose and Insulin Concentrations in Man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Apostolopoulou, M.; Gordillo, R.; Koliaki, C.; Gancheva, S.; Jelenik, T.; De Filippo, E.; Herder, C.; Markgraf, D.; Jankowiak, F.; Esposito, I.; et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care 2018, 41, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Carlier, A.; Phan, F.; Szpigel, A.; Hajduch, E.; Salem, J.-E.; Gautheron, J.; Le Goff, W.; Guérin, M.; Lachkar, F.; Ratziu, V.; et al. Dihydroceramides in Triglyceride-Enriched VLDL Are Associated with Nonalcoholic Fatty Liver Disease Severity in Type 2 Diabetes. Cell Rep. Med. 2020, 1, 100154. [Google Scholar] [CrossRef]
- Promrat, K.; Longato, L.; Wands, J.R.; de la Monte, S.M. Weight Loss Amelioration of Non-Alcoholic Steatohepatitis Linked to Shifts in Hepatic Ceramide Expression and Serum Ceramide Levels. Hepatol. Res. 2011, 41, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Mamtani, M.; Meikle, P.J.; Kulkarni, H.; Weir, J.M.; Barlow, C.K.; Jowett, J.B.; Bellis, C.; Dyer, T.D.; Almasy, L.; Mahaney, M.C.; et al. Plasma Dihydroceramide Species Associate with Waist Circumference in Mexican American Families. Obesity 2014, 22, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Lopez, X.; Goldfine, A.B.; Holland, W.L.; Gordillo, R.; Scherer, P.E. Plasma Ceramides Are Elevated in Female Children and Adolescents with Type 2 Diabetes. J. Pediatr. Endocrinol. Metab. 2013, 26, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Szpigel, A.; Hainault, I.; Carlier, A.; Venteclef, N.; Batto, A.F.; Hajduch, E.; Bernard, C.; Ktorza, A.; Gautier, J.F.; Ferré, P.; et al. Lipid Environment Induces ER Stress, TXNIP Expression and Inflammation in Immune Cells of Individuals with Type 2 Diabetes. Diabetologia 2018, 61, 399–412. [Google Scholar] [CrossRef]
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A.; et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep. 2017, 18, 2269–2279. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol. Metab. TEM 2017, 28, 497–505. [Google Scholar] [CrossRef]
- Litherland, G.; Hajduch, E.; Hundal, H. Intracellular Signalling Mechanisms Regulating Glucose Transport in Insulin-Sensitive Tissues. Mol. Membr. Biol. 2001, 18, 195–204. [Google Scholar]
- Bandet, C.L.; Tan-Chen, S.; Bourron, O.; Stunff, H.L.; Hajduch, E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int. J. Mol. Sci. 2019, 20, 479. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Talbot, C.L.; Chaurasia, B. Ceramides in Adipose Tissue. Front. Endocrinol. 2020, 11, 407. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, T.O.; Lass, A. DAG Tales: The Multiple Faces of Diacylglycerol--Stereochemistry, Metabolism, and Signaling. Cell. Mol. Life Sci. CMLS 2015, 72, 3931–3952. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci. 2017, 38, 649–665. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Brown, S.H.J.; Lim, X.Y.; Fiveash, C.E.; Osborne, B.; Bentley, N.L.; Braude, J.P.; Mitchell, T.W.; Coster, A.C.F.; Don, A.S.; et al. Regulation of Glucose Homeostasis and Insulin Action by Ceramide Acyl-Chain Length: A Beneficial Role for Very Long-Chain Sphingolipid Species. Biochim. Biophys. Acta 2016, 1861, 1828–1839. [Google Scholar] [CrossRef]
- Park, J.W.; Park, W.J.; Kuperman, Y.; Boura-Halfon, S.; Pewzner-Jung, Y.; Futerman, A.H. Ablation of Very Long Acyl Chain Sphingolipids Causes Hepatic Insulin Resistance in Mice Due to Altered Detergent-Resistant Membranes. Hepatology 2013, 57, 525–532. [Google Scholar] [CrossRef]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Bröneke, H.S.; et al. Obesity-Induced CerS6-Dependent C16:0 Ceramide Production Promotes Weight Gain and Glucose Intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef]
- Chaurasia, B.; Tippetts, T.S.; Mayoral Monibas, R.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a Ceramide Double Bond Improves Insulin Resistance and Hepatic Steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef]
- Zigdon, H.; Kogot-Levin, A.; Park, J.W.; Goldschmidt, R.; Kelly, S.; Merrill, A.H.; Scherz, A.; Pewzner-Jung, Y.; Saada, A.; Futerman, A.H. Ablation of Ceramide Synthase 2 Causes Chronic Oxidative Stress Due to Disruption of the Mitochondrial Respiratory Chain. J. Biol. Chem. 2013, 288, 4947–4956. [Google Scholar] [CrossRef]
- Pewzner-Jung, Y.; Brenner, O.; Braun, S.; Laviad, E.L.; Ben-Dor, S.; Feldmesser, E.; Horn-Saban, S.; Amann-Zalcenstein, D.; Raanan, C.; Berkutzki, T.; et al. A Critical Role for Ceramide Synthase 2 in Liver Homeostasis: II. Insights into Molecular Changes Leading to Hepatopathy. J. Biol. Chem. 2010, 285, 10911–10923. [Google Scholar] [CrossRef]
- Turban, S.; Hajduch, E. Protein Kinase C Isoforms: Mediators of Reactive Lipid Metabolites in the Development of Insulin Resistance. FEBS Lett. 2011, 585, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.M.; Prado, C.; Takane, K.K.; Lasnier, F.; Garcia-Ocana, A.; Ferre, P.; Dugail, I.; Hajduch, E. Plasma Membrane Subdomain Compartmentalization Contributes to Distinct Mechanisms of Ceramide Action on Insulin Signaling. Diabetes 2010, 59, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Hajduch, E.; Kular, G.; Hundal, H.S. Ceramide Disables 3-Phosphoinositide Binding to the Pleckstrin Homology Domain of Protein Kinase B (PKB)/Akt by a PKC Zeta-Dependent Mechanism. Mol. Cell. Biol. 2003, 23, 7794–7808. [Google Scholar] [CrossRef]
- Fox, T.E.; Houck, K.L.; O’Neill, S.M.; Nagarajan, M.; Stover, T.C.; Pomianowski, P.T.; Unal, O.; Yun, J.K.; Naides, S.J.; Kester, M. Ceramide Recruits and Activates Protein Kinase C Zeta (PKC Zeta) within Structured Membrane Microdomains. J. Biol. Chem. 2007, 282, 12450–12457. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Saddoughi, S.A.; Song, P.; Sultan, I.; Ponnusamy, S.; Senkal, C.E.; Snook, C.F.; Arnold, H.K.; Sears, R.C.; Hannun, Y.A.; et al. Direct Interaction between the Inhibitor 2 and Ceramide via Sphingolipid-Protein Binding Is Involved in the Regulation of Protein Phosphatase 2A Activity and Signaling. FASEB J. 2009, 23, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Cazzolli, R.; Carpenter, L.; Biden, T.J.; Schmitz-Peiffer, C. A Role for Protein Phosphatase 2A-like Activity, but Not Atypical Protein Kinase Czeta, in the Inhibition of Protein Kinase B/Akt and Glycogen Synthesis by Palmitate. Diabetes 2001, 50, 2210–2218. [Google Scholar] [CrossRef]
- Calvo, M.; Tebar, F.; Lopez-Iglesias, C.; Enrich, C. Morphologic and Functional Characterization of Caveolae in Rat Liver Hepatocytes. Hepatology 2001, 33, 1259–1269. [Google Scholar] [CrossRef]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-Stranded RNA-Dependent Protein Kinase Links Pathogen Sensing with Stress and Metabolic Homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef]
- Yang, X.; Nath, A.; Opperman, M.J.; Chan, C. The Double-Stranded RNA-Dependent Protein Kinase Differentially Regulates Insulin Receptor Substrates 1 and 2 in HepG2 Cells. Mol. Biol. Cell 2010, 21, 3449–3458. [Google Scholar] [CrossRef]
- Hage Hassan, R.; Pacheco de Sousa, A.C.; Mahfouz, R.; Hainault, I.; Blachnio-Zabielska, A.; Bourron, O.; Koskas, F.; Gorski, J.; Ferre, P.; Foufelle, F.; et al. Sustained Action of Ceramide on the Insulin Signaling Pathway in Muscle Cells: Implication of the double-stranded rna-activated protein kinase. J. Biol. Chem. 2016, 291, 3019–3029. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A Lipidomic Analysis of Nonalcoholic Fatty Liver Disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Holt, H.B.; Wild, S.H.; Wood, P.J.; Zhang, J.; Darekar, A.A.; Dewbury, K.; Poole, R.B.; Holt, R.I.G.; Phillips, D.I.; Byrne, C.D. Non-Esterified Fatty Acid Concentrations Are Independently Associated with Hepatic Steatosis in Obese Subjects. Diabetologia 2006, 49, 141–148. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, I.T.; Cortez-Pinto, H.; Fidalgo, G.; Rodrigues, D.; Camilo, M.E. Plasma Total and Free Fatty Acids Composition in Human Non-Alcoholic Steatohepatitis. Clin. Nutr. Edinb. Scotl. 2002, 21, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Diraison, F.; Moulin, P.; Beylot, M. Contribution of Hepatic de Novo Lipogenesis and Reesterification of Plasma Non Esterified Fatty Acids to Plasma Triglyceride Synthesis during Non-Alcoholic Fatty Liver Disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de Novo Lipogenesis Is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of Hepatic Triglyceride Accumulation in Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Adiels, M.; Taskinen, M.-R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Häkkinen, A.; Olofsson, S.-O.; Yki-Järvinen, H.; et al. Overproduction of Large VLDL Particles Is Driven by Increased Liver Fat Content in Man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef]
- Taskinen, M.R. Diabetic Dyslipidaemia: From Basic Research to Clinical Practice. Diabetologia 2003, 46, 733–749. [Google Scholar] [CrossRef]
- Poulsen, M.K.; Nellemann, B.; Stødkilde-Jørgensen, H.; Pedersen, S.B.; Grønbæk, H.; Nielsen, S. Impaired Insulin Suppression of VLDL-Triglyceride Kinetics in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2016, 101, 1637–1646. [Google Scholar] [CrossRef]
- Lytle, K.A.; Bush, N.C.; Triay, J.M.; Kellogg, T.A.; Kendrick, M.L.; Swain, J.M.; Gathaiya, N.W.; Hames, K.C.; Jensen, M.D. Hepatic Fatty Acid Balance and Hepatic Fat Content in Humans With Severe Obesity. J. Clin. Endocrinol. Metab. 2019, 104, 6171–6181. [Google Scholar] [CrossRef]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin Resistance Drives Hepatic de Novo Lipogenesis in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Ferre, P.; Foufelle, F. Hepatic Steatosis: A Role for de Novo Lipogenesis and the Transcription Factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 83–92. [Google Scholar] [CrossRef] [PubMed]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferre, P.; Foufelle, F. GRP78 Expression Inhibits Insulin and ER Stress-Induced SREBP-1c Activation and Reduces Hepatic Steatosis in Mice. J. Clin. Investig. 2009, 119, 1201–1215. [Google Scholar] [CrossRef]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and Increased SREBP-1c Lead to Mixed Insulin Resistance and Sensitivity in Livers of Lipodystrophic and Ob/Ob Mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef]
- Auguet, T.; Berlanga, A.; Guiu-Jurado, E.; Martinez, S.; Porras, J.A.; Aragonès, G.; Sabench, F.; Hernandez, M.; Aguilar, C.; Sirvent, J.J.; et al. Altered Fatty Acid Metabolism-Related Gene Expression in Liver from Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 22173–22187. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-X.; Shen, W.; Sun, H. Effects of Nuclear Receptor FXR on the Regulation of Liver Lipid Metabolism in Patients with Non-Alcoholic Fatty Liver Disease. Hepatol. Int. 2010, 4, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Pettinelli, P.; Videla, L.A. Up-Regulation of PPAR-Gamma MRNA Expression in the Liver of Obese Patients: An Additional Reinforcing Lipogenic Mechanism to SREBP-1c Induction. J. Clin. Endocrinol. Metab. 2011, 96, 1424–1430. [Google Scholar] [CrossRef]
- Worgall, T.S.; Juliano, R.A.; Seo, T.; Deckelbaum, R.J. Ceramide Synthesis Correlates with the Posttranscriptional Regulation of the Sterol-Regulatory Element-Binding Protein. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.-Z.; Takahashi, S.; et al. Intestinal Farnesoid X Receptor Signaling Promotes Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-R.; Lee, E.-J.; Shin, K.-O.; Kim, M.H.; Pewzner-Jung, Y.; Lee, Y.-M.; Park, J.-W.; Futerman, A.H.; Park, W.-J. Hepatic Triglyceride Accumulation via Endoplasmic Reticulum Stress-Induced SREBP-1 Activation Is Regulated by Ceramide Synthases. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Kondo, T.; Sajan, M.; Luo, J.; Bronson, R.; Asano, T.; Farese, R.; Cantley, L.C.; Kahn, C.R. Divergent Regulation of Hepatic Glucose and Lipid Metabolism by Phosphoinositide 3-Kinase via Akt and PKClambda/Zeta. Cell Metab. 2006, 3, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Kammoun, H.L.; Hainault, I.; Ferre, P.; Foufelle, F. Endoplasmic Reticulum Stress: A New Actor in the Development of Hepatic Steatosis. Curr. Opin. Lipidol. 2010, 21, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant Lipid Metabolism Disrupts Calcium Homeostasis Causing Liver Endoplasmic Reticulum Stress in Obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.-G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell 2019, 177, 1536–1552. [Google Scholar] [CrossRef]
- Smith, M.E.; Tippetts, T.S.; Brassfield, E.S.; Tucker, B.J.; Ockey, A.; Swensen, A.C.; Anthonymuthu, T.S.; Washburn, T.D.; Kane, D.A.; Prince, J.T.; et al. Mitochondrial Fission Mediates Ceramide-Induced Metabolic Disruption in Skeletal Muscle. Biochem. J. 2013, 456, 427–439. [Google Scholar] [CrossRef]
- Baiceanu, A.; Mesdom, P.; Lagouge, M.; Foufelle, F. Endoplasmic Reticulum Proteostasis in Hepatic Steatosis. Nat. Rev. Endocrinol. 2016, 12, 710–722. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated Fatty Acids Induce Endoplasmic Reticulum Stress and Apoptosis Independently of Ceramide in Liver Cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; Von Montfort, C.; Nuño, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase Is Required for Chronic Alcohol Induced Hepatic Endoplasmic Reticulum Stress and Mitochondrial Cholesterol Loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.D.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER Stress Induces NLRP3 Inflammasome Activation and Hepatocyte Death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- Dasgupta, D.; Nakao, Y.; Mauer, A.S.; Thompson, J.M.; Sehrawat, T.S.; Liao, C.-Y.; Krishnan, A.; Lucien, F.; Guo, Q.; Liu, M.; et al. IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice With Steatohepatitis. Gastroenterology 2020, 159, 1487–1503. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Luedde, T. Apoptosis and Necroptosis in the Liver: A Matter of Life and Death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef] [PubMed]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.-K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-Caspase Inhibitor VX-166 Reduces Fibrosis in an Animal Model of Nonalcoholic Steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Concas, D.; Kudo, H.; Levene, A.; Pollard, J.; Charlton, P.; Thomas, H.C.; Thursz, M.R.; Goldin, R.D. Impact of Pan-Caspase Inhibition in Animal Models of Established Steatosis and Non-Alcoholic Steatohepatitis. J. Hepatol. 2010, 53, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed Cell Death Induced by Ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide Channels Increase the Permeability of the Mitochondrial Outer Membrane to Small Proteins. J. Biol. Chem. 2002, 277, 26796–26803. [Google Scholar] [CrossRef] [PubMed]
- Kogot-Levin, A.; Saada, A. Ceramide and the Mitochondrial Respiratory Chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Müller, D.; Schneider, M.; et al. Ceramides Bind VDAC2 to Trigger Mitochondrial Apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Yin, X.-M. Dissection of the Multiple Mechanisms of TNF-Alpha-Induced Apoptosis in Liver Injury. J. Cell. Mol. Med. 2004, 8, 445–454. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Defective TNF-Alpha-Mediated Hepatocellular Apoptosis and Liver Damage in Acidic Sphingomyelinase Knockout Mice. J. Clin. Investig. 2003, 111, 197–208. [Google Scholar] [CrossRef]
- Lawler, J.F.; Yin, M.; Diehl, A.M.; Roberts, E.; Chatterjee, S. Tumor Necrosis Factor-Alpha Stimulates the Maturation of Sterol Regulatory Element Binding Protein-1 in Human Hepatocytes through the Action of Neutral Sphingomyelinase. J. Biol. Chem. 1998, 273, 5053–5059. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-Induced Insulin Resistance Mediated by the Proinflammatory Receptor TLR4 Requires Saturated Fatty Acid-Induced Ceramide Biosynthesis in Mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E. Adipose Tissue: From Lipid Storage Compartment to Endocrine Organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the Past Two Decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-Mediated Activation of Ceramidase Activity Initiates the Pleiotropic Actions of Adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef]
- Vasiliauskaité-Brooks, I.; Sounier, R.; Rochaix, P.; Bellot, G.; Fortier, M.; Hoh, F.; De Colibus, L.; Bechara, C.; Saied, E.M.; Arenz, C.; et al. Structural Insights into Adiponectin Receptors Suggest Ceramidase Activity. Nature 2017, 544, 120–123. [Google Scholar] [CrossRef]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2020, 33, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Shmarakov, I.O.; Jiang, H.; Liu, J.; Fernandez, E.J.; Blaner, W.S. Hepatic Stellate Cell Activation: A Source for Bioactive Lipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 629–642. [Google Scholar] [CrossRef]
- Moles, A.; Tarrats, N.; Morales, A.; Domínguez, M.; Bataller, R.; Caballería, J.; García-Ruiz, C.; Fernández-Checa, J.C.; Marí, M. Acidic Sphingomyelinase Controls Hepatic Stellate Cell Activation and in Vivo Liver Fibrogenesis. Am. J. Pathol. 2010, 177, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Muñoz, I.; de la Torre, P.; Sánchez-Alcázar, J.A.; García, I.; Santiago, E.; Muñoz-Yagüe, M.T.; Solís-Herruzo, J.A. Tumor Necrosis Factor Alpha Inhibits Collagen Alpha 1(I) Gene Expression in Rat Hepatic Stellate Cells through a G Protein. Gastroenterology 1997, 113, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Markiewicz, M.; Yamanaka, M.; Bielawska, A.; Mao, C.; Obeid, L.M.; Hannun, Y.A.; Trojanowska, M. Modulation of Transforming Growth Factor-Beta (TGF-Beta) Signaling by Endogenous Sphingolipid Mediators. J. Biol. Chem. 2003, 278, 9276–9282. [Google Scholar] [CrossRef]
- Chen, Q.; Lee, C.-E.; Denard, B.; Ye, J. Sustained Induction of Collagen Synthesis by TGF-β Requires Regulated Intramembrane Proteolysis of CREB3L1. PLoS ONE 2014, 9, e108528. [Google Scholar] [CrossRef]
- Yamamoto, A.; Morioki, H.; Nakae, T.; Miyake, Y.; Harada, T.; Noda, S.; Mitsuoka, S.; Matsumoto, K.; Tomimatsu, M.; Kanemoto, S.; et al. Transcription Factor Old Astrocyte Specifically Induced Substance Is a Novel Regulator of Kidney Fibrosis. FASEB J. 2021, 35, e21158. [Google Scholar] [CrossRef] [PubMed]
- Quillin, R.C.; Wilson, G.C.; Nojima, H.; Freeman, C.M.; Wang, J.; Schuster, R.M.; Blanchard, J.A.; Edwards, M.J.; Gandhi, C.R.; Gulbins, E.; et al. Inhibition of Acidic Sphingomyelinase Reduces Established Hepatic Fibrosis in Mice. Hepatol. Res. 2015, 45, 305–314. [Google Scholar] [CrossRef]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsins B and D Drive Hepatic Stellate Cell Proliferation and Promote Their Fibrogenic Potential. Hepatology 2009, 49, 1297–1307. [Google Scholar] [CrossRef]
- Chen, J.Y.; Newcomb, B.; Zhou, C.; Pondick, J.V.; Ghoshal, S.; York, S.R.; Motola, D.L.; Coant, N.; Yi, J.K.; Mao, C.; et al. Tricyclic Antidepressants Promote Ceramide Accumulation to Regulate Collagen Production in Human Hepatic Stellate Cells. Sci. Rep. 2017, 7, 44867. [Google Scholar] [CrossRef]
- Alsamman, S.; Christenson, S.A.; Yu, A.; Ayad, N.M.E.; Mooring, M.S.; Segal, J.M.; Hu, J.K.-H.; Schaub, J.R.; Ho, S.S.; Rao, V.; et al. Targeting Acid Ceramidase Inhibits YAP/TAZ Signaling to Reduce Fibrosis in Mice. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Norred, W.P.; Bacon, C.W.; Riley, R.T.; Merrill, A.H. Inhibition of Sphingolipid Biosynthesis by Fumonisins. Implications for Diseases Associated with Fusarium Moniliforme. J. Biol. Chem. 1991, 266, 14486–14490. [Google Scholar] [CrossRef]
- Gelderblom, W.C.; Abel, S.; Smuts, C.M.; Marnewick, J.; Marasas, W.F.; Lemmer, E.R.; Ramljak, D. Fumonisin-Induced Hepatocarcinogenesis: Mechanisms Related to Cancer Initiation and Promotion. Environ. Health Perspect. 2001, 109 (Suppl. 2), 291–300. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, D.; Hafler, D.A. Fingolimod for Multiple Sclerosis. N. Engl. J. Med. 2012, 366, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Blitzer, J.T.; Wang, L.; Summers, S.A. DES1: A Key Driver of Lipotoxicity in Metabolic Disease. DNA Cell Biol. 2020, 39, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Preitner, F.; Mody, N.; Graham, T.E.; Peroni, O.D.; Kahn, B.B. Long-Term Fenretinide Treatment Prevents High-Fat Diet-Induced Obesity, Insulin Resistance, and Hepatic Steatosis. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1420–E1429. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant Lipid Metabolism in Hepatocellular Carcinoma Revealed by Liver Lipidomics. Int. J. Mol. Sci. 2017, 18, 2550. [Google Scholar] [CrossRef] [PubMed]
- Cabot, M.C.; Han, T.Y.; Giuliano, A.E. The Multidrug Resistance Modulator SDZ PSC 833 Is a Potent Activator of Cellular Ceramide Formation. FEBS Lett. 1998, 431, 185–188. [Google Scholar] [CrossRef]
- Hajj, C.; Becker-Flegler, K.A.; Haimovitz-Friedman, A. Novel Mechanisms of Action of Classical Chemotherapeutic Agents on Sphingolipid Pathways. Biol. Chem. 2015, 396, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-Ceramide Increases the Anti-Tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 2018, 154, 1024–1036. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajduch, E.; Lachkar, F.; Ferré, P.; Foufelle, F. Roles of Ceramides in Non-Alcoholic Fatty Liver Disease. J. Clin. Med. 2021, 10, 792. https://doi.org/10.3390/jcm10040792
Hajduch E, Lachkar F, Ferré P, Foufelle F. Roles of Ceramides in Non-Alcoholic Fatty Liver Disease. Journal of Clinical Medicine. 2021; 10(4):792. https://doi.org/10.3390/jcm10040792
Chicago/Turabian StyleHajduch, Eric, Floriane Lachkar, Pascal Ferré, and Fabienne Foufelle. 2021. "Roles of Ceramides in Non-Alcoholic Fatty Liver Disease" Journal of Clinical Medicine 10, no. 4: 792. https://doi.org/10.3390/jcm10040792
APA StyleHajduch, E., Lachkar, F., Ferré, P., & Foufelle, F. (2021). Roles of Ceramides in Non-Alcoholic Fatty Liver Disease. Journal of Clinical Medicine, 10(4), 792. https://doi.org/10.3390/jcm10040792