
Cognitive Dysfunction in Non-Alcoholic Fatty Liver Disease—Current Knowledge, Mechanisms and Perspectives

 , ,
, , 
Abstract
1. Introduction
2. Evidence for Cognitive Dysfunction in NAFLD
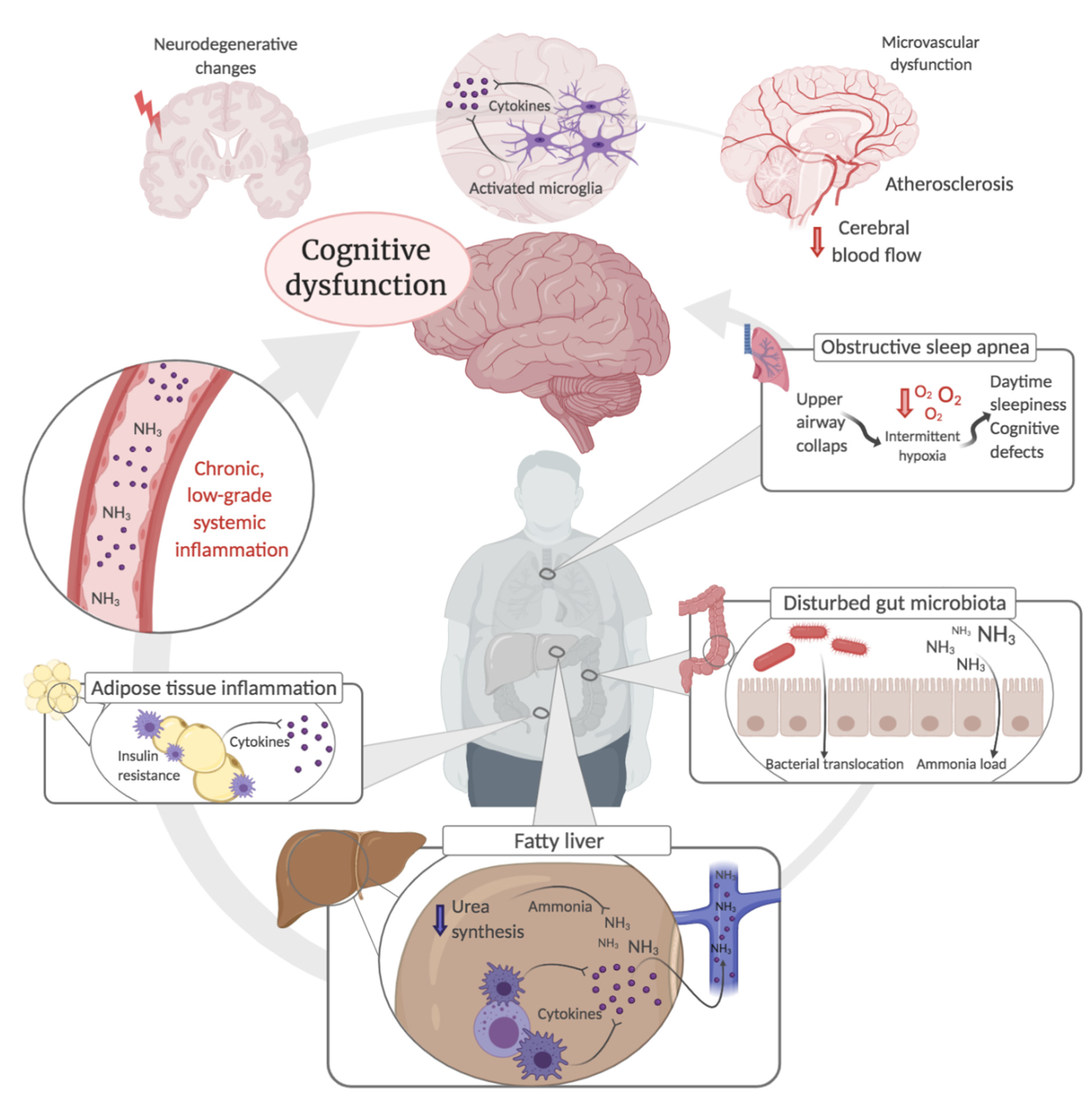
3. Possible Mechanisms behind Cognitive Dysfunction in NAFLD
3.1. Systemic- and Neuroinflammation
3.2. Metabolic Liver Dysfunction and Ammonia
3.3. Disturbed Gut Microbiota
3.4. Atherosclerosis and Cerebrovascular Dysfunction
3.5. Neurodegeneration
3.6. Obstructive Sleep Apnoea
4. Methods Used to Characterize Cognition in NAFLD
5. Clinical Considerations and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P. Utility and appropriateness of the fatty liver inhibition of progression (FLIP) algorithm and steatosis, activity, and fibrosis (SAF) score in the evaluation of biopsies of nonalcoholic fatty liver disease. Hepatology 2014, 60, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef]
- Rafiq, N.; Bai, C.; Fang, Y.; Srishord, M.; McCullough, A.; Gramlich, T.; Younossi, Z.M. Long-term follow-up of patients with nonalcoholic fatty liver. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2009, 7, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e310. [Google Scholar] [CrossRef] [PubMed]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Colognesi, M.; Gabbia, D.; De Martin, S. Depression and Cognitive Impairment-Extrahepatic Manifestations of NAFLD and NASH. Biomedicines 2020, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Fargion, S.; Fracanzani, A.L. Brain involvement in non-alcoholic fatty liver disease (NAFLD): A systematic review. Dig. Liver Dis. 2019, 51, 1214–1222. [Google Scholar] [CrossRef]
- Elliott, C.; Frith, J.; Day, C.P.; Jones, D.E.; Newton, J.L. Functional impairment in alcoholic liver disease and non-alcoholic fatty liver disease is significant and persists over 3 years of follow-up. Dig. Dis. Sci. 2013, 58, 2383–2391. [Google Scholar] [CrossRef]
- Doward, L.C.; Balp, M.M.; Twiss, J.; Slota, C.; Cryer, D.; Brass, C.A.; Anstee, Q.M.; Sanyal, A.J. Development of a Patient-Reported Outcome Measure for Non-Alcoholic Steatohepatitis (NASH-CHECK): Results of a Qualitative Study. Patient 2020, 1–11. [Google Scholar] [CrossRef]
- Newton, J.L. Systemic symptoms in non-alcoholic fatty liver disease. Dig. Dis. (Basel Switz.) 2010, 28, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Martin, T.; Bae, J.P.; Paczkowski, R.; Freeman, E. Health-related quality of life burden of nonalcoholic steatohepatitis: A robust pragmatic literature review. J. Patient-Rep. Outcomes 2018, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef]
- Frisardi, V.; Solfrizzi, V.; Seripa, D.; Capurso, C.; Santamato, A.; Sancarlo, D.; Vendemiale, G.; Pilotto, A.; Panza, F. Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-alcoholic fatty liver disease and its relationship with cardiovascular disease and other extrahepatic diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef] [PubMed]
- Ndumele, C.E.; Nasir, K.; Conceiçao, R.D.; Carvalho, J.A.; Blumenthal, R.S.; Santos, R.D. Hepatic steatosis, obesity, and the metabolic syndrome are independently and additively associated with increased systemic inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1927–1932. [Google Scholar] [CrossRef]
- Lykke Eriksen, P.; Sorensen, M.; Gronbaek, H.; Hamilton-Dutoit, S.; Vilstrup, H.; Thomsen, K.L. Non-alcoholic fatty liver disease causes dissociated changes in metabolic liver functions. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 551–560. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Aldridge, D.R.; Tranah, E.J.; Shawcross, D.L. Pathogenesis of hepatic encephalopathy: Role of ammonia and systemic inflammation. J. Clin. Exp. Hepatol. 2015, 5, S7–S20. [Google Scholar] [CrossRef]
- Bajaj, J.S. The role of microbiota in hepatic encephalopathy. Gut Microbes 2014, 5, 397–403. [Google Scholar] [CrossRef]
- Butterworth, R.F. The liver-brain axis in liver failure: Neuroinflammation and encephalopathy. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 522–528. [Google Scholar] [CrossRef]
- Rose, C.F.; Amodio, P.; Bajaj, J.S.; Dhiman, R.K.; Montagnese, S.; Taylor-Robinson, S.D.; Vilstrup, H.; Jalan, R. Hepatic encephalopathy: Novel insights into classification, pathophysiology and therapy. J. Hepatol. 2020, 73, 1526–1547. [Google Scholar] [CrossRef]
- Seo, S.W.; Gottesman, R.F.; Clark, J.M.; Hernaez, R.; Chang, Y.; Kim, C.; Ha, K.H.; Guallar, E.; Lazo, M. Nonalcoholic fatty liver disease is associated with cognitive function in adults. Neurology 2016, 86, 1136–1142. [Google Scholar] [CrossRef]
- Weinstein, A.A.; de Avila, L.; Paik, J.; Golabi, P.; Escheik, C.; Gerber, L.; Younossi, Z.M. Cognitive Performance in Individuals With Non-Alcoholic Fatty Liver Disease and/or Type 2 Diabetes Mellitus. Psychosomatics 2018, 59, 567–574. [Google Scholar] [CrossRef]
- McCrimmon, R.J.; Ryan, C.M.; Frier, B.M. Diabetes and cognitive dysfunction. Lancet 2012, 379, 2291–2299. [Google Scholar] [CrossRef]
- Weinstein, G.; Davis-Plourde, K.; Himali, J.J.; Zelber-Sagi, S.; Beiser, A.S.; Seshadri, S. Non-alcoholic fatty liver disease, liver fibrosis score and cognitive function in middle-aged adults: The Framingham Study. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Petta, S.; Casuccio, A.; Maida, C.; Corte, V.D.; Daidone, M.; Di Raimondo, D.; Pecoraro, R.; Fonte, R.; Cirrincione, A.; et al. Reactive hyperemia index (RHI) and cognitive performance indexes are associated with histologic markers of liver disease in subjects with non-alcoholic fatty liver disease (NAFLD): A case control study. Cardiovasc. Diabetol. 2018, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, B.; Markovic, O.; Duric, V.; Filipovic, B. Cognitive Changes and Brain Volume Reduction in Patients with Nonalcoholic Fatty Liver Disease. Can. J. Gastroenterol. Hepatol. 2018, 2018, 9638797. [Google Scholar] [CrossRef] [PubMed]
- Celikbilek, A.; Celikbilek, M.; Bozkurt, G. Cognitive assessment of patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2018, 30, 944–950. [Google Scholar] [CrossRef] [PubMed]
- An, K.; Starkweather, A.; Sturgill, J.; Salyer, J.; Sterling, R.K. Association of CTRP13 With Liver Enzymes and Cognitive Symptoms in Nonalcoholic Fatty Liver Disease. Nurs. Res. 2019, 68, 29–38. [Google Scholar] [CrossRef]
- Felipo, V.; Urios, A.; Montesinos, E.; Molina, I.; Garcia-Torres, M.L.; Civera, M.; Olmo, J.A.; Ortega, J.; Martinez-Valls, J.; Serra, M.A.; et al. Contribution of hyperammonemia and inflammatory factors to cognitive impairment in minimal hepatic encephalopathy. Metab. Brain Dis. 2012, 27, 51–58. [Google Scholar] [CrossRef]
- Takahashi, A.; Kono, S.; Wada, A.; Oshima, S.; Abe, K.; Imaizumi, H.; Fujita, M.; Hayashi, M.; Okai, K.; Miura, I.; et al. Reduced brain activity in female patients with non-alcoholic fatty liver disease as measured by near-infrared spectroscopy. PLoS ONE 2017, 12, e0174169. [Google Scholar] [CrossRef]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The Fatty Liver Index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Haukeland, J.W.; Damås, J.K.; Konopski, Z.; Løberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjøro, K.; Aukrust, P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef]
- Fricker, Z.P.; Pedley, A.; Massaro, J.M.; Vasan, R.S.; Hoffmann, U.; Benjamin, E.J.; Long, M.T. Liver Fat Is Associated With Markers of Inflammation and Oxidative Stress in Analysis of Data From the Framingham Heart Study. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 1157–1164.e1154. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef]
- Dixon, L.J.; Flask, C.A.; Papouchado, B.G.; Feldstein, A.E.; Nagy, L.E. Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS ONE 2013, 8, e56100. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Catrysse, L.; van Loo, G. Adipose tissue macrophages and their polarization in health and obesity. Cell Immunol. 2018, 330, 114–119. [Google Scholar] [CrossRef]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. Jhep Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef]
- Narayanan, S.; Surette, F.A.; Hahn, Y.S. The Immune Landscape in Nonalcoholic Steatohepatitis. Immune Netw. 2016, 16, 147–158. [Google Scholar] [CrossRef]
- Miller, A.A.; Spencer, S.J. Obesity and neuroinflammation: A pathway to cognitive impairment. Brain Behav. Immun. 2014, 42, 10–21. [Google Scholar] [CrossRef]
- Viscogliosi, G.; Andreozzi, P.; Chiriac, I.M.; Cipriani, E.; Servello, A.; Marigliano, B.; Ettorre, E.; Marigliano, V. Depressive symptoms in older people with metabolic syndrome: Is there a relationship with inflammation? Int. J. Geriatr. Psychiatry 2013, 28, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.P.; Becher, B.; Ludwin, S.K.; Prat, A.; Quintana, F.J. Glial Cells as Regulators of Neuroimmune Interactions in the Central Nervous System. J. Immunol. 2020, 204, 251. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Williams, L.M. Hypothalamic dysfunction in obesity. Proc. Nutr. Soc. 2012, 71, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.L.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J. Neurosci. 2009, 29, 359. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Liu, T. Inflammatory cause of metabolic syndrome via brain stress and NF-κB. Aging 2012, 4, 98–115. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Veniaminova, E.; Oplatchikova, M.; Bettendorff, L.; Kotenkova, E.; Lysko, A.; Vasilevskaya, E.; Kalueff, A.V.; Fedulova, L.; Umriukhin, A.; Lesch, K.P.; et al. Prefrontal cortex inflammation and liver pathologies accompany cognitive and motor deficits following Western diet consumption in non-obese female mice. Life Sci. 2020, 241, 117163. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Bose, D.; Saha, P.; Sarkar, S.; Seth, R.; Kimono, D.; Albadrani, M.; Nagarkatti, M.; Nagarkatti, P.; Chatterjee, S. Lipocalin 2 induces neuroinflammation and blood-brain barrier dysfunction through liver-brain axis in murine model of nonalcoholic steatohepatitis. J. Neuroinflammation 2020, 17, 201. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Sheng, L.; Nguyen, M.; Di Lucente, J.; Hu, Y.; Li, Y.; Maezawa, I.; Jin, L.W.; Wan, Y.Y. Dysregulated bile acid receptor-mediated signaling and IL-17A induction are implicated in diet-associated hepatic health and cognitive function. Biomark Res. 2020, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflammation 2016, 13, 1. [Google Scholar] [CrossRef]
- Li, X.; Cheng, X.; Wang, X.; Liu, Q.; Ma, H.; Li, M. Dyslipidemic Diet Induces Mobilization of Peripheral Neutrophils and Monocytes That Exacerbate Hemorrhagic Brain Injury and Neuroinflammation. Front. Cell Neurosci. 2020, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Balzano, T.; Forteza, J.; Molina, P.; Giner, J.; Monzo, A.; Sancho-Jimenez, J.; Urios, A.; Montoliu, C.; Felipo, V. The Cerebellum of Patients with Steatohepatitis Shows Lymphocyte Infiltration, Microglial Activation and Loss of Purkinje and Granular Neurons. Sci. Rep. 2018, 8, 3004. [Google Scholar] [CrossRef]
- Balzano, T.; Forteza, J.; Borreda, I.; Molina, P.; Giner, J.; Leone, P.; Urios, A.; Montoliu, C.; Felipo, V. Histological Features of Cerebellar Neuropathology in Patients With Alcoholic and Nonalcoholic Steatohepatitis. J. Neuropathol. Exp. Neurol. 2018, 77, 837–845. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Bak-Fredslund, K.P.; Lykke Eriksen, P.; Munk, O.L.; Villadsen, G.E.; Keiding, S.; Sørensen, M. Metabolic liver function in humans measured by 2-18F-fluoro-2-deoxy-D-galactose PET/CT-reproducibility and clinical potential. EJNMMI Res. 2017, 7, 71. [Google Scholar] [CrossRef]
- Vilstrup, H. Synthesis of urea after stimulation with amino acids: Relation to liver function. Gut 1980, 21, 990–995. [Google Scholar] [CrossRef]
- Anthony, P.P.; Ishak, K.G.; Nayak, N.C.; Poulsen, H.E.; Scheuer, P.J.; Sobin, L.H. The morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization. J. Clin. Pathol. 1978, 31, 395–414. [Google Scholar] [CrossRef]
- Thomsen, K.L.; Gronbaek, H.; Glavind, E.; Hebbard, L.; Jessen, N.; Clouston, A.; George, J.; Vilstrup, H. Experimental nonalcoholic steatohepatitis compromises ureagenesis, an essential hepatic metabolic function. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G295–G301. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, F.; Heeboll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Gronbaek, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, P.L.; Vilstrup, H.; Rigbolt, K.; Suppli, M.P.; Sorensen, M.; Heeboll, S.; Veidal, S.S.; Knop, F.K.; Thomsen, K.L. Non-alcoholic fatty liver disease alters expression of genes governing hepatic nitrogen conversion. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, F.; Thomsen, K.L.; Habtesion, A.; Jones, H.; Davies, N.; Gracia-Sancho, J.; Manicardi, N.; Hall, A.; Andreola, F.; Paish, H.L.; et al. Ammonia Scavenging Prevents Progression of Fibrosis in Experimental Nonalcoholic Fatty Liver Disease. Hepatology 2020, 71, 874–892. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, K.L.; De Chiara, F.; Rombouts, K.; Vilstrup, H.; Andreola, F.; Mookerjee, R.P.; Jalan, R. Ammonia: A novel target for the treatment of non-alcoholic steatohepatitis. Med. Hypotheses 2018, 113, 91–97. [Google Scholar] [CrossRef]
- Marini, J.C.; Broussard, S.R. Hyperammonemia increases sensitivity to LPS. Mol. Genet. Metab. 2006, 88, 131–137. [Google Scholar] [CrossRef]
- Jover, R.; Rodrigo, R.; Felipo, V.; Insausti, R.; Saez-Valero, J.; Garcia-Ayllon, M.S.; Suarez, I.; Candela, A.; Compan, A.; Esteban, A.; et al. Brain edema and inflammatory activation in bile duct ligated rats with diet-induced hyperammonemia: A model of hepatic encephalopathy in cirrhosis. Hepatology 2006, 43, 1257–1266. [Google Scholar] [CrossRef]
- Wright, G.; Davies, N.A.; Shawcross, D.L.; Hodges, S.J.; Zwingmann, C.; Brooks, H.F.; Mani, A.R.; Harry, D.; Stadlbauer, V.; Zou, Z.; et al. Endotoxemia produces coma and brain swelling in bile duct ligated rats. Hepatology 2007, 45, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Jalan, R. Ammonia and inflammation in the pathogenesis of hepatic encephalopathy: Pandora’s box? Hepatology 2007, 46, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Haussinger, D.; Kircheis, G.; Fischer, R.; Schliess, F.; vom Dahl, S. Hepatic encephalopathy in chronic liver disease: A clinical manifestation of astrocyte swelling and low-grade cerebral edema? J. Hepatol. 2000, 32, 1035–1038. [Google Scholar] [CrossRef]
- Kimelberg, H.K. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 2005, 50, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Cauli, O.; Gomez-Pinedo, U.; Agusti, A.; Hernandez-Rabaza, V.; Garcia-Verdugo, J.M.; Felipo, V. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology 2010, 139, 675–684. [Google Scholar] [CrossRef]
- Zemtsova, I.; Gorg, B.; Keitel, V.; Bidmon, H.J.; Schror, K.; Haussinger, D. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology 2011, 54, 204–215. [Google Scholar] [CrossRef]
- Cagnin, A.; Taylor-Robinson, S.D.; Forton, D.M.; Banati, R.B. In vivo imaging of cerebral "peripheral benzodiazepine binding sites" in patients with hepatic encephalopathy. Gut 2006, 55, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Balzano, T.; Dadsetan, S.; Forteza, J.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Malaguarnera, M.; Gil-Perotin, S.; Cubas-Nuñez, L.; Casanova, B.; Castro-Quintas, A.; et al. Chronic hyperammonemia induces peripheral inflammation that leads to cognitive impairment in rats: Reversed by anti-TNF-α treatment. J. Hepatol. 2020, 73, 582–592. [Google Scholar] [CrossRef]
- Higarza, S.G.; Arboleya, S.; Gueimonde, M.; Gomez-Lazaro, E.; Arias, J.L.; Arias, N. Neurobehavioral dysfunction in non-alcoholic steatohepatitis is associated with hyperammonemia, gut dysbiosis, and metabolic and functional brain regional deficits. PLoS ONE 2019, 14, e0223019. [Google Scholar] [CrossRef]
- Ahluwalia, V.; Betrapally, N.S.; Hylemon, P.B.; White, M.B.; Gillevet, P.M.; Unser, A.B.; Fagan, A.; Daita, K.; Heuman, D.M.; Zhou, H.; et al. Impaired Gut-Liver-Brain Axis in Patients with Cirrhosis. Sci. Rep. 2016, 6, 26800. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Kassam, Z.; Fagan, A.; Gavis, E.A.; Liu, E.; Cox, I.J.; Kheradman, R.; Heuman, D.; Wang, J.; Gurry, T.; et al. Fecal microbiota transplant from a rational stool donor improves hepatic encephalopathy: A randomized clinical trial. Hepatology 2017, 66, 1727–1738. [Google Scholar] [CrossRef]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; White, M.B.; Unser, A.; Thacker, L.R.; Sanyal, A.J.; Kang, D.J.; et al. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology 2015, 62, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Yang, M.; Edwards, S.; Ye, S.Q. Nonalcoholic fatty liver disease: For better or worse, blame the gut microbiota? JPEN J. Parenter Enter. Nutr. 2013, 37, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875.e1–3. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Ilan, Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2609–2618. [Google Scholar] [CrossRef]
- Liu, R.; Kang, J.D.; Sartor, R.B.; Sikaroodi, M.; Fagan, A.; Gavis, E.A.; Zhou, H.; Hylemon, P.B.; Herzog, J.W.; Li, X.; et al. Neuroinflammation in Murine Cirrhosis Is Dependent on the Gut Microbiome and Is Attenuated by Fecal Transplant. Hepatology 2020, 71, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.K.; Magdy, Y.M.; El-Waseef, D.A.; Nabih, E.S.; Hamouda, M.A.; El-Kharashi, O.A. Modulation of hippocampal TLR4/BDNF signal pathway using probiotics is a step closer towards treating cognitive impairment in NASH model. Physiol. Behav. 2020, 214, 112762. [Google Scholar] [CrossRef] [PubMed]
- Fargion, S.; Porzio, M.; Fracanzani, A.L. Nonalcoholic fatty liver disease and vascular disease: State-of-the-art. World J. Gastroenterol. 2014, 20, 13306–13324. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C. Cerebral hemodynamics and vascular risk factors: Setting the stage for Alzheimer’s disease. J. Alzheimers Dis. 2012, 32, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Oni, E.T.; Agatston, A.S.; Blaha, M.J.; Fialkow, J.; Cury, R.; Sposito, A.; Erbel, R.; Blankstein, R.; Feldman, T.; Al-Mallah, M.H.; et al. A systematic review: Burden and severity of subclinical cardiovascular disease among those with nonalcoholic fatty liver; should we care? Atherosclerosis 2013, 230, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Wang, C.C.; Lin, S.K.; Tseng, Y.F.; Hsu, C.S.; Tseng, T.C.; Lin, H.H.; Wang, L.Y.; Kao, J.H. Elevation of serum aminotransferase activity increases risk of carotid atherosclerosis in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2009, 24, 1411–1416. [Google Scholar] [CrossRef]
- Tripodi, A.; Fracanzani, A.L.; Primignani, M.; Chantarangkul, V.; Clerici, M.; Mannucci, P.M.; Peyvandi, F.; Bertelli, C.; Valenti, L.; Fargion, S. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 61, 148–154. [Google Scholar] [CrossRef]
- Long, M.T.; Wang, N.; Larson, M.G.; Mitchell, G.F.; Palmisano, J.; Vasan, R.S.; Hoffmann, U.; Speliotes, E.K.; Vita, J.A.; Benjamin, E.J.; et al. Nonalcoholic fatty liver disease and vascular function: Cross-sectional analysis in the Framingham heart study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1284–1291. [Google Scholar] [CrossRef]
- Mitchell, G.F.; van Buchem, M.A.; Sigurdsson, S.; Gotal, J.D.; Jonsdottir, M.K.; Kjartansson, Ó.; Garcia, M.; Aspelund, T.; Harris, T.B.; Gudnason, V.; et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: The Age, Gene/Environment Susceptibility—Reykjavik study. Brain 2011, 134, 3398–3407. [Google Scholar] [CrossRef]
- Silbert, L.C.; Nelson, C.; Howieson, D.B.; Moore, M.M.; Kaye, J.A. Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline. Neurology 2008, 71, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Egorova, S.; Blacker, D.; Killiany, R.J.; Muzikansky, A.; Dickerson, B.C.; Tanzi, R.E.; Albert, M.S.; Greenberg, S.M.; Guttmann, C.R. Magnetic resonance imaging white matter hyperintensities and brain volume in the prediction of mild cognitive impairment and dementia. Arch. Neurol. 2008, 65, 94–100. [Google Scholar] [CrossRef]
- Debette, S.; Beiser, A.; DeCarli, C.; Au, R.; Himali, J.J.; Kelly-Hayes, M.; Romero, J.R.; Kase, C.S.; Wolf, P.A.; Seshadri, S. Association of MRI markers of vascular brain injury with incident stroke, mild cognitive impairment, dementia, and mortality: The Framingham Offspring Study. Stroke 2010, 41, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Zelber-Sagi, S.; Preis, S.R.; Beiser, A.S.; DeCarli, C.; Speliotes, E.K.; Satizabal, C.L.; Vasan, R.S.; Seshadri, S. Association of Nonalcoholic Fatty Liver Disease With Lower Brain Volume in Healthy Middle-aged Adults in the Framingham Study. JAMA Neurol. 2018, 75, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Tuttolomondo, A.; Gagliardo, C.; Zafonte, R.; Brancatelli, G.; Cabibi, D.; Cammà, C.; Di Marco, V.; Galvano, L.; La Tona, G.; et al. The Presence of White Matter Lesions Is Associated With the Fibrosis Severity of Nonalcoholic Fatty Liver Disease. Medicine 2016, 95, e3446. [Google Scholar] [CrossRef] [PubMed]
- Wolters, F.J.; Zonneveld, H.I.; Hofman, A.; van der Lugt, A.; Koudstaal, P.J.; Vernooij, M.W.; Ikram, M.A. Cerebral Perfusion and the Risk of Dementia: A Population-Based Study. Circulation 2017, 136, 719–728. [Google Scholar] [CrossRef]
- VanWagner, L.B.; Terry, J.G.; Chow, L.S.; Alman, A.C.; Kang, H.; Ingram, K.H.; Shay, C.; Lewis, C.E.; Bryan, R.N.; Launer, L.J.; et al. Nonalcoholic fatty liver disease and measures of early brain health in middle-aged adults: The CARDIA study. Obesity 2017, 25, 642–651. [Google Scholar] [CrossRef]
- Airaghi, L.; Rango, M.; Maira, D.; Barbieri, V.; Valenti, L.; Lombardi, R.; Biondetti, P.; Fargion, S.; Fracanzani, A.L. Subclinical cerebrovascular disease in NAFLD without overt risk factors for atherosclerosis. Atherosclerosis 2018, 268, 27–31. [Google Scholar] [CrossRef]
- Ng, T.P.; Feng, L.; Nyunt, M.S.; Feng, L.; Gao, Q.; Lim, M.L.; Collinson, S.L.; Chong, M.S.; Lim, W.S.; Lee, T.S.; et al. Metabolic Syndrome and the Risk of Mild Cognitive Impairment and Progression to Dementia: Follow-up of the Singapore Longitudinal Ageing Study Cohort. JAMA Neurol. 2016, 73, 456–463. [Google Scholar] [CrossRef]
- Vanhanen, M.; Koivisto, K.; Moilanen, L.; Helkala, E.L.; Hänninen, T.; Soininen, H.; Kervinen, K.; Kesäniemi, Y.A.; Laakso, M.; Kuusisto, J. Association of metabolic syndrome with Alzheimer disease: A population-based study. Neurology 2006, 67, 843–847. [Google Scholar] [CrossRef]
- Atti, A.R.; Valente, S.; Iodice, A.; Caramella, I.; Ferrari, B.; Albert, U.; Mandelli, L.; De Ronchi, D. Metabolic Syndrome, Mild Cognitive Impairment, and Dementia: A Meta-Analysis of Longitudinal Studies. Am. J. Geriatr. Psychiatry 2019, 27, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Pedditzi, E.; Peters, R.; Beckett, N. The risk of overweight/obesity in mid-life and late life for the development of dementia: A systematic review and meta-analysis of longitudinal studies. Age Ageing 2016, 45, 14–21. [Google Scholar] [CrossRef]
- Han, E.; Lee, J.Y.; Han, K.D.; Cho, H.; Kim, K.J.; Lee, B.W.; Kang, E.S.; Cha, B.S.; Younossi, Z.M.; Lee, Y.H. Gamma glutamyltransferase and risk of dementia in prediabetes and diabetes. Sci. Rep. 2020, 10, 6800. [Google Scholar] [CrossRef]
- Solfrizzi, V.; Scafato, E.; Custodero, C.; Loparco, F.; Ciavarella, A.; Panza, F.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Napoli, N.; et al. Liver fibrosis score, physical frailty, and the risk of dementia in older adults: The Italian Longitudinal Study on Aging. Alzheimers Dement. 2020, 6, e12065. [Google Scholar] [CrossRef]
- Labenz, C.; Kostev, K.; Kaps, L.; Galle, P.R.; Schattenberg, J.M. Incident Dementia in Elderly Patients with Nonalcoholic Fatty Liver Disease in Germany. Dig. Dis. Sci. 2020. [Google Scholar] [CrossRef]
- Zeltser, N.; Meyer, I.; Hernandez, G.V.; Trahan, M.J.; Fanter, R.K.; Abo-Ismail, M.; Glanz, H.; Strand, C.R.; Burrin, D.G.; La Frano, M.R.; et al. Neurodegeneration in juvenile Iberian pigs with diet-induced nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab 2020, 319, E592–E606. [Google Scholar] [CrossRef]
- Karbalaei, R.; Allahyari, M.; Rezaei-Tavirani, M.; Asadzadeh-Aghdaei, H.; Zali, M.R. Protein-protein interaction analysis of Alzheimer`s disease and NAFLD based on systems biology methods unhide common ancestor pathways. Gastroenterol. Hepatol. Bed Bench 2018, 11, 27–33. [Google Scholar]
- de la Monte, S.M.; Longato, L.; Tong, M.; Wands, J.R. Insulin resistance and neurodegeneration: Roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr. Opin. Investig. Drugs 2009, 10, 1049–1060. [Google Scholar]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Karakashian, M. Alcoholic and non-alcoholic fatty liver disease: Focus on ceramide. Adv. Biol. Regul. 2018, 70, 40–50. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Øie, C.I.; Appa, R.S.; Hilden, I.; Petersen, H.H.; Gruhler, A.; Smedsrød, B.; Hansen, J.-B. Rat liver sinusoidal endothelial cells (LSECs) express functional low density lipoprotein receptor-related protein-1 (LRP-1). J. Hepatol. 2011, 55, 1346–1352. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Wang, Q.-H.; Zhang, T.; Liu, Y.-H.; Yao, X.-Q.; Zeng, F.; Li, J.; Zhou, F.-Y.; Wang, L.; Yan, J.-C.; et al. Associations Between Hepatic Functions and Plasma Amyloid-Beta Levels—Implications for the Capacity of Liver in Peripheral Amyloid-Beta Clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, C.; Ohtsuki, S.; Terasaki, T. Insulin Facilitates the Hepatic Clearance of Plasma Amyloid β-Peptide (1–40) by Intracellular Translocation of Low-Density Lipoprotein Receptor-Related Protein 1 (LRP-1) to the Plasma Membrane in Hepatocytes. Mol. Pharmacol. 2007, 72, 850–855. [Google Scholar] [CrossRef]
- Pinçon, A.; De Montgolfier, O.; Akkoyunlu, N.; Daneault, C.; Pouliot, P.; Villeneuve, L.; Lesage, F.; Levy, B.I.; Thorin-Trescases, N.; Thorin, É.; et al. Non-Alcoholic Fatty Liver Disease, and the Underlying Altered Fatty Acid Metabolism, Reveals Brain Hypoperfusion and Contributes to the Cognitive Decline in APP/PS1 Mice. Metabolites 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, X.; Lu, Y. Obstructive Sleep Apnea Syndrome and Metabolic Diseases. Endocrinology 2018, 159, 2670–2675. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Olivetti, C.; Rosina, F.; Carbone, G.; Gambino, R. Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obes. Rev. 2013, 14, 417–431. [Google Scholar] [CrossRef]
- Mir, H.M.; Stepanova, M.; Afendy, H.; Cable, R.; Younossi, Z.M. Association of Sleep Disorders with Nonalcoholic Fatty Liver Disease (NAFLD): A Population-based Study. J. Clin. Exp. Hepatol. 2013, 3, 181–185. [Google Scholar] [CrossRef]
- Jordan, A.S.; McSharry, D.G.; Malhotra, A. Adult obstructive sleep apnoea. Lancet 2014, 383, 736–747. [Google Scholar] [CrossRef]
- Young, T.; Palta, M.; Dempsey, J.; Peppard, P.E.; Nieto, F.J.; Hla, K.M. Burden of sleep apnea: Rationale, design, and major findings of the Wisconsin Sleep Cohort study. WMJ 2009, 108, 246–249. [Google Scholar]
- Wallace, A.; Bucks, R.S. Memory and obstructive sleep apnea: A meta-analysis. Sleep 2013, 36, 203–220. [Google Scholar] [CrossRef]
- Olaithe, M.; Bucks, R.S. Executive dysfunction in OSA before and after treatment: A meta-analysis. Sleep 2013, 36, 1297–1305. [Google Scholar] [CrossRef]
- Leng, Y.; McEvoy, C.T.; Allen, I.E.; Yaffe, K. Association of Sleep-Disordered Breathing With Cognitive Function and Risk of Cognitive Impairment: A Systematic Review and Meta-analysis. JAMA Neurol. 2017, 74, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Olivetti, C.; Cassader, M.; Gambino, R. Obstructive sleep apnea-hypopnea syndrome and nonalcoholic fatty liver disease: Emerging evidence and mechanisms. Semin Liver Dis. 2012, 32, 49–64. [Google Scholar] [CrossRef]
- Savransky, V.; Bevans, S.; Nanayakkara, A.; Li, J.; Smith, P.L.; Torbenson, M.S.; Polotsky, V.Y. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G871–G877. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Minville, C.; Tordjman, J.; Lévy, P.; Bouillot, J.L.; Basdevant, A.; Bedossa, P.; Clément, K.; Pépin, J.L. Chronic intermittent hypoxia is a major trigger for non-alcoholic fatty liver disease in morbid obese. J. Hepatol. 2012, 56, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Benotti, P.; Wood, G.C.; Argyropoulos, G.; Pack, A.; Keenan, B.T.; Gao, X.; Gerhard, G.; Still, C. The impact of obstructive sleep apnea on nonalcoholic fatty liver disease in patients with severe obesity. Obesity 2016, 24, 871–877. [Google Scholar] [CrossRef]
- Weissenborn, K. Minimal/Covert Hepatic Encephalopathy—Impact of Comorbid Conditions. J. Clin. Exp. Hepatol. 2019, 9, 109–111. [Google Scholar] [CrossRef]
- Newton, J.L.; Jones, D.E.; Henderson, E.; Kane, L.; Wilton, K.; Burt, A.D.; Day, C.P. Fatigue in non-alcoholic fatty liver disease (NAFLD) is significant and associates with inactivity and excessive daytime sleepiness but not with liver disease severity or insulin resistance. Gut 2008, 57, 807–813. [Google Scholar] [CrossRef]
- Gerber, L.H.; Weinstein, A.A.; Mehta, R.; Younossi, Z.M. Importance of fatigue and its measurement in chronic liver disease. World J. Gastroenterol. 2019, 25, 3669–3683. [Google Scholar] [CrossRef]
- Capuron, L.; Welberg, L.; Heim, C.; Wagner, D.; Solomon, L.; Papanicolaou, D.A.; Craddock, R.C.; Miller, A.H.; Reeves, W.C. Cognitive dysfunction relates to subjective report of mental fatigue in patients with chronic fatigue syndrome. Neuropsychopharmacology 2006, 31, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Labenz, C.; Huber, Y.; Michel, M.; Nagel, M.; Galle, P.R.; Kostev, K.; Schattenberg, J.M. Nonalcoholic Fatty Liver Disease Increases the Risk of Anxiety and Depression. Hepatol. Commun. 2020, 4, 1293–1301. [Google Scholar] [CrossRef]
- Macavei, B.; Baban, A.; Dumitrascu, D.L. Psychological factors associated with NAFLD/NASH: A systematic review. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5081–5097. [Google Scholar] [PubMed]
- Rock, P.L.; Roiser, J.P.; Riedel, W.J.; Blackwell, A.D. Cognitive impairment in depression: a systematic review and meta-analysis. Psychol. Med. 2014, 44, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Heyman, A.; Mohs, R.C.; Hughes, J.P.; van Belle, G.; Fillenbaum, G.; Mellits, E.D.; Clark, C. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 1989, 39, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Balp, M.M.; Krieger, N.; Przybysz, R.; Way, N.; Cai, J.; Zappe, D.; McKenna, S.J.; Wall, G.; Janssens, N.; Tapper, E. The burden of non-alcoholic steatohepatitis (NASH) among patients from Europe: A real-world patient-reported outcomes study. JHEP Rep. 2019, 1, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Grieco, M.; Giorgi, A.; Gentile, M.C.; D’Erme, M.; Morano, S.; Maras, B.; Filardi, T. Glucagon-Like Peptide-1: A Focus on Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1112. [Google Scholar] [CrossRef]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell Mol. Neurobiol. 2018, 38, 121–132. [Google Scholar] [CrossRef]


{kind=link}
| Design and Study Population | Controlling for Important Confounders | Diagnosis of NAFLD and Fibrosis | Neuropsychological Tests | Cognitive Domains Assessed | Main Findings | Conclusion | Important Limitations | |
|---|---|---|---|---|---|---|---|---|
| Felipo 2012 (Spain) [38] | Cross-sectional. 40 NAFLD (n = 29 steatosis/n = 11 NASH), 54 controls. | None. | Liver biopsy. | Digit Symbol Substitution Test (DST). | Visuospatial function and psychomotor speed. | 5/11 Patients with pre-cirrhotic NASH were classified as having minimal hepatic encephalopathy (MHE) on the PSE-test 1 and performed poorly on the NCT-A and NCT-B, LTT (all p < 0.001), and SDT (p < 0.01), compared with healthy controls. NASH subgroup with MHE had higher levels of ammonia and IL-6 compared to other NASH, NAFLD, and controls. | Suggests MHE-related cognitive deficits in pre-cirrhotic NASH, but not simple steatosis. | Small sample size with subgroup analysis. All NAFLD patients undergoing surgery for morbid obesity (no diabetes status). Raw data on cognitive tests missing. |
| Trailmaking A test (NCT-A). | Attention and psychomotor speed. | |||||||
| Trailmaking B test (NCT-B). | Executive function. | |||||||
| Serial Dotting Test (SDT). | Attention and working memory. | |||||||
| Line Tracing Test (LTT). | Visuospatial function. | |||||||
| Seo 2016 (USA) [30] | Cross-sectional, population-based. 874 NAFLD, 3598 controls. | Age, education, diabetes, BMI, cardiovascular disease. | Ultrasound. NAFL fibrosis score (NFS *). | Simple Reaction Time Test (SRTT). | Psychomotor speed. | NAFLD patients had poor performance on the SDLT (β, 95% CI: 0.105 to 1.347) and also worse performance on the SRTT and SDST, but non-significantly so after adjusting for life-style related confounders (β, 95% CI: −0.496 to 14.679; −0.009 to 0.211). Poor performance on the SDST and SDLT scores were associated with increasing blood transaminases. | Suggests problems with memory and attention in NAFLD. | No biopsy-proven NAFLD. Persons aged > 59 years not included. |
| Digit Symbol Substitution Test (SDST). | Visuospatial function and psychomotor speed. | |||||||
| Serial Digit Learning Test (SDLT). | Memory and attention. | |||||||
| Takahashi 2017 (Japan) [39] | Cross-sectional. 24 female NAFLD, 15 age-matched controls. | None. | Ultrasound. | Verbal Fluency Task (VFT). | Executive function, verbal fluency. | NAFLD patients performed significantly worse on the VFT than controls, listing on average 2 words fewer during the test (p = 0.03). | Suggests problems with executive function and semantic fluency in NAFLD. | Small sample size, no adjustment for confounding. No biopsy-proven NAFLD. Limited cognitive assessment. |
| Tuttolomondo 2018 (Italy) [34] | Cross-sectional. 83 NAFLD (7,5% cirrhosis, 52% NASH), 80 controls. | Age, diabetes, BMI, cardiovascular disease. | Liver biopsy (in 65%). Ultrasound, liver stiffness (transient elastography). | Mini Mental State Examination (MMSE) 2. | Visuospatial function, executive function, memory, attention, language, and orientation. | NAFLD group performed worse on the MMSE than controls, independent of confounders (mean ± SD, 26.9 ± 1.6 vs. 28.0 ± 1.4; p < 0.0001). In NASH patients, poor performance on the MMSE 2 was associated with ballooning (β, 95% CI: −2.65 to −0.037; p = 0.044). No difference between NASH vs. non-NASH or low fibrosis vs. high fibrosis. | Suggests global reduction of cognitive function in NAFLD. | Small sample size. Limited cognitive assessment. |
| Filipovic 2018 (Serbia) [35] | Cross-sectional. 40 NAFLD, 30 controls with functional dyspepsia or irritable bowel syndrome. | Age, diabetes equally distributed between groups, but not otherwise controlled for. | Ultrasound (+ elevated ALT or AST). | Montreal Cognitive Assessment (MoCA) 3. | Visuospatial function, executive function, memory, attention, language, and orientation. | MoCA score was lower in NAFLD patients (mean ± SD, 24.07 ± 3.18 vs. 27.17 ± 2.35; p < 0.001), and NAFLD patients had a 4-fold increased risk of having an abnormal MoCA 3 score, compared with controls (RR, 95% CI: 1.815 to 8.381; p = 0.0005). | Suggests global reduction of cognitive function in NAFLD. | Small sample size, no adjustment for confounding. No biopsy-proven NAFLD. |
| Celikbilek 2018 (Turkey) [36] | Cross-sectional. 70 NAFLD, 73 controls. | Age, education, diabetes, metabolic syndrome. | Ultrasound, FIB-4 score **. | Montreal Cognitive Assessment (MoCA) 3. | Visuospatial function, executive function, memory, attention, language, and orientation. | NAFLD was associated with lower MoCA score on univariate regression analysis (OR = 2.99; p = 0.002), but not after adjusting for confounders (multivariate). MoCA score was negatively correlated with FIB-4 ** score. | Suggests global reduction of cognitive function in NAFLD (mostly executive and visuospatial function). | No biopsy-proven NAFLD. Patients with morbid obesity not included. |
| Weinstein 2018 (USA) [31] | Cross-sectional, population-based. 413 NAFLD (174 +T2DM), 689 controls (142 +T2DM). Age > 60 years. | Age, education, obesity, cardiovascular disease. Diabetes controlled for in subgroup analysis. | Presence of fatty liver index score *** ≥ 60. | Consortium to Establish a Registry for Alzheimer Disease – Word Learning subset (CERAD-WL). | Verbal memory (immediate and delayed recall). | NAFLD patients without T2DM did not demonstrate cognitive dysfunction, but NAFLD + T2DM performed worse than T2DM only and healthy controls on the DSST (mean ± SE, 47.1 ± 1.7 vs. 56.0 ± 1.1 and 53.6 ± 1.2). NAFLD + T2DM was associated with poor performance on DSST after adjusting for confounders (β, 95% CI: −6.75 to −0.12; p < 0.01). | Suggests no specific cognitive impairments in NAFLD. | No biopsy-proven NAFLD. Not generalizable to younger individuals. |
| Animal Fluency Test (AFT). | Executive function, verbal fluency. | |||||||
| Digit Symbol Substitution Test (DSST). | Visuospatial function, psychomotor speed. | |||||||
| An 2019 (USA) [37] | Cross-sectional. 23 NAFLD, 21 sex-matched controls. | None. 8/23 NAFLD patients with diabetes. | Liver biopsy (2/23 by transient elastography). | The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) 4. | Immediate and delayed memory, attention, language, and visuospatial memory. | Mean RBANS total score for NAFLD patients was below mean, but within the normative range after adjusting for age and educational level. | Suggests no specific cognitive impairments in NAFLD. | No control group for cognitive assessment. Small sample size, no adjustment for confounding. |
| Weinstein 2019 (USA) [33] | Cross-sectional, population-based. 378 NAFLD, 1278 total. | Age, education, diabetes, BMI, cardiovascular disease. | Multi-detector CT and NAFLD fibrosis score (NFS *). | WAIS-R 5 subtest: Logical memory delayed (LMd). | Verbal memory (delayed recall). | No significant association between NAFLD and cognitive performance on any tests after adjusting for confounders, but advanced fibrosis (NFS *) was associated with poor performance on TrA – TrB (β, mean ± SE, −0.11 ± 0.05; p = 0.028) and SIM (β, mean ± SE, −2.22 ± 0.83; p = 0.009). | Suggests problems with executive function in NAFLD with fibrosis. | No biopsy-proven NAFLD. |
| WAIS-R 5 subtest: Visual reproduction (VRd). | Visual memory (delayed recall). | |||||||
| WAIS-R 5 subtest: The Similarities test (SIM). | Abstract reasoning. | |||||||
| Trailmaking A – B test (TrA-TrB). | Executive function. | |||||||
| The Hooper Visual Organization Test (HVOT). | Visual perception. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kjærgaard, K.; Mikkelsen, A.C.D.; Wernberg, C.W.; Grønkjær, L.L.; Eriksen, P.L.; Damholdt, M.F.; Mookerjee, R.P.; Vilstrup, H.; Lauridsen, M.M.; Thomsen, K.L. Cognitive Dysfunction in Non-Alcoholic Fatty Liver Disease—Current Knowledge, Mechanisms and Perspectives. J. Clin. Med. 2021, 10, 673. https://doi.org/10.3390/jcm10040673
Kjærgaard K, Mikkelsen ACD, Wernberg CW, Grønkjær LL, Eriksen PL, Damholdt MF, Mookerjee RP, Vilstrup H, Lauridsen MM, Thomsen KL. Cognitive Dysfunction in Non-Alcoholic Fatty Liver Disease—Current Knowledge, Mechanisms and Perspectives. Journal of Clinical Medicine. 2021; 10(4):673. https://doi.org/10.3390/jcm10040673
Chicago/Turabian StyleKjærgaard, Kristoffer, Anne Catrine Daugaard Mikkelsen, Charlotte Wilhelmina Wernberg, Lea Ladegaard Grønkjær, Peter Lykke Eriksen, Malene Flensborg Damholdt, Rajeshwar Prosad Mookerjee, Hendrik Vilstrup, Mette Munk Lauridsen, and Karen Louise Thomsen. 2021. "Cognitive Dysfunction in Non-Alcoholic Fatty Liver Disease—Current Knowledge, Mechanisms and Perspectives" Journal of Clinical Medicine 10, no. 4: 673. https://doi.org/10.3390/jcm10040673
APA StyleKjærgaard, K., Mikkelsen, A. C. D., Wernberg, C. W., Grønkjær, L. L., Eriksen, P. L., Damholdt, M. F., Mookerjee, R. P., Vilstrup, H., Lauridsen, M. M., & Thomsen, K. L. (2021). Cognitive Dysfunction in Non-Alcoholic Fatty Liver Disease—Current Knowledge, Mechanisms and Perspectives. Journal of Clinical Medicine, 10(4), 673. https://doi.org/10.3390/jcm10040673