
Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach


Abstract
1. Introduction
2. Autoinflammation and Autoimmunity
2.1. Pathogenesis Part I: Who Started the Fire
2.2. Pathogenesis Part II: What Keeps the Fire Burning
2.3. Pathogenesis Part III: Why Is Firefighting so Hard
3. Clinical Symptoms
4. Laboratory Findings and Biomarkers
5. Diagnostic Criteria
6. The Course of the Disease Splits in Two
7. Complications
7.1. Cytokine Storm
7.2. Parenchymal Lung Disease and PAH
7.3. Coagulation Disorders
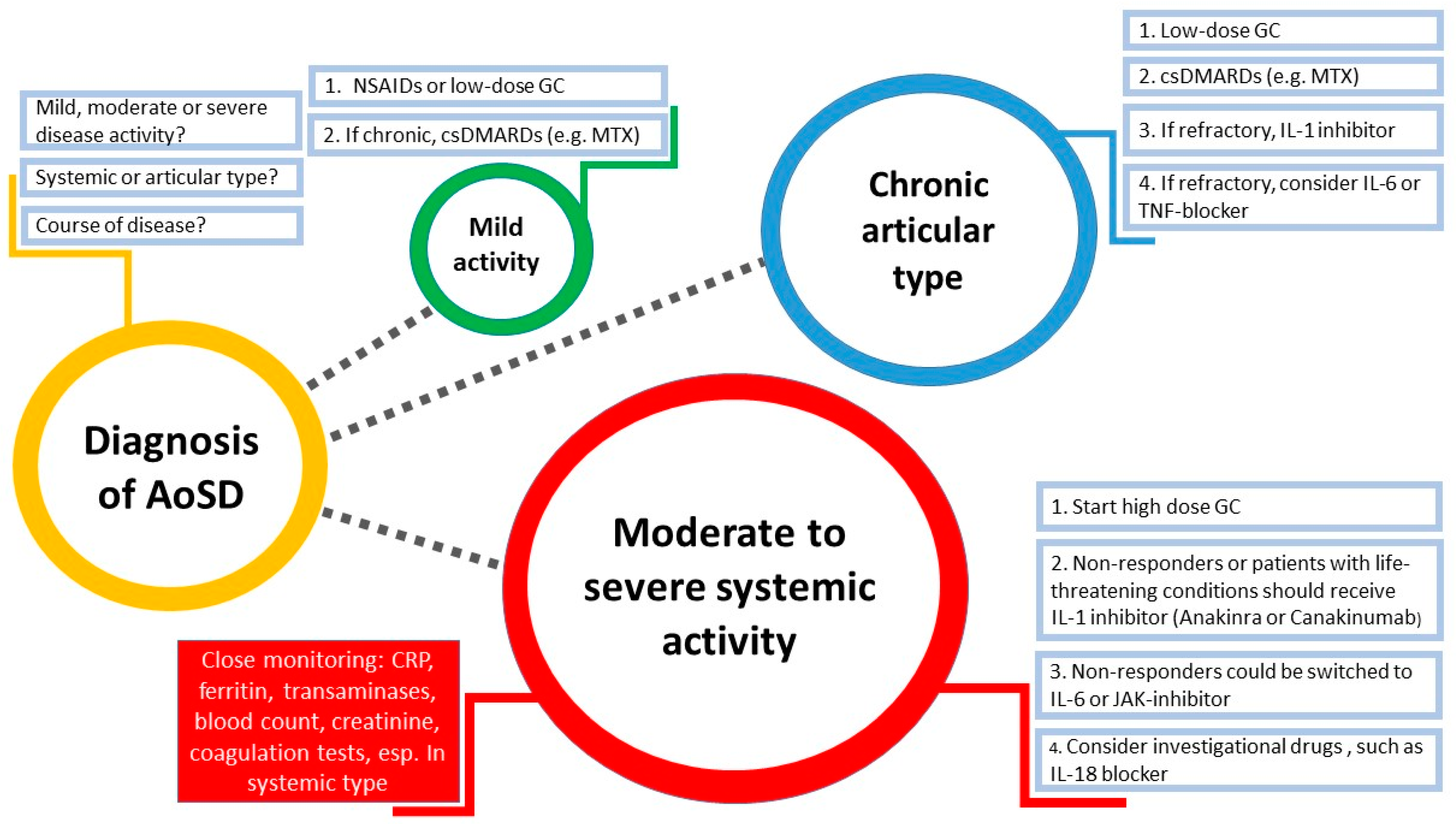
8. Treatment Management
8.1. Anti-TNF Therapy
8.2. Anti-IL-1 Therapy
8.3. Anti-IL-6 Therapy
8.4. JAK Inhibitors
8.5. Anti-IL-18 Therapy
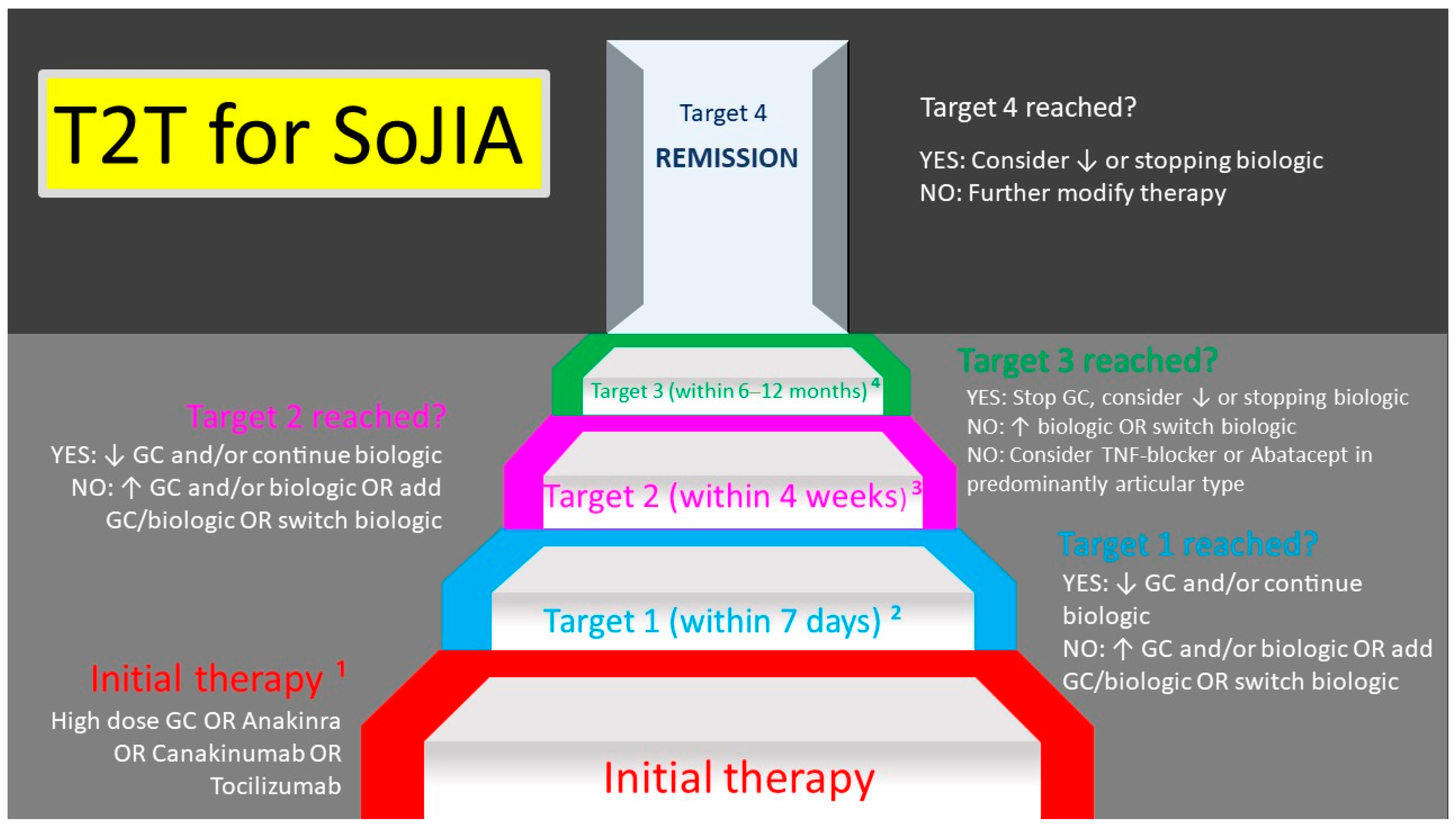
9. Still’s Disease in Children and Adults—Is There a Difference?
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AoSD | Adult-onset Still’s disease |
| SoJIA | Systemic-onset juvenile idiopathic arthritis |
| MAS | Macrophage activation syndrome |
| HLH | Haemophagocytic lymphohistiocytosis |
| PRRs | Pattern recognition receptors |
| NLR | Leucine-rich repeat containing family |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| GF | Glycosylated ferritin |
| DIC | Disseminated intravascular coagulation |
| TMA | Thrombotic microangiopathy |
| PET/CT | Positron Emission Tomography/Computer Tomography |
| ILAR | International League of Associations for Rheumatology |
References
- Fautrel, B. Adult-onset Still disease. Best Pract. Res. Clin. Rheumatol. 2008, 22, 773–792. [Google Scholar] [CrossRef]
- Bywaters, E.G. Still’s disease in the adult. Ann. Rheum. Dis. 1971, 30, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Nirmala, N.; Brachat, A.; Feist, E.; Blank, N.; Specker, C.; Witt, M.; Zernicke, J.; Martini, A.; Junge, G. Gene-expression analysis of adult-onset Still’s disease and systemic juvenile idiopathic arthritis is consistent with a continuum of a single disease entity. Pediatr Rheumatol. Online J. 2015, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Gerfaud-Valentin, M.; Jamilloux, Y.; Iwaz, J.; Sève, P. Adult-onset Still’s disease. Autoimmun. Rev. 2014, 13, 708–722. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297. [Google Scholar] [CrossRef]
- McGonagle, D.; Aziz, A.; Dickie, L.J.; McDermott, M.F. An integrated classification of pediatric inflammatory diseases, based on the concepts of autoinflammation and the immunological disease continuum. Pediatr Res. 2009, 65, 38r–45r. [Google Scholar] [CrossRef]
- Ruscitti, P.; Cipriani, P.; Masedu, F.; Iacono, D.; Ciccia, F.; Liakouli, V.; Guggino, G.; Carubbi, F.; Berardicurti, O.; Di Benedetto, P.; et al. Adult-onset Still’s disease: Evaluation of prognostic tools and validation of the systemic score by analysis of 100 cases from three centers. BMC Med. 2016, 14, 194. [Google Scholar] [CrossRef]
- Gordon, S. Pattern recognition receptors: Doubling up for the innate immune response. Cell 2002, 111, 927–930. [Google Scholar] [CrossRef]
- McGonagle, D.; Savic, S.; McDermott, M.F. The NLR network and the immunological disease continuum of adaptive and innate immune-mediated inflammation against self. Semin. Immunopathol. 2007, 29, 303–313. [Google Scholar] [CrossRef]
- Caso, F.; Costa, L.; Nucera, V.; Barilaro, G.; Masala, I.F.; Talotta, R.; Caso, P.; Scarpa, R.; Sarzi-Puttini, P.; Atzeni, F. From autoinflammation to autoimmunity: Old and recent findings. Clin. Rheumatol. 2018, 37, 2305–2321. [Google Scholar] [CrossRef]
- Savic, S.; Mistry, A.; Wilson, A.G.; Barcenas-Morales, G.; Doffinger, R.; Emery, P.; McGonagle, D. Autoimmune-autoinflammatory rheumatoid arthritis overlaps: A rare but potentially important subgroup of diseases. RMD Open 2017, 3, e000550. [Google Scholar] [CrossRef]
- Feist, E.; Mitrovic, S.; Fautrel, B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat. Rev. Rheumatol. 2018, 14, 603–618. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.L.; Chen, J.; Zeng, T.; He, L.; Li, M.; Luo, C.; Liu, S.; Ding, T.T.; Yimaiti, K.; et al. Both HLA class I and II regions identified as genome-wide significant susceptibility loci for adult-onset Still’s disease in Chinese individuals. Ann. Rheum. Dis. 2020, 79, 161–163. [Google Scholar] [CrossRef]
- Jia, J.; Shi, H.; Liu, M.; Liu, T.; Gu, J.; Wan, L.; Teng, J.; Liu, H.; Cheng, X.; Ye, J.; et al. Cytomegalovirus Infection May Trigger Adult-Onset Still’s Disease Onset or Relapses. Front. Immunol 2019, 10, 898. [Google Scholar] [CrossRef]
- Scirè, C.A.; Cavagna, L.; Perotti, C.; Bruschi, E.; Caporali, R.; Montecucco, C. Diagnostic value of procalcitonin measurement in febrile patients with systemic autoimmune diseases. Clin. Exp. Rheumatol. 2006, 24, 123–128. [Google Scholar]
- Fukuoka, K.; Miyamoto, A.; Ozawa, Y.; Ikegaya, N.; Maesono, T.; Komagata, Y.; Kaname, S.; Arimura, Y. Adult-onset Still’s disease-like manifestation accompanied by the cancer recurrence after long-term resting state. Mod. Rheumatol. 2019, 29, 704–707. [Google Scholar] [CrossRef]
- Neishi, J.; Tsukada, Y.; Maehara, T.; Ueki, K.; Maezawa, A.; Nojima, Y. Adult Still’s disease as a paraneoplastic manifestation of breast cancer. Scand. J. Rheumatol. 2000, 29, 328–330. [Google Scholar] [CrossRef]
- Ahn, J.K.; Oh, J.M.; Lee, J.; Kim, S.W.; Cha, H.S.; Koh, E.M. Adult onset Still’s disease diagnosed concomitantly with occult papillary thyroid cancer: Paraneoplastic manifestation or coincidence? Clin. Rheumatol. 2010, 29, 221–224. [Google Scholar] [CrossRef]
- Liozon, E.; Ly, K.H.; Vidal-Cathala, E.; Fauchais, A.L. Adult-onset Still’s disease as a manifestation of malignancy: Report of a patient with melanoma and literature review. La Rev. De Med. Interne 2014, 35, 60–64. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, S.I.; Hong, K.W.; Kang, M.W. Diagnostic value of 18F-FDG PET/CT in patients with fever of unknown origin. Intern. Med. J. 2012, 42, 834–837. [Google Scholar] [CrossRef]
- Church, L.D.; Cook, G.P.; McDermott, M.F. Primer: Inflammasomes and interleukin 1beta in inflammatory disorders. Nat. Clin. Pract. Rheumatol. 2008, 4, 34–42. [Google Scholar] [CrossRef]
- Wang, M.Y.; Jia, J.C.; Yang, C.D.; Hu, Q.Y. Pathogenesis, disease course, and prognosis of adult-onset Still’s disease: An update and review. Chin. Med. J. 2019, 132, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.W.; Chen, Y.M.; Lin, C.C.; Tang, K.T.; Chen, H.H.; Hung, W.T.; Lai, K.L.; Chen, D.Y. Elevated Expression of the NLRP3 Inflammasome and Its Correlation with Disease Activity in Adult-onset Still Disease. J. Rheumatol. 2017, 44, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Gerfaud-Valentin, M.; Martinon, F.; Belot, A.; Henry, T.; Sève, P. Pathogenesis of adult-onset Still’s disease: New insights from the juvenile counterpart. Immunol. Res. 2015, 61, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Ruddell, R.G.; Hoang-Le, D.; Barwood, J.M.; Rutherford, P.S.; Piva, T.J.; Watters, D.J.; Santambrogio, P.; Arosio, P.; Ramm, G.A. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology 2009, 49, 887–900. [Google Scholar] [CrossRef]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef]
- Foell, D.; Roth, J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheum. 2004, 50, 3762–3771. [Google Scholar] [CrossRef]
- Chen, D.Y.; Lan, J.L.; Lin, F.J.; Hsieh, T.Y.; Wen, M.C. Predominance of Th1 cytokine in peripheral blood and pathological tissues of patients with active untreated adult onset Still’s disease. Ann. Rheum. Dis. 2004, 63, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Chen, Y.M.; Lan, J.L.; Lin, C.C.; Chen, H.H.; Hsieh, C.W. Potential role of Th17 cells in the pathogenesis of adult-onset Still’s disease. Rheumatology 2010, 49, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.R.; Kubasch, A.S.; Ioannidis, C.; Rösen-Wolff, A.; Girschick, H.J.; Morbach, H.; Hedrich, C.M. Altered expression of IL-10 family cytokines in monocytes from CRMO patients result in enhanced IL-1β expression and release. Clin. Immunol. 2015, 161, 300–307. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chen, Y.M.; Lin, C.C.; Hsieh, C.W.; Wu, Y.C.; Hung, W.T.; Chen, H.H.; Lan, J.L. The potential role of advanced glycation end products (AGEs) and soluble receptors for AGEs (sRAGE) in the pathogenesis of adult-onset still’s disease. BMC Musculoskelet. Disord. 2015, 16, 111. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, Y.; Kishida, D.; Ueno, K.I.; Ushiyama, S.; Ichikawa, T.; Sekijima, Y. Characteristics of Circulating Natural Killer Cells and Their Interferon-γ Production in Active Adult-onset Still Disease. J. Rheumatol. 2019, 46, 1268–1276. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, H.S.; Lee, J.S.; Kim, J.J.; Jung, K.H.; Park, Y.W.; Yoo, D.H. Natural killer cell cytolytic function in Korean patients with adult-onset Still’s disease. J. Rheumatol. 2012, 39, 2000–2007. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Z.; Chi, H.; Hu, Q.; Ye, J.; Liu, H.; Cheng, X.; Shi, H.; Zhou, Z.; Teng, J.; et al. Elevated serum levels of interleukin-10 in adult-onset Still’s disease are associated with disease activity. Clin. Rheumatol. 2019, 38, 3205–3210. [Google Scholar] [CrossRef]
- Sfriso, P.; Priori, R.; Valesini, G.; Rossi, S.; Montecucco, C.M.; D’Ascanio, A.; Carli, L.; Bombardieri, S.; LaSelva, G.; Iannone, F.; et al. Adult-onset Still’s disease: An Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin. Rheumatol. 2016, 35, 1683–1689. [Google Scholar] [CrossRef]
- Zhou, G.; Zhou, Y.; Zhong, C.; Ye, H.; Liu, Z.; Liu, Y.; Tang, G.; Qu, J.; Lv, X. Retrospective analysis of 1,641 cases of classic fever of unknown origin. Ann. Transl Med. 2020, 8, 690. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.F.; Auwaerter, P.G. Fever and Fever of Unknown Origin: Review, Recent Advances, and Lingering Dogma. Open Forum Infect. Dis. 2020, 7, ofaa132. [Google Scholar] [CrossRef]
- Bagnari, V.; Colina, M.; Ciancio, G.; Govoni, M.; Trotta, F. Adult-onset Still’s disease. Rheumatol. Int. 2010, 30, 855–862. [Google Scholar] [CrossRef]
- Elkon, K.B.; Hughes, G.R.; Bywaters, E.G.; Ryan, P.F.; Inman, R.D.; Bowley, N.B.; James, M.P.; Eady, R.A. Adult-onset Still’s disease. Twenty-year followup and further studies of patients with active disease. Arthritis Rheum. 1982, 25, 647–654. [Google Scholar] [CrossRef]
- Di Benedetto, P.; Cipriani, P.; Iacono, D.; Pantano, I.; Caso, F.; Emmi, G.; Grembiale, R.D.; Cantatore, F.P.; Atzeni, F.; Perosa, F.; et al. Ferritin and C-reactive protein are predictive biomarkers of mortality and macrophage activation syndrome in adult onset Still’s disease. Analysis of the multicentre Gruppo Italiano di Ricerca in Reumatologia Clinica e Sperimentale (GIRRCS) cohort. PLoS ONE 2020, 15, e0235326. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.Y.; Zeng, T.; Sun, C.Y.; Luo, C.N.; Liu, S.; Ding, T.T.; Ji, Z.F.; Lu, A.; Yimaiti, K.; Teng, J.L.; et al. Clinical features and current treatments of adult-onset Still’s disease: A multicentre survey of 517 patients in China. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 121), 52–57. [Google Scholar]
- Gerfaud-Valentin, M.; Maucort-Boulch, D.; Hot, A.; Iwaz, J.; Ninet, J.; Durieu, I.; Broussolle, C.; Sève, P. Adult-onset still disease: Manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine 2014, 93, 91–99. [Google Scholar] [CrossRef]
- Fautrel, B.; Zing, E.; Golmard, J.L.; Le Moel, G.; Bissery, A.; Rioux, C.; Rozenberg, S.; Piette, J.C.; Bourgeois, P. Proposal for a new set of classification criteria for adult-onset still disease. Medicine 2002, 81, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.Y.; Lee, J.H.; Yu, H.H.; Wang, L.C.; Yang, Y.H.; Chiang, B.L. Initial manifestations and clinical course of systemic onset juvenile idiopathic arthritis: A ten-year retrospective study. J. Formos. Med. Assoc. 2012, 111, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Beukelman, T.; Gallo, L.; Spangler, J.; Rosenkranz, M.; Arkachaisri, T.; Ayala, R.; Groh, B.; Finkel, T.H.; Cron, R.Q. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: Data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry (PASOJAR). J. Rheumatol. 2008, 35, 343–348. [Google Scholar]
- Giacomelli, R.; Ruscitti, P.; Shoenfeld, Y. A comprehensive review on adult onset Still’s disease. J. Autoimmun 2018, 93, 24–36. [Google Scholar] [CrossRef]
- Fautrel, B.; Le Moël, G.; Saint-Marcoux, B.; Taupin, P.; Vignes, S.; Rozenberg, S.; Koeger, A.C.; Meyer, O.; Guillevin, L.; Piette, J.C.; et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J. Rheumatol. 2001, 28, 322–329. [Google Scholar]
- Schiller, D.; Mittermayer, H. Hyperferritinemia as a marker of Still’s disease. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1998, 26, 534–535. [Google Scholar] [CrossRef] [PubMed]
- Rosário, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.P.; Shoenfeld, Y. The hyperferritinemic syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Fardet, L.; Coppo, P.; Kettaneh, A.; Dehoux, M.; Cabane, J.; Lambotte, O. Low glycosylated ferritin, a good marker for the diagnosis of hemophagocytic syndrome. Arthritis Rheum. 2008, 58, 1521–1527. [Google Scholar] [CrossRef]
- Vignes, S.; Le Moël, G.; Fautrel, B.; Wechsler, B.; Godeau, P.; Piette, J.C. Percentage of glycosylated serum ferritin remains low throughout the course of adult onset Still’s disease. Ann. Rheum. Dis. 2000, 59, 347–350. [Google Scholar] [CrossRef]
- Kudela, H.; Drynda, S.; Lux, A.; Horneff, G.; Kekow, J. Comparative study of Interleukin-18 (IL-18) serum levels in adult onset Still’s disease (AOSD) and systemic onset juvenile idiopathic arthritis (sJIA) and its use as a biomarker for diagnosis and evaluation of disease activity. BMC Rheumatol. 2019, 3, 4. [Google Scholar] [CrossRef]
- Mitrovic, S.; Fautrel, B. New Markers for Adult-Onset Still’s Disease. Jt. Bone Spine 2018, 85, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Rau, M.; Schiller, M.; Krienke, S.; Heyder, P.; Lorenz, H.; Blank, N. Clinical manifestations but not cytokine profiles differentiate adult-onset Still’s disease and sepsis. J. Rheumatol. 2010, 37, 2369–2376. [Google Scholar] [CrossRef]
- Kim, H.A.; An, J.M.; Nam, J.Y.; Jeon, J.Y.; Suh, C.H. Serum S100A8/A9, but not follistatin-like protein 1 and interleukin 18, may be a useful biomarker of disease activity in adult-onset Still’s disease. J. Rheumatol. 2012, 39, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.B.; Suh, C.H.; An, J.M.; Jung, J.Y.; Jeon, J.Y.; Nam, J.Y.; Kim, H.A. Serum S100A12 may be a useful biomarker of disease activity in adult-onset Still’s disease. J. Rheumatol. 2014, 41, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T.; et al. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 19, 424–430. [Google Scholar]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar]
- Lebrun, D.; Mestrallet, S.; Dehoux, M.; Golmard, J.L.; Granger, B.; Georgin-Lavialle, S.; Arnaud, L.; Grateau, G.; Pouchot, J.; Fautrel, B. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin. Arthritis Rheum. 2018, 47, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Pouchot, J.; Sampalis, J.S.; Beaudet, F.; Carette, S.; Décary, F.; Salusinsky-Sternbach, M.; Hill, R.O.; Gutkowski, A.; Harth, M.; Myhal, D.; et al. Adult Still’s disease: Manifestations, disease course, and outcome in 62 patients. Medicine 1991, 70, 118–136. [Google Scholar] [CrossRef]
- Maria, A.T.; Le Quellec, A.; Jorgensen, C.; Touitou, I.; Rivière, S.; Guilpain, P. Adult onset Still’s disease (AOSD) in the era of biologic therapies: Dichotomous view for cytokine and clinical expressions. Autoimmun. Rev. 2014, 13, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Franchini, S.; Dagna, L.; Salvo, F.; Aiello, P.; Baldissera, E.; Sabbadini, M.G. Adult onset Still’s disease: Clinical presentation in a large cohort of Italian patients. Clin. Exp. Rheumatol. 2010, 28, 41–48. [Google Scholar]
- Ichida, H.; Kawaguchi, Y.; Sugiura, T.; Takagi, K.; Katsumata, Y.; Gono, T.; Ota, Y.; Kataoka, S.; Kawasumi, H.; Yamanaka, H. Clinical manifestations of Adult-onset Still’s disease presenting with erosive arthritis: Association with low levels of ferritin and Interleukin-18. Arthritis Care Res. 2014, 66, 642–646. [Google Scholar] [CrossRef]
- Hot, A.; Toh, M.L.; Coppéré, B.; Perard, L.; Madoux, M.H.; Mausservey, C.; Desmurs-Clavel, H.; Ffrench, M.; Ninet, J. Reactive hemophagocytic syndrome in adult-onset Still disease: Clinical features and long-term outcome: A case-control study of 8 patients. Medicine 2010, 89, 37–46. [Google Scholar] [CrossRef]
- Chen, D.Y.; Lan, J.L.; Lin, F.J.; Hsieh, T.Y. Proinflammatory cytokine profiles in sera and pathological tissues of patients with active untreated adult onset Still’s disease. J. Rheumatol. 2004, 31, 2189–2198. [Google Scholar] [PubMed]
- Fujii, T.; Nojima, T.; Yasuoka, H.; Satoh, S.; Nakamura, K.; Kuwana, M.; Suwa, A.; Hirakata, M.; Mimori, T. Cytokine and immunogenetic profiles in Japanese patients with adult Still’s disease. Association with chronic articular disease. Rheumatology 2001, 40, 1398–1404. [Google Scholar] [CrossRef]
- Franchini, S.; Dagna, L.; Salvo, F.; Aiello, P.; Baldissera, E.; Sabbadini, M.G. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010, 62, 2530–2535. [Google Scholar] [CrossRef]
- Pouchot, J.; Arlet, J.B. Biological treatment in adult-onset Still’s disease. Best Pract. Res. Clin. Rheumatol. 2012, 26, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Cron, R.Q.; Hartwell, J.; Manson, J.J.; Tattersall, R.S. Silencing the cytokine storm: The use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020, 2, e358–e367. [Google Scholar] [CrossRef]
- Carter, S.J.; Tattersall, R.S.; Ramanan, A.V. Macrophage activation syndrome in adults: Recent advances in pathophysiology, diagnosis and treatment. Rheumatology 2019, 58, 5–17. [Google Scholar] [CrossRef]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.F.; Ferlic-Stark, L.L.; Allen, C.E.; Kozinetz, C.A.; McClain, K.L. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatric Blood Cancer 2011, 56, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Saper, V.E.; Chen, G.; Deutsch, G.H.; Guillerman, R.P.; Birgmeier, J.; Jagadeesh, K.; Canna, S.; Schulert, G.; Deterding, R.; Xu, J.; et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann. Rheum. Dis. 2019, 78, 1722–1731. [Google Scholar] [CrossRef]
- Schulert, G.S.; Yasin, S.; Carey, B.; Chalk, C.; Do, T.; Schapiro, A.H.; Husami, A.; Watts, A.; Brunner, H.I.; Huggins, J.; et al. Systemic Juvenile Idiopathic Arthritis-Associated Lung Disease: Characterization and Risk Factors. Arthritis Rheumatol. 2019, 71, 1943–1954. [Google Scholar] [CrossRef]
- Ruscitti, P.; Berardicurti, O.; Iacono, D.; Pantano, I.; Liakouli, V.; Caso, F.; Emmi, G.; Grembiale, R.D.; Cantatore, F.P.; Atzeni, F.; et al. Parenchymal lung disease in adult onset Still’s disease: An emergent marker of disease severity-characterisation and predictive factors from Gruppo Italiano di Ricerca in Reumatologia Clinica e Sperimentale (GIRRCS) cohort of patients. Arthritis Res. Ther. 2020, 22, 151. [Google Scholar] [CrossRef]
- Ruscitti, P.; Cipriani, P.; Di Benedetto, P.; Liakouli, V.; Carubbi, F.; Berardicurti, O.; Ciccia, F.; Guggino, G.; Triolo, G.; Giacomelli, R. Advances in immunopathogenesis of macrophage activation syndrome during rheumatic inflammatory diseases: Toward new therapeutic targets? Expert Rev. Clin. Immunol. 2017, 13, 1041–1047. [Google Scholar] [CrossRef]
- Grom, A.A.; Horne, A.; De Benedetti, F. Macrophage activation syndrome in the era of biologic therapy. Nat. Rev. Rheumatol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Gerfaud-Valentin, M.; Cottin, V.; Jamilloux, Y.; Hot, A.; Gaillard-Coadon, A.; Durieu, I.; Broussolle, C.; Iwaz, J.; Sève, P. Parenchymal lung involvement in adult-onset Still disease: A STROBE-compliant case series and literature review. Medicine 2016, 95, e4258. [Google Scholar] [CrossRef]
- Mehta, M.V.; Manson, D.K.; Horn, E.M.; Haythe, J. An atypical presentation of adult-onset Still’s disease complicated by pulmonary hypertension and macrophage activation syndrome treated with immunosuppression: A case-based review of the literature. Pulm. Circ. 2016, 6, 136–142. [Google Scholar] [CrossRef]
- Efthimiou, P.; Kadavath, S.; Mehta, B. Life-threatening complications of adult-onset Still’s disease. Clin. Rheumatol. 2014, 33, 305–314. [Google Scholar] [CrossRef]
- Mitrovic, S.; Fautrel, B. Complications of adult-onset Still’s disease and their management. Expert Rev. Clin. Immunol. 2018, 14, 351–365. [Google Scholar] [CrossRef]
- Gando, S.; Levi, M.; Toh, C.H. Disseminated intravascular coagulation. Nat. Rev. Dis Primers 2016, 2, 16037. [Google Scholar] [CrossRef]
- El Karoui, K.; Karras, A.; Lebrun, G.; Charles, P.; Arlet, J.B.; Jacquot, C.; Orssaud, C.; Nochy, D.; Pouchot, J. Thrombotic microangiopathy and purtscher-like retinopathy associated with adult-onset Still’s disease: A role for glomerular vascular endothelial growth factor? Arthritis Rheum. 2009, 61, 1609–1613. [Google Scholar] [CrossRef] [PubMed]
- Kontzias, A.; Efthimiou, P. Adult-onset Still’s disease: Pathogenesis, clinical manifestations and therapeutic advances. Drugs 2008, 68, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Fautrel, B.; Sibilia, J.; Mariette, X.; Combe, B. Tumour necrosis factor alpha blocking agents in refractory adult Still’s disease: An observational study of 20 cases. Ann. Rheum. Dis. 2005, 64, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Kraetsch, H.G.; Antoni, C.; Kalden, J.R.; Manger, B. Successful treatment of a small cohort of patients with adult onset of Still’s disease with infliximab: First experiences. Ann. Rheum. Dis. 2001, 60 (Suppl. 3), iii55–iii57. [Google Scholar] [CrossRef]
- Cavagna, L.; Caporali, R.; Epis, O.; Bobbio-Pallavicini, F.; Montecucco, C. Infliximab in the treatment of adult Still’s disease refractory to conventional therapy. Clin. Exp. Rheumatol. 2001, 19, 329–332. [Google Scholar]
- Kötter, I.; Wacker, A.; Koch, S.; Henes, J.; Richter, C.; Engel, A.; Günaydin, I.; Kanz, L. Anakinra in patients with treatment-resistant adult-onset Still’s disease: Four case reports with serial cytokine measurements and a review of the literature. Semin. Arthritis Rheum. 2007, 37, 189–197. [Google Scholar] [CrossRef]
- Giampietro, C.; Ridene, M.; Lequerre, T.; Costedoat Chalumeau, N.; Amoura, Z.; Sellam, J.; Sibilia, J.; Bourgeois, P.; Fautrel, B. Anakinra in adult-onset Still’s disease: Long-term treatment in patients resistant to conventional therapy. Arthritis Care Res. 2013, 65, 822–826. [Google Scholar] [CrossRef]
- Ortiz-Sanjuán, F.; Blanco, R.; Riancho-Zarrabeitia, L.; Castañeda, S.; Olivé, A.; Riveros, A.; Velloso-Feijoo, M.L.; Narváez, J.; Jiménez-Moleón, I.; Maiz-Alonso, O.; et al. Efficacy of Anakinra in Refractory Adult-Onset Still’s Disease: Multicenter Study of 41 Patients and Literature Review. Medicine 2015, 94, e1554. [Google Scholar] [CrossRef] [PubMed]
- Colafrancesco, S.; Priori, R.; Valesini, G.; Argolini, L.; Baldissera, E.; Bartoloni, E.; Cammelli, D.; Canestrari, G.; Cantarini, L.; Cavallaro, E.; et al. Response to Interleukin-1 Inhibitors in 140 Italian Patients with Adult-Onset Still’s Disease: A Multicentre Retrospective Observational Study. Front. Pharm. 2017, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- Feist, E.; Burmester, G.R. Canakinumab for treatment of cryopyrin-associated periodic syndrome. Expert Opin. Biol. Ther. 2010, 10, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Sfriso, P.; Bindoli, S.; Doria, A.; Feist, E.; Galozzi, P. Canakinumab for the treatment of adult-onset Still’s disease. Expert Rev. Clin. Immunol. 2020, 16, 129–138. [Google Scholar] [CrossRef]
- Feist, E.; Quartier, P.; Fautrel, B.; Schneider, R.; Sfriso, P.; Efthimiou, P.; Cantarini, L.; Lheritier, K.; Leon, K.; Karyekar, C.S.; et al. Efficacy and safety of canakinumab in patients with Still’s disease: Exposure-response analysis of pooled systemic juvenile idiopathic arthritis data by age groups. Clin. Exp. Rheumatol. 2018, 36, 668–675. [Google Scholar] [PubMed]
- Kontzias, A.; Efthimiou, P. The use of Canakinumab, a novel IL-1β long-acting inhibitor, in refractory adult-onset Still’s disease. Semin. Arthritis Rheum. 2012, 42, 201–205. [Google Scholar] [CrossRef]
- Kedor, C.; Listing, J.; Zernicke, J.; Weiß, A.; Behrens, F.; Blank, N.; Henes, J.C.; Kekow, J.; Rubbert-Roth, A.; Schulze-Koops, H.; et al. Canakinumab for Treatment of Adult-Onset Still’s Disease to Achieve Reduction of Arthritic Manifestation (CONSIDER): Phase II, randomised, double-blind, placebo-controlled, multicentre, investigator-initiated trial. Ann. Rheum. Dis. 2020, 79, 1090–1097. [Google Scholar] [CrossRef]
- Kedor, C.; Listing, J.; Zernicke, J.; Feist, E. Response to: ‘Changing the outcome measures, changing the results? The urgent need of a specific Disease Activity Score to adult-onset Still’s disease’ by Ruscitti et al. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Kedor, C.; Zernicke, J.; Feist, E. Response to: ‘Correspondence on ‘Changing the outcome measures, changing the results? The urgent need of a specific disease activity score to adult-onset Still’s disease’’ by Muraviov and Muraviova. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ilowite, N.T.; Prather, K.; Lokhnygina, Y.; Schanberg, L.E.; Elder, M.; Milojevic, D.; Verbsky, J.W.; Spalding, S.J.; Kimura, Y.; Imundo, L.F.; et al. Randomized, double-blind, placebo-controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014, 66, 2570–2579. [Google Scholar] [CrossRef]
- Song, S.T.; Kim, J.J.; Lee, S.; Kim, H.A.; Lee, E.Y.; Shin, K.C.; Lee, J.H.; Lee, K.H.; Choi, S.T.; Cha, H.S.; et al. Efficacy of tocilizumab therapy in Korean patients with adult-onset Still’s disease: A multicentre retrospective study of 22 cases. Clin. Exp. Rheumatol. 2016, 34, S64–S71. [Google Scholar]
- Li, T.; Gu, L.; Wang, X.; Guo, L.; Shi, H.; Yang, C.; Chen, S. A Pilot Study on Tocilizumab for Treating Refractory Adult-Onset Still’s Disease. Sci Rep. 2017, 7, 13477. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kameda, H.; Ikeda, K.; Ishii, T.; Murakami, K.; Takamatsu, H.; Tanaka, Y.; Abe, T.; Takeuchi, T. Tocilizumab in patients with adult-onset still’s disease refractory to glucocorticoid treatment: A randomised, double-blind, placebo-controlled phase III trial. Ann. Rheum. Dis. 2018, 77, 1720–1729. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, M.; Zhang, X.; Xia, Q.; Yang, J.; Xu, S.; Pan, F. Efficacy and safety of tocilizumab with inhibition of interleukin-6 in adult-onset Still’s disease: A meta-analysis. Mod. Rheumatol. 2018, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Simeni Njonnou, S.R.; Soyfoo, M.S.; Vandergheynst, F.A. Efficacy of sarilumab in adult-onset Still’s disease as a corticosteroid-sparing agent. Rheumatology 2019, 58, 1878–1879. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, M.; Jia, J.; Teng, J.; Chi, H.; Liu, T.; Liu, H.L.; Cheng, X.; Ye, J.; Su, Y.; et al. Tofacitinib in refractory adult-onset Still’s disease: 14 cases from a single centre in China. Ann. Rheum. Dis. 2020, 79, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Moriyama, M.; Kondo, M.; Kumakura, S.; Murakawa, Y. Tofacitinib-induced remission in refractory adult-onset Still’s disease complicated by macrophage activation syndrome. Scand. J. Rheumatol. 2020, 49, 336–338. [Google Scholar] [CrossRef]
- Hoff, P.; Walther, M.; Wesselmann, H.; Weinerth, J.; Feist, E.; Ohrndorf, S. Successful treatment of adult Still’s disease with tofacitinib in a HIV-2 positive female patient. Z. Fur Rheumatol. 2020. [Google Scholar] [CrossRef]
- Kacar, M.; Fitton, J.; Gough, A.K.; Buch, M.H.; McGonagle, D.G.; Savic, S. Mixed results with baricitinib in biological-resistant adult-onset Still’s disease and undifferentiated systemic autoinflammatory disease. RMD Open 2020, 6. [Google Scholar] [CrossRef]
- Gabay, C.; Fautrel, B.; Rech, J.; Spertini, F.; Feist, E.; Kötter, I.; Hachulla, E.; Morel, J.; Schaeverbeke, T.; Hamidou, M.A.; et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann. Rheum. Dis. 2018, 77, 840–847. [Google Scholar] [CrossRef]
- Still, G.F. On a Form of Chronic Joint Disease in Children. Med. Chir. Trans. 1897, 80, 47-60.9. [Google Scholar] [CrossRef]
- Schaller, J.G.; Ochs, H.D.; Thomas, E.D.; Nisperos, B.; Feigl, P.; Wedgwood, R.J. Histocompatibility antigens in childhood-onset arthritis. J. Pediatr. 1976, 88, 926–930. [Google Scholar] [CrossRef]
- Ombrello, M.J.; Arthur, V.L.; Remmers, E.F.; Hinks, A.; Tachmazidou, I.; Grom, A.A.; Foell, D.; Martini, A.; Gattorno, M.; Özen, S.; et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: Clinical and therapeutic implications. Ann. Rheum. Dis. 2017, 76, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, M.J.; Remmers, E.F.; Tachmazidou, I.; Grom, A.; Foell, D.; Haas, J.P.; Martini, A.; Gattorno, M.; Özen, S.; Prahalad, S.; et al. HLA-DRB1*11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 15970–15975. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.M.; Monies, D.M.; Abouelhoda, M.; Al-Tassan, N.; Al-Dusery, H.; Naim, E.A.; Al-Younes, B.; Shinwari, J.; Al-Mohanna, F.A.; Meyer, B.F.; et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2015, 67, 288–295. [Google Scholar] [CrossRef]
- Singh, A.; Suri, D.; Vignesh, P.; Anjani, G.; Jacob, P.; Girisha, K.M. LACC1 gene mutation in three sisters with polyarthritis without systemic features. Ann. Rheum. Dis. 2020, 79, 425–426. [Google Scholar] [CrossRef]
- Sulliman Ommar Omarjee, A.-L.M.; Quiniou, G.; Moreews, M.; Ainouze, M.; Frachette, C.; Melki, I.; Dumaine, C.; Gerfaud-Valentin, M.; Duquesne, A.; Tilmann, K.; et al. LACC1 deficiency defines a novel form of juvenile arthritis associated with reduced autophagy and metabolism in macrophages. J. Exp. Med. 2020, accepted. [Google Scholar]
- Reiff, A. Treatment of Systemic Juvenile Idiopathic Arthritis with Tocilizumab—the Role of Anti-Interleukin-6 Therapy After a Decade of Treatment. Biol. Ther. 2012, 2, 1. [Google Scholar] [CrossRef]
- Holzinger, D.; Foell, D.; Kessel, C. The role of S100 proteins in the pathogenesis and monitoring of autoinflammatory diseases. Mol. Cell Pediatr. 2018, 5, 7. [Google Scholar] [CrossRef]
- Kessel, C.; Holzinger, D.; Foell, D. Phagocyte-derived S100 proteins in autoinflammation: Putative role in pathogenesis and usefulness as biomarkers. Clin. Immunol. 2013, 147, 229–241. [Google Scholar] [CrossRef]
- Cepika, A.M.; Banchereau, R.; Segura, E.; Ohouo, M.; Cantarel, B.; Goller, K.; Cantrell, V.; Ruchaud, E.; Gatewood, E.; Nguyen, P.; et al. A multidimensional blood stimulation assay reveals immune alterations underlying systemic juvenile idiopathic arthritis. J. Exp. Med. 2017, 214, 3449–3466. [Google Scholar] [CrossRef]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Hügle, B.; Schippers, A.; Fischer, N.; Ohl, K.; Denecke, B.; Ticconi, F.; Vastert, B.; Costa, I.G.; Haas, J.P.; Tenbrock, K. Transcription factor motif enrichment in whole transcriptome analysis identifies STAT4 and BCL6 as the most prominent binding motif in systemic juvenile idiopathic arthritis. Arthritis Res. Ther. 2018, 20, 98. [Google Scholar] [CrossRef]
- Gohar, F.; McArdle, A.; Jones, M.; Callan, N.; Hernandez, B.; Kessel, C.; Miranda-Garcia, M.; Lavric, M.; Holzinger, D.; Pretzer, C.; et al. Molecular signature characterisation of different inflammatory phenotypes of systemic juvenile idiopathic arthritis. Ann. Rheum. Dis. 2019, 78, 1107–1113. [Google Scholar] [CrossRef]
- Weiss, E.S.; Girard-Guyonvarc’h, C.; Holzinger, D.; de Jesus, A.A.; Tariq, Z.; Picarsic, J.; Schiffrin, E.J.; Foell, D.; Grom, A.A.; Ammann, S.; et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018, 131, 1442–1455. [Google Scholar] [CrossRef]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; I Ivanov, I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Henderson, L.A.; Hoyt, K.J.; Lee, P.Y.; Rao, D.A.; Jonsson, A.H.; Nguyen, J.P.; Rutherford, K.; Julé, A.M.; Charbonnier, L.M.; Case, S.; et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Kessel, C.; Lippitz, K.; Weinhage, T.; Hinze, C.; Wittkowski, H.; Holzinger, D.; Fall, N.; Grom, A.A.; Gruen, N.; Foell, D. Proinflammatory Cytokine Environments Can Drive Interleukin-17 Overexpression by γ/δ T Cells in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2017, 69, 1480–1494. [Google Scholar] [CrossRef]
- Vandenhaute, J.; Avau, A.; Filtjens, J.; Malengier-Devlies, B.; Imbrechts, M.; Van den Berghe, N.; Ahmadzadeh, K.; Mitera, T.; Boon, L.; Leclercq, G.; et al. Regulatory Role for NK Cells in a Mouse Model of Systemic Juvenile Idiopathic Arthritis. J. Immunol. 2019, 203, 3339–3348. [Google Scholar] [CrossRef]
- Wulffraat, N.M.; Rijkers, G.T.; Elst, E.; Brooimans, R.; Kuis, W. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatology 2003, 42, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Put, K.; Vandenhaute, J.; Avau, A.; van Nieuwenhuijze, A.; Brisse, E.; Dierckx, T.; Rutgeerts, O.; Garcia-Perez, J.E.; Toelen, J.; Waer, M.; et al. Inflammatory Gene Expression Profile and Defective Interferon-γ and Granzyme K in Natural Killer Cells From Systemic Juvenile Idiopathic Arthritis Patients. Arthritis Rheumatol. 2017, 69, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, K.M.; Linghu, B.; Szustakowski, J.D.; Husami, A.; Yang, F.; Zhang, K.; Filipovich, A.H.; Fall, N.; Harley, J.B.; Nirmala, N.R.; et al. Whole-Exome Sequencing Reveals Overlap Between Macrophage Activation Syndrome in Systemic Juvenile Idiopathic Arthritis and Familial Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. 2014, 66, 3486–3495. [Google Scholar] [CrossRef]
- Ravelli, A.; Minoia, F.; Davì, S.; Horne, A.; Bovis, F.; Pistorio, A.; Aricò, M.; Avcin, T.; Behrens, E.M.; De Benedetti, F.; et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016, 68, 566–576. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Schneider, R. Chapter 16—Systemic Juvenile Idiopathic Arthritis. In Textbook of Pediatric Rheumatology, 7th ed.; Petty, R.E., Laxer, R.M., Lindsley, C.B., Wedderburn, L.R., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2016; pp. 205–216.e206. [Google Scholar]
- Kumar, S.; Kunhiraman, D.S.; Rajam, L. Application of the Yamaguchi criteria for classification of "suspected" systemic juvenile idiopathic arthritis (sJIA). Pediatr. Rheumatol. Online J. 2012, 10, 40. [Google Scholar] [CrossRef]
- Silva, J.R.; Brito, I. Systemic juvenile idiopathic arthritis versus adult-onset Still´s disease: The pertinence of changing the current classification criteria. Acta Reum. Port. 2020, 45, 150–151. [Google Scholar]
- Föll, D.; Wittkowski, H.; Hinze, C. Still’s disease as biphasic disorder: Current knowledge on pathogenesis and novel treatment approaches. Z. Rheumatol. 2020, 79, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Hinze, C.H.; Holzinger, D.; Lainka, E.; Haas, J.P.; Speth, F.; Kallinich, T.; Rieber, N.; Hufnagel, M.; Jansson, A.F.; Hedrich, C.; et al. Practice and consensus-based strategies in diagnosing and managing systemic juvenile idiopathic arthritis in Germany. Pediatr. Rheumatol. Online J. 2018, 16, 7. [Google Scholar] [CrossRef]
- DeWitt, E.M.; Kimura, Y.; Beukelman, T.; Nigrovic, P.A.; Onel, K.; Prahalad, S.; Schneider, R.; Stoll, M.L.; Angeles-Han, S.; Milojevic, D.; et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res. 2012, 64, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Ruperto, N.; Brunner, H.I.; Quartier, P.; Constantin, T.; Wulffraat, N.; Horneff, G.; Brik, R.; McCann, L.; Kasapcopur, O.; Rutkowska-Sak, L.; et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2396–2406. [Google Scholar] [CrossRef]
- De Benedetti, F.; Brunner, H.I.; Ruperto, N.; Kenwright, A.; Wright, S.; Calvo, I.; Cuttica, R.; Ravelli, A.; Schneider, R.; Woo, P.; et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2385–2395. [Google Scholar] [CrossRef] [PubMed]
- Quartier, P.; Allantaz, F.; Cimaz, R.; Pillet, P.; Messiaen, C.; Bardin, C.; Bossuyt, X.; Boutten, A.; Bienvenu, J.; Duquesne, A.; et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann. Rheum. Dis. 2011, 70, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Ter Haar, N.M.; van Dijkhuizen, E.H.P.; Swart, J.F.; van Royen-Kerkhof, A.; El Idrissi, A.; Leek, A.P.; de Jager, W.; de Groot, M.C.H.; Haitjema, S.; Holzinger, D.; et al. Treatment to Target Using Recombinant Interleukin-1 Receptor Antagonist as First-Line Monotherapy in New-Onset Systemic Juvenile Idiopathic Arthritis: Results From a Five-Year Follow-Up Study. Arthritis Rheumatol. 2019, 71, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Klotsche, J.; Raab, A.; Niewerth, M.; Sengler, C.; Ganser, G.; Kallinich, T.; Niehues, T.; Hufnagel, M.; Thon, A.; Hospach, T.; et al. Outcome and Trends in Treatment of Systemic Juvenile Idiopathic Arthritis in the German National Pediatric Rheumatologic Database, 2000–2013. Arthritis Rheumatol 2016, 68, 3023–3034. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Infections | Tuberculosis, toxoplasmosis, brucellosis, yersiniosis HIV, Epstein-Barr, cytomegalovirus, hepatitis, herpes, influenza, parvovirus B19, measles, rubella |
| Malignancies | Lymphoma, Castleman disease, myeloproliferative disorders, melanoma and colon, breast, lung, kidney and thyroid cancer In pediatrics also: leukemia |
| Systemic diseases | Systemic lupus erythematosus, idiopathic inflammatory myopathies, vasculitis, hereditary autoinflammatory syndromes, neutrophilic dermatosis, Sweet syndrome, reactive arthritis, sarcoidosis, Schnitzler syndrome, Kikuchi-Fujimoto disease In pediatrics also: other types of inflammatory arthritis |
| Di Benedetto P, Cipriani P, Iacono D, et al. (2020) [40] | Hu QY, Zeng T, Sun CY et al. (2019) [41] | Sfriso P, Priori R, Valesini G, et al. (2016) [35] | Gerfaud-Valentin M, Maucort-Boulch D, Hot A, et al. (2014) [42] | Fautrel B. et al. (2002) [43] | Tsai H. et al. (2012) [44] | Behrens E. D. et al. (2008) [45] | |
|---|---|---|---|---|---|---|---|
| Case number | 147 | 517 | 245 | 57 | 72 | 28 | 136 |
| Nationality | Italy | China | Italy | France | France | Taiwan | United States |
| Female | 39.5 | 72 | 47.3 | 53 | nk | 53.6 | 54 |
| Average age at onset | 45.2 | 37.7 | 38.8 | 36 | 35.2 | 8.7 | 5.7 Median 2 |
| Fever ≥ 39 °C | 100 | 91.3 | 92.6 | 95 | 84.7 | 100 | 98 |
| Rash | 74.8 | 79.9 | 67.7 | 77 | 70.8 | 67.9 | 81 |
| Arthralgia/arthritis | 88.4 | 73.1 | 93 | 95 | 88.8 | 89.3 | 88 |
| Sore throat | 56.5 | 60.5 | 62 | 53 | 52.7 | nk | nk |
| Lymphadenopathy | 54.4 | 51.1 | 60.4 * | 60 | 44.4 * | 46.4 | 31 |
| Hepatomegaly | nk | 6.6 | 41.7 | 21 | nk | nk | ~7 |
| Splenomegaly | 66.7 | 34.4 | 60.4 * | 30 | 44.4 * | 21.4 * | ~5 |
| Pericarditis | 21.1 | 14.1 | 17.3 | 19 | 20.8 | nk | 10 |
| Pleuritis | 19.7 | nk | nk | 18 | nk | 7.1 * | nk |
| Myalgia | 64.6 | 32.5 | nk | 44 | nk | nk | nk |
| AoSD pneumonia | 12.2 | nk | nk | nk | nk | nk | nk |
| Abdominal pain | 13.6 | nk | nk | 18 | nk | nk | nk |
| Criteria | 1992 Yamaguchi [57] | 2002 Fautrel [47] | 2004 ILAR [58] |
|---|---|---|---|
| Major |
|
|
|
| Minor |
|
|
|
| Exclusion criteria | Infection, malignancy or other rheumatic disorders than mimic AoSD | None | Other forms of JIA must be excluded |
| Algorithm | Five criteria, at least two major ones AND no exclusion criteria | Four major criteria OR three majors with two minor ones | All major criteria AND at least one minor criteria |
| Sensitivity | 96.2% | 80.6% | Not applicable |
| Specificity | 92.1% | 98.5% | Not applicable |
| Variable | Number of Points |
|---|---|
| Temperature | |
| <38.4 °C | 0 |
| 38.4–39.4 °C | 33 |
| >39.4 °C | 49 |
| Organomegaly | |
| None | 0 |
| Hepatomegaly or splenomegaly | 23 |
| Hepatomegaly and splenomegaly | 38 |
| Cytopenia | |
| One lineage | 0 |
| Two lineages | 24 |
| Three lineages | 34 |
| Triglycerides (mmol/L) | |
| <1.5 | 0 |
| 1.5–4.0 | 44 |
| >4.0 | 64 |
| Fibrinogen (g/L) | |
| >2.5 | 0 |
| ≤2.5 | 30 |
| Ferritin (ng/mL) | |
| <2000 | 0 |
| 2000–6000 | 35 |
| >6000 | 50 |
| Serum aspartate aminotransferase (IU/L) | |
| <30 | 0 |
| ≥30 | 19 |
| Haemophagocytosis on bone marrow aspirate | |
| No | 0 |
| Yes | 35 |
| Known immunosuppression | |
| No | 0 |
| Yes | 18 |
| Variables | Points |
|---|---|
| Platelet count (/µL) | |
| 50,000–100,000 | 1 |
| <50,000 | 2 |
| Prolongation of PT (seconds) | |
| 3–6 | 1 |
| >6 | 2 |
| Fibrinogen (mg/dL) | |
| <100 | 1 |
| D-dimer (µg/mL) | |
| 0.5–1 | 1 |
| 1–2 | 2 |
| >2 | 3 |
| If score ≥ 5: compatible with DIC. Repeat daily. If score < 5: suggestive of DIC. Repeat after 1–2 days. | |
| Major criteria |
|
| Minor criteria |
|
| Algorithm | Both major criteria with at least two minor criteria |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomaras, S.; Goetzke, C.C.; Kallinich, T.; Feist, E. Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. J. Clin. Med. 2021, 10, 733. https://doi.org/10.3390/jcm10040733
Tomaras S, Goetzke CC, Kallinich T, Feist E. Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. Journal of Clinical Medicine. 2021; 10(4):733. https://doi.org/10.3390/jcm10040733
Chicago/Turabian StyleTomaras, Stylianos, Carl Christoph Goetzke, Tilmann Kallinich, and Eugen Feist. 2021. "Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach" Journal of Clinical Medicine 10, no. 4: 733. https://doi.org/10.3390/jcm10040733
APA StyleTomaras, S., Goetzke, C. C., Kallinich, T., & Feist, E. (2021). Adult-Onset Still’s Disease: Clinical Aspects and Therapeutic Approach. Journal of Clinical Medicine, 10(4), 733. https://doi.org/10.3390/jcm10040733