
Assessing Cognitive Function in Neuromuscular Diseases: A Pilot Study in a Sample of Children and Adolescents

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Participants and Procedures
2.3. Assessments
2.3.1. Cognitive Functioning
- Wechsler Preschool and Primary Scale of Intelligence, Third Edition (WPPSI-III), for children aged between 2 years and 6 months and 7 years and 3 months.
- Wechsler Intelligence Scale for Children, Fourth Edition (WISC-IV), for patients aged between 6 years and 16 years and 11 months.
2.3.2. Comparator Group for Cognitive Assessment
- WISC-IV: n = 2200 subjects (1100 males and 1100 females), aged between 6–16 years, 11 months, and 30 days, attending primary, middle and high-school.
- WPSSI-III: n = 987 subjects, aged 2 years and 6 months–7 years and 3 months, attending public school and kindergarten, proportioned to the Italian general population.
2.3.3. Motor Function Assessment
2.4. Data Analysis
3. Results
3.1. Sample Demographics, Clinical and Genetics Characteristics
3.2. Cognitive Assessment and Correlation Analyses
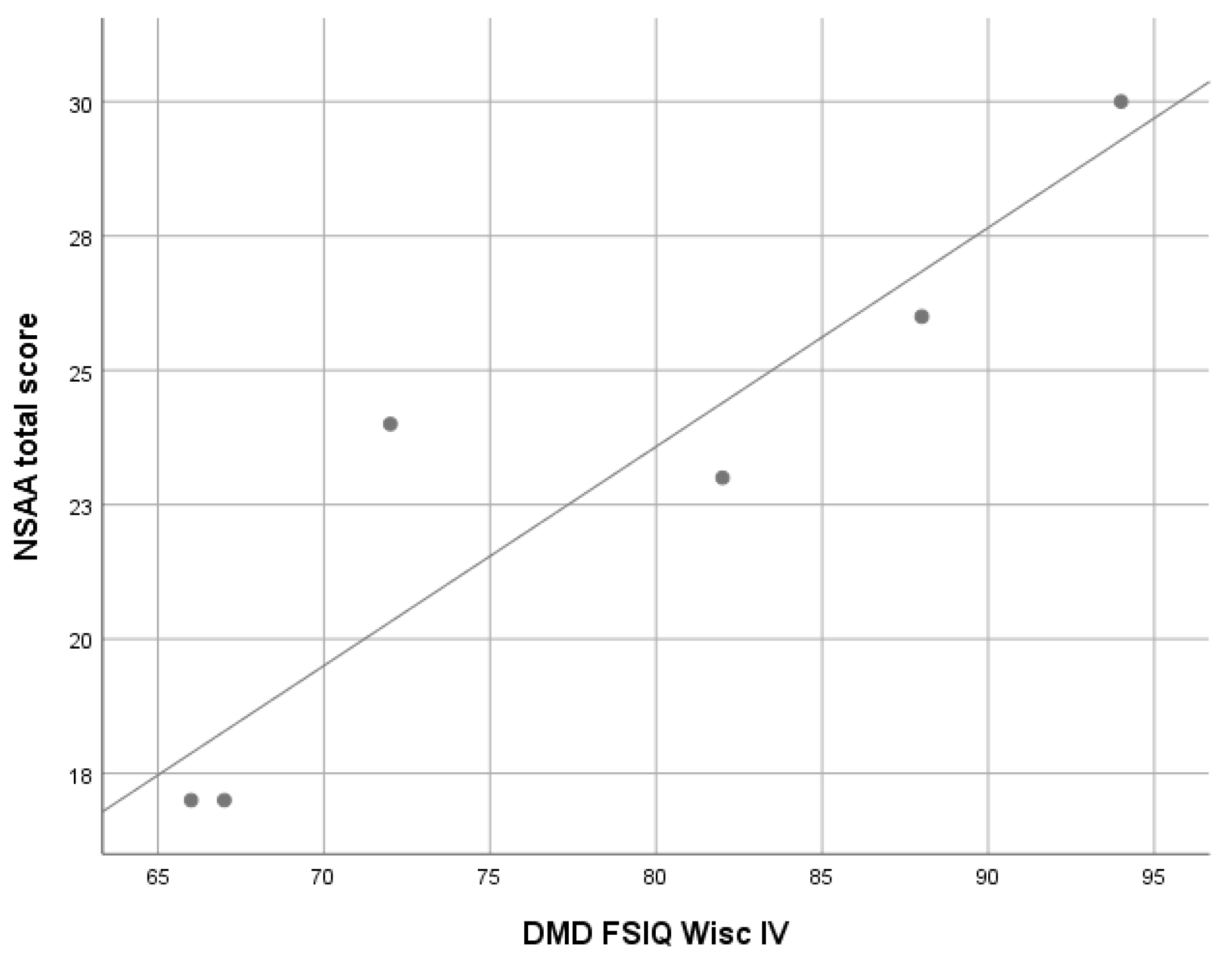
3.2.1. Duchenne Muscular Dystrophy (DMD)
3.2.2. Becker Muscular Dystrophy (BMD)
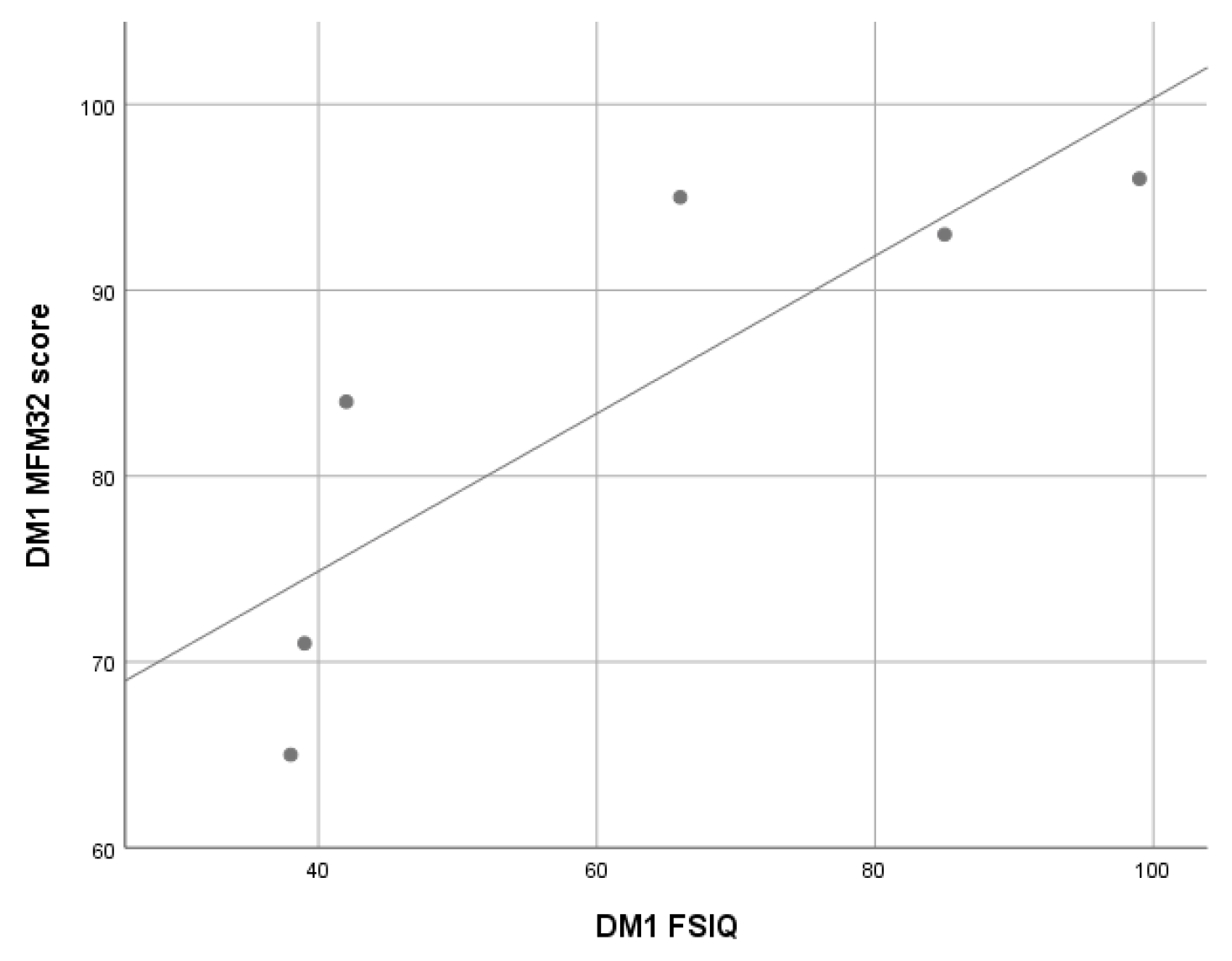
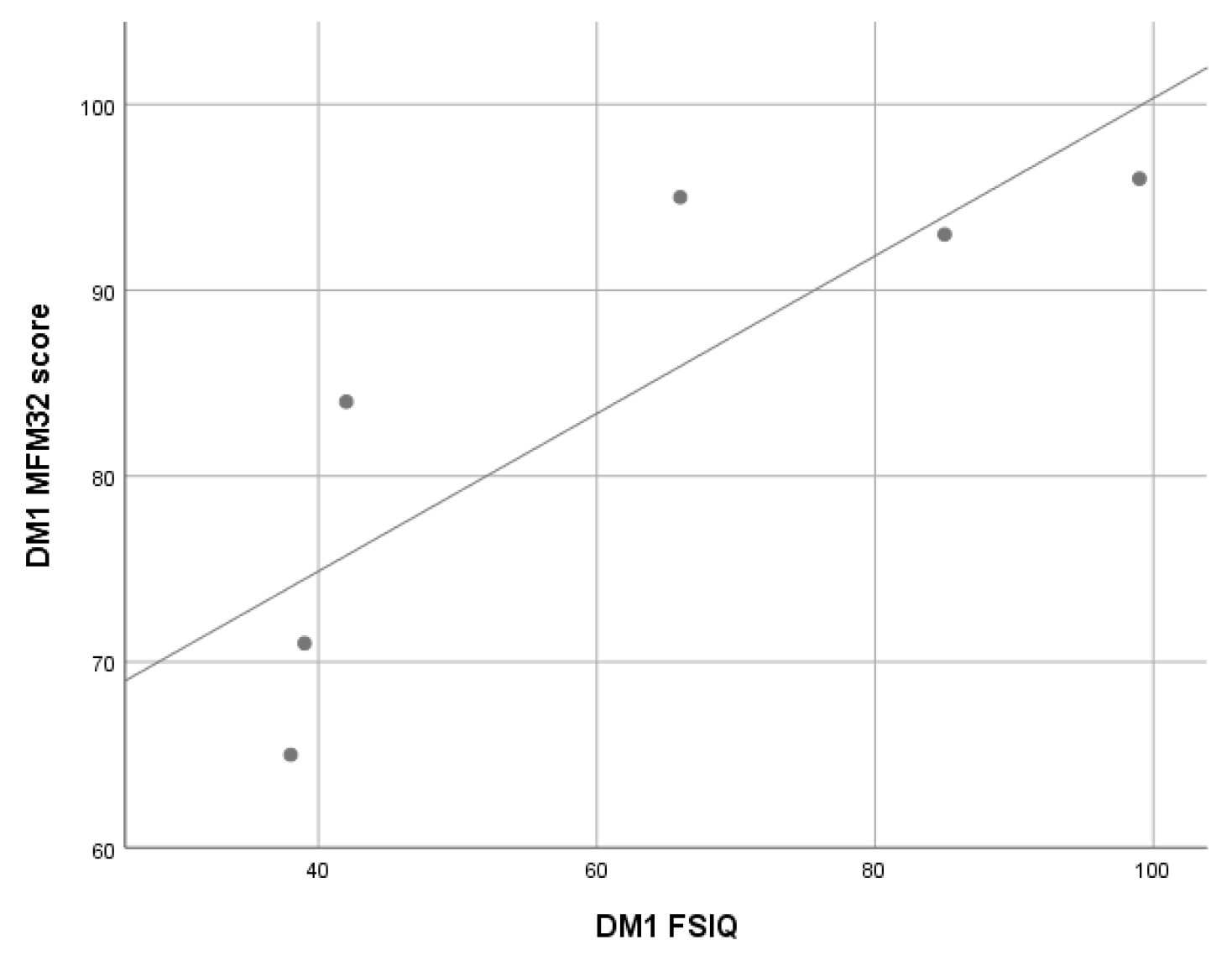
3.2.3. Myotonic Dystrophy Type 1 (DM1) Patients
3.2.4. Glycogen Storage Disease Type 2 (GSD2) Patients
3.2.5. Spinal Muscular Atrophy (SMA) Patients (Type 2 e 3)
3.2.6. Hereditary Motor Sensory Neuropathy (HMSN) Patients
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; Dealwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Corzo, D.; Nicolino, M.; Byrne, B.; Mandel, H.; Hwu, W.-L.; Leslie, N.; Levine, J.; Spencer, C.; McDonald, M.; et al. Recombinant human acid -glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology 2006, 68, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef]
- Case, L.E.; Beckemeyer, A.A.; Kishnani, P.S. Infantile Pompe disease on ERT-Update on clinical presentation, musculoskeletal management, and exercise considerations. Am. J. Med Genet. Part C Semin. Med Genet. 2012, 160, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Neurodevelopmental, Emotional, and Behavioural Problems in Duchenne Muscular Dystrophy in Relation to Underlying Dystrophin Gene Mutations—PubMed. Available online: https://pubmed-ncbi-nlm-nih-gov.bibliopass.unito.it/26365034/ (accessed on 19 July 2021).
- D’Angelo, M.G.; Bresolin, N. Cognitive impairment in neuromuscular disorders. Muscle Nerve 2006, 34, 16–33. [Google Scholar] [CrossRef]
- Orsini, M.; Ferreira, A.C.A.D.F.; De Assis, A.C.D.; Magalhães, T.; Teixeira, S.; Bastos, V.H.; Marinho, V.; Oliveira, T.; Fiorelli, R.; Oliveira, A.B.; et al. Cognitive Impairment in Neuromuscular Diseases: A Systematic Review. Neurol. Int. 2018, 10, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Ciafaloni, E.; Fox, D.J.; Pandya, S.; Westfield, C.P.; Puzhankara, S.; Romitti, P.A.; Mathews, K.D.; Miller, T.M.; Matthews, D.J.; Miller, L.A.; et al. Delayed Diagnosis in Duchenne Muscular Dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J. Pediatr. 2009, 155, 380–385. [Google Scholar] [CrossRef] [Green Version]
- Cotton, S.; Voudouris, N.J.; Greenwood, K.M. Intelligence and Duchenne muscular dystrophy: Full-Scale, Verbal, and Performance intelligence quotients. Dev. Med. Child Neurol. 2001, 43, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Ricotti, V.; Roberts, R.G.; Muntoni, F. Dystrophin and the brain. Dev. Med. Child Neurol. 2010, 53, 12. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.G.; Lorusso, M.L.; Civati, F.; Comi, G.; Magri, F.; Del Bo, R.; Guglieri, M.; Molteni, M.; Turconi, A.C.; Bresolin, N. Neurocognitive Profiles in Duchenne Muscular Dystrophy and Gene Mutation Site. Pediatr. Neurol. 2011, 45, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and Mutations: One Gene, Several Proteins, Multiple Phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Felisari, G.; Boneschi, F.M.; Bardoni, A.; Sironi, M.; Comi, G.; Robotti, M.; Turconi, A.C.; Lai, M.; Corrao, G.; Bresolin, N. Loss of Dp140 dystrophin isoform and intellectual impairment in Duchenne dystrophy. Neurology 2000, 55, 559–564. [Google Scholar] [CrossRef]
- Chieffo, D.; Brogna, C.; Berardinelli, A.; D’Angelo, G.; Mallardi, M.; D’Amico, A.; Alfieri, P.; Mercuri, E.; Pane, M. Early Neurodevelopmental Findings Predict School Age Cognitive Abilities in Duchenne Muscular Dystrophy: A Longitudinal Study. PLoS ONE 2015, 10, e0133214. [Google Scholar] [CrossRef] [Green Version]
- Doorenweerd, N.; Straathof, C.S.; Dumas, E.M.; Spitali, P.; Ginjaar, I.B.; Wokke, B.H.; Schrans, D.G.; Bergen, J.C.V.D.; van Zwet, E.W.; Webb, A.; et al. Reduced cerebral gray matter and altered white matter in boys with Duchenne muscular dystrophy. Ann. Neurol. 2014, 76, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Young, H.K.; Barton, B.A.; Waisbren, S.; Dale, L.P.; Ryan, M.M.; Webster, R.I.; North, K.N. Cognitive and Psychological Profile of Males With Becker Muscular Dystrophy. J. Child Neurol. 2007, 23, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.G.; A Wahl, R. Duchenne and Becker muscular dystrophy in adolescents: Current perspectives. Adolesc. Heal. Med. Ther. 2018, 9, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douniol, M.; Jacquette, A.; Cohen, D.; Bodeau, N.; Rachidi, L.; Angeard, N.; Cuisset, J.-M.; Vallée, L.; Eymard, B.; Plaza, M.; et al. Psychiatric and cognitive phenotype of childhood myotonic dystrophy type 1. Dev. Med. Child Neurol. 2012, 54, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Pinzan, E. Advances in imaging of brain abnormalities in neuromuscular disease. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419845567. [Google Scholar] [CrossRef]
- Steyaert, J.; Umans, S.; Willekens, D.; Legius, E.; Pijkels, E.; de Die-Smulders, C.; Berghe, H.V.D.; Fryns, J.-P. A study of the cognitive and psychological profile in 16 children with congenital or juvenile myotonic dystrophy. Clin. Genet. 2008, 52, 135–141. [Google Scholar] [CrossRef]
- Angeard, N.; Jacquette, A.; Gargiulo, M.; Radvanyi, H.; Moutier, S.; Eymard, B.; Héron, D. A new window on neurocognitive dysfunction in the childhood form of myotonic dystrophy type 1 (DM1). Neuromuscul. Disord. 2011, 21, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Filli, L.; Schwegler, S.; Meyer, C.; Killeen, T.; Easthope, C.S.; Broicher, S.D.; Curt, A.; Zörner, B.; Bolliger, M.; Jung, H.H.; et al. Characterizing cognitive-motor impairments in patients with myotonic dystrophy type 1. Neuromuscul. Disord. 2020, 30, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Modoni, A.; Silvestri, G.; Pomponi, M.G.; Mangiola, F.; Tonali, P.A.; Marra, C. Characterization of the Pattern of Cognitive Impairment in Myotonic Dystrophy Type 1. Arch. Neurol. 2004, 61, 1943–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winblad, S.; Lindberg, C.; Hansen, S. Cognitive deficits and CTG repeat expansion size in classical myotonic dystrophy type 1 (DM1). Behav. Brain Funct. 2006, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Palmer, B.; Boone, K.B.; Chang, L.; Lee, A.; Black, S. Cognitive deficits and personality patterns in maternally versus paternally inherited myotonic dystrophy. J. Clin. Exp. Neuropsychol. 1994, 16, 784–795. [Google Scholar] [CrossRef]
- Censori, B.; Danni, M.; Del Pesce, M.; Provinciali, L. Neuropsychological profile in myotonic dystrophy. J. Neurol. 1990, 237, 251–256. [Google Scholar] [CrossRef]
- Spiridigliozzi, G.A.; Heller, J.H.; Kishnani, P.S.; Van der Ploeg, A.T.; Ebbink, B.J.; Aarsen, F.K.; van Gelder, C.M.; Hout, J.M.P.V.D. Cognitive outcome of patients with classic infantile Pompe disease receiving enzyme therapy. Neurology 2013, 80, 1173. [Google Scholar] [CrossRef] [Green Version]
- Korlimarla, A.; Spiridigliozzi, G.A.; Crisp, K.; Herbert, M.; Chen, S.; Malinzak, M.; Stefanescu, M.; Austin, S.L.; Cope, H.; Zimmerman, K.; et al. Novel Approaches to Quantify CNS Involvement in Children with Pompe Disease. Neurology 2020, 95, e718–e732. [Google Scholar] [CrossRef]
- Spiridigliozzi, G.A.; Keeling, L.A.; Stefanescu, M.; Li, C.; Austin, S.; Kishnani, P.S. Cognitive and Academic Outcomes in Long-Term Survivors of Infantile-Onset Pompe Disease: A Longitudinal Follow-Up. Mol. Genet. Metab. 2017, 121, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Von Gontard, A.; Zerres, K.; Backes, M.; Laufersweiler-Plass, C.; Wendland, C.; Melchers, P.; Lehmkuhl, G.; Rudnik-Schöneborn, S. Intelligence and cognitive function in children and adolescents with spinal muscular atrophy. Neuromuscul. Disord. 2002, 12, 130–136. [Google Scholar] [CrossRef]
- Masson, R.; Brusa, C.; Scoto, M.; Baranello, G. Brain, cognition, and language development in spinal muscular atrophy type 1: A scoping review. Dev. Med. Child Neurol. 2021, 63, 527–536. [Google Scholar] [CrossRef]
- Polido, G.J.; De Miranda, M.M.V.; Junior, N.C.; Mendonça, R.D.H.; Caromano, F.A.; Reed, U.C.; Zanoteli, E.; Voos, M.C.; Carvas, N. Cognitive performance of children with spinal muscular atrophy: A systematic review. Dement. Neuropsychol. 2019, 13, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Polido, G.J.; Barbosa, A.F.; Morimoto, C.H.; Caromano, F.A.; Favero, F.M.; Zanoteli, E.; Reed, U.C.; Voos, M.C. Matching pairs difficulty in children with spinal muscular atrophy type I. Neuromuscul. Disord. 2017, 27, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Chanson, J.-B.; Echaniz-Laguna, A.; Blanc, F.; Lacour, A.; Ballonzoli, L.; Kremer, S.; Namer, I.J.; Lannes, B.; Tranchant, C.; Vermersch, P.; et al. Central nervous system abnormalities in patients with PMP22 gene mutations: A prospective study. J. Neurol. Neurosurg. Psychiatry 2012, 84, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Ferri, R.; Orsini, A.; Rea, M. Bayley: Scales of Infant and Toddler Development-Terza Edizione; Contributo alla taratura italiana; Giunti O.S.: Firenze, Italy, 2015. [Google Scholar]
- Orsini, Pezzuti & Picone. WISC-IV: Contributo alla Taratura Italiana. [WISC-IV Italian Edition]; Giunti O.S.: Florence, Italy, 2012. [Google Scholar]
- Desguerre, I.; Christov, C.; Mayer, M.; Zeller, R.; Becane, H.-M.; Bastuji-Garin, S.; Leturcq, F.; Chiron, C.; Chelly, J.; Gherardi, R.K. Clinical Heterogeneity of Duchenne Muscular Dystrophy (DMD): Definition of Sub-Phenotypes and Predictive Criteria by Long-Term Follow-Up. PLoS ONE 2009, 4, e4347. [Google Scholar] [CrossRef] [Green Version]
- Bushby, K.M.D.; Appleton, R.; Anderson, L.V.B.; Welch, J.L.; Kelly, P.; Gardner-Medwin, D. Deletion status and intellectual impairment in duchenne muscular dystrophy. Dev. Med. Child Neurol. 1995, 37, 260–269. [Google Scholar] [CrossRef]
- Specific Profiles of Neurocognitive and Reading Functions in a Sample of 42 Italian Boys with Duchenne Muscular Dystrophy—PubMed. Available online: https://pubmed-ncbi-nlm-nih-gov.bibliopass.unito.it/22385039/ (accessed on 19 July 2021).
- Bird, T.D.; Follett, C.; Griep, E. Cognitive and personality function in myotonic muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1983, 46, 971–980. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Variables | DMD (n = 15) | BMD (n = 4) | DM1 (n = 8) | GSD2 (n = 6) | SMA Type 2 or 3 (n = 3) | HMSN (n = 7) |
|---|---|---|---|---|---|---|
| Males, n (%) | 15.0 (100) | 4.0 (100) | 4.0 (50.0) | 4.0 (67.0) | 2.0 (67.0) | 4.0 (57.0) |
| Age (years), median (IQR or range) | ||||||
| at symptoms onset | 4.0 (2.0) | * | 0.0 (8.0) | ** | 0.5 (0.3–2.0) | 7.0 (5.5) |
| at diagnosis | 4.5 (1.5) | 7.1 (6.7–7.6) | 0.2 (0.3) | (EO) 1.5 month (LO) 4.3 years | *** | 7.1 (7.5) |
| at assessment | 6.7 (4.3) | 12.1 (6.1) | 12.9 (6.6) | 8.4 (13.0) | 10.7 (1.0) | 11.5 (5.8) |
| Family history of NMDs, n (%) | 9.0 (60.0) | 3.0 (100) | - | - | - | 6.0 (100) |
| Resuscitation at birth, n (%) | 1.0 (7.0) | 1 (25.0) | 2.0 (25.0) | 1.0 (20.0) | 0.0 (0.0) | 1.0 (14.0) |
| Hypotonia at birth, n (%) | 0.0 (0.0) | 0.0 (0.0) | 5.0 (71.0) | 1.0 (25.0) | 0.0 (0.0) | 0.0 (0.0) |
| Walking delay, n (%) | 5.0 (33.0) | 1.0 (25.0) | 2.0 (40.0) | 0.0 (0.0) | **** | 0.0 (0.0) |
| Speech delay, n (%) | 8.0 (53.0) | 0.0 (0.0) | 5.0 (71.0) | 2.0 (33.0) | 0.0 (0.0) | 1.0 (14.0) |
| Gestational Age | ||||||
| Preterm, n (%) | 2.0 (14.0) | 0.0 (0.0) | 0.0 (0.0) | 2.0 (40.0) | 1.0 (33.0) | 1.0 (14.0) |
| Full term, n (%) | 12.0 (86.0) | 4.0 (100) | 7.0 (100) | 3.0 (60.0) | 2.0 (67.0) | 6.0 (86.0) |
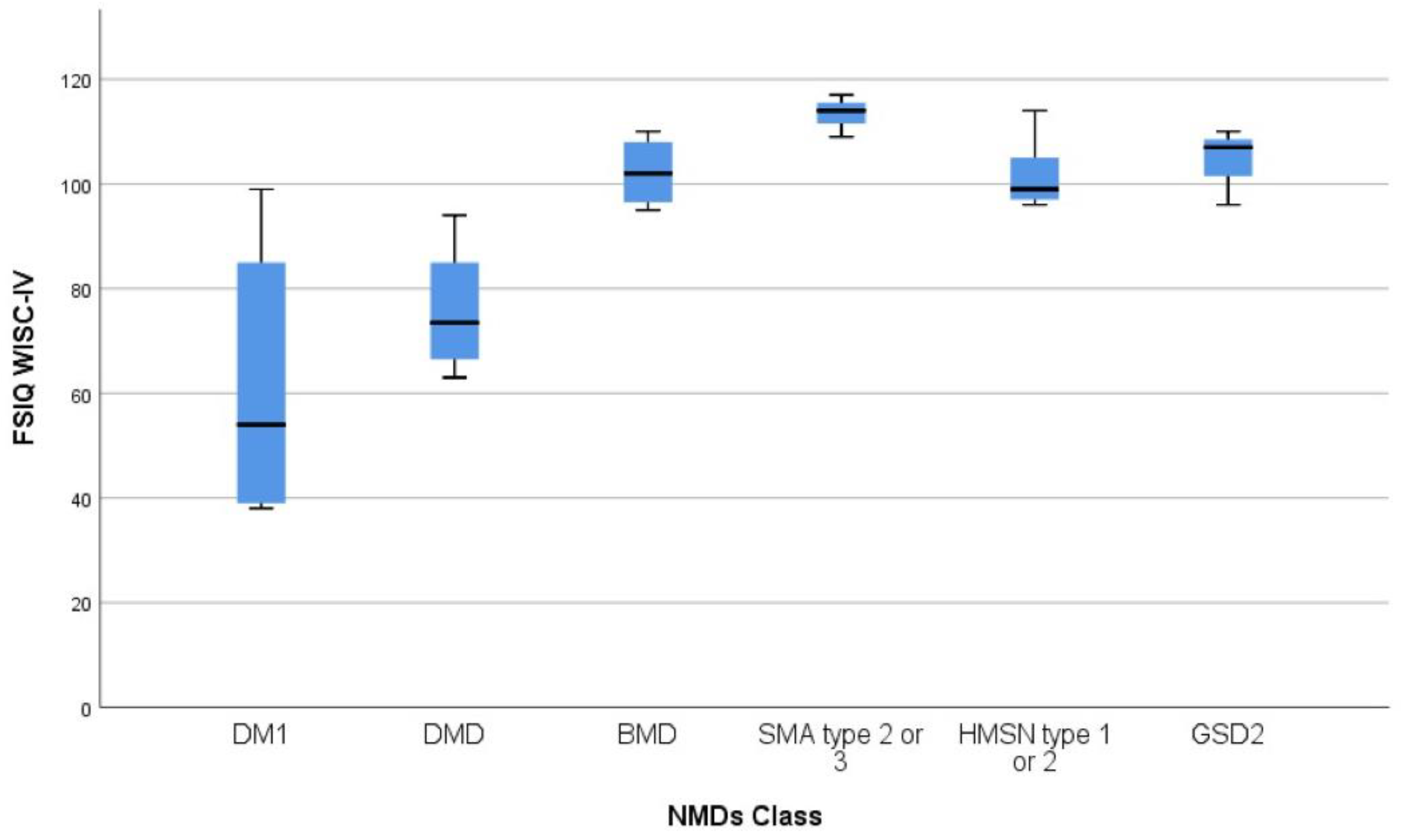
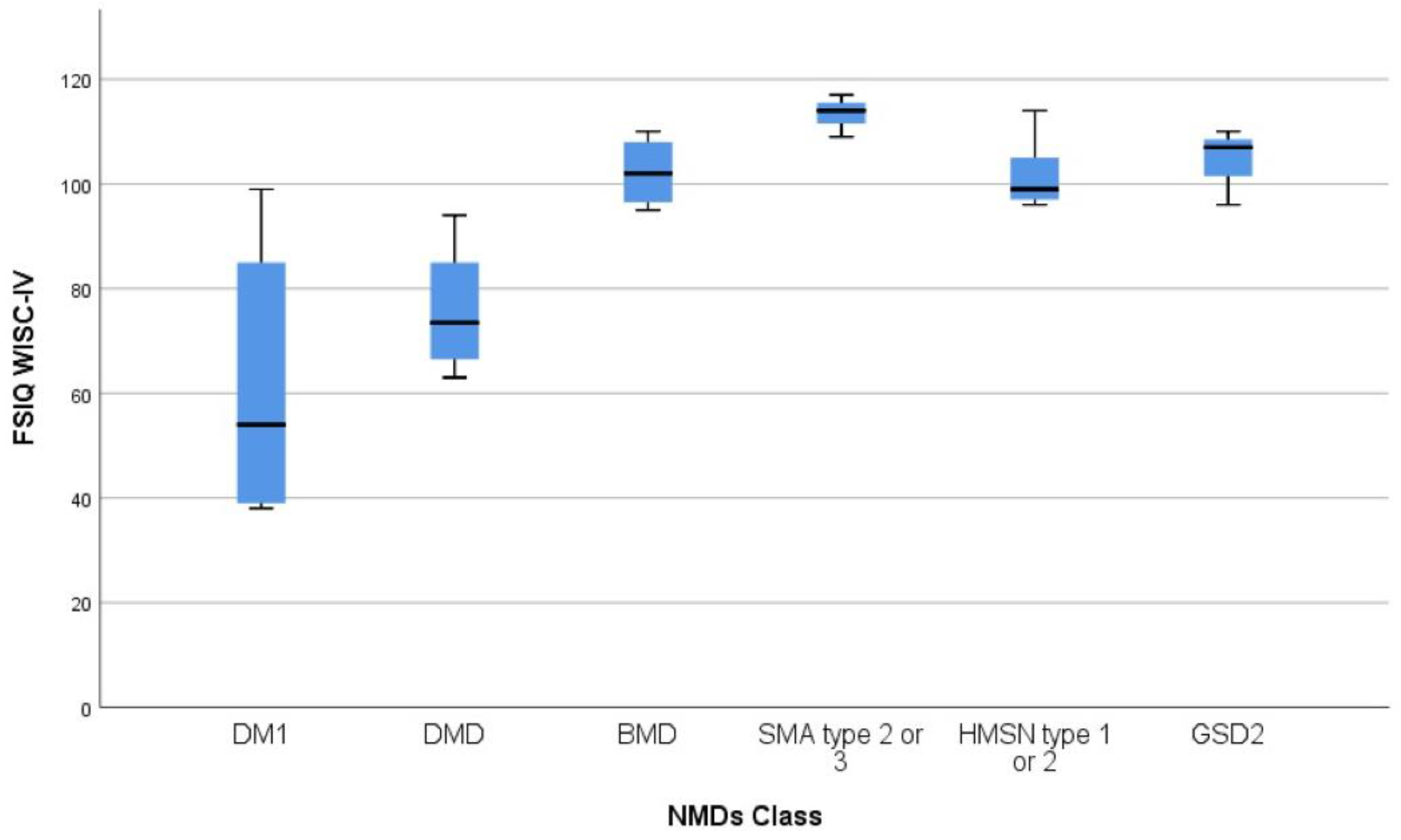
| NMD Diagnosis | WISC-IV Scales | |||||
|---|---|---|---|---|---|---|
| VCI Mean (SD) | PRI Mean (SD) | WMI Mean (SD) | PSI Mean (SD) | FSIQ Mean (SD) Range | ||
| DMD (n = 8) | 78.7 (7.7) | 82.9 (8.4) | 80.9 (9.7) | 82.2 (14.2) | 75.9 (SD 11.1) | 63–94 |
| BMD (n = 4) | 100.5 (8.7) | 105.2 (19.0) | 109.0 (7.3) | 91.0 (2.4) | 102.2 (6.9) | 95–110 |
| DM1 (n = 6) | 73.7 (19.2) | 67. 3 (27.8) | 70.0 (27.3) | 66.7 (15.8) | 61.5 (26.1) | 38–99 |
| GSD2 (n = 4) | 104.7 (8.1) | 110.3 (12.0) | 95.0 (4.6) | 100.0 (5.2) | 105 (6.2) | 96–110 |
| SMA (n = 3) | 119.3 (4.1) | 110.0 (9.5) | 108.0 (6.9) | 98.0 (1.7) | 113.3 (4.0) | 109–117 |
| HMSN (n = 6) | 99.7 (5.0) | 104.7 (7.6) | 102.0 (13.9) | 98.0 (7.0) | 101.7 (6.8) | 96–114 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Alessandro, R.; Ragusa, N.; Vacchetti, M.; Rolle, E.; Rossi, F.; Brusa, C.; Davico, C.; Vitiello, B.; Mongini, T.; Ricci, F.S. Assessing Cognitive Function in Neuromuscular Diseases: A Pilot Study in a Sample of Children and Adolescents. J. Clin. Med. 2021, 10, 4777. https://doi.org/10.3390/jcm10204777
D’Alessandro R, Ragusa N, Vacchetti M, Rolle E, Rossi F, Brusa C, Davico C, Vitiello B, Mongini T, Ricci FS. Assessing Cognitive Function in Neuromuscular Diseases: A Pilot Study in a Sample of Children and Adolescents. Journal of Clinical Medicine. 2021; 10(20):4777. https://doi.org/10.3390/jcm10204777
Chicago/Turabian StyleD’Alessandro, Rossella, Neftj Ragusa, Martina Vacchetti, Enrica Rolle, Francesca Rossi, Chiara Brusa, Chiara Davico, Benedetto Vitiello, Tiziana Mongini, and Federica S. Ricci. 2021. "Assessing Cognitive Function in Neuromuscular Diseases: A Pilot Study in a Sample of Children and Adolescents" Journal of Clinical Medicine 10, no. 20: 4777. https://doi.org/10.3390/jcm10204777
APA StyleD’Alessandro, R., Ragusa, N., Vacchetti, M., Rolle, E., Rossi, F., Brusa, C., Davico, C., Vitiello, B., Mongini, T., & Ricci, F. S. (2021). Assessing Cognitive Function in Neuromuscular Diseases: A Pilot Study in a Sample of Children and Adolescents. Journal of Clinical Medicine, 10(20), 4777. https://doi.org/10.3390/jcm10204777