Inhibition of MLKL Attenuates Necroptotic Cell Death in a Murine Cell Model of Hepatic Ischaemia Injury


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Free Fatty Acid Treatment
2.3. Oxygen Glucose Deprivation (OGD) Treatment
2.4. Drug Administration
2.5. Cell Viability Assay
2.6. Oil Red-O Sstaining and Quantification
2.7. Quantitative Real-Time PCR
2.8. Western Blot Assay
2.9. Antibodies
2.10. Flow Cytometry
2.11. Knockdown of MLKL and RIPK3 Expression Using Transient Transfection of MLKL siRNA and RIPK3 siRNA Respectively
2.12. Enzyme-Linked Immunosorbent Assays and Lactate Dehydrogenase Assay
2.13. Statistical Analysis
3. Results
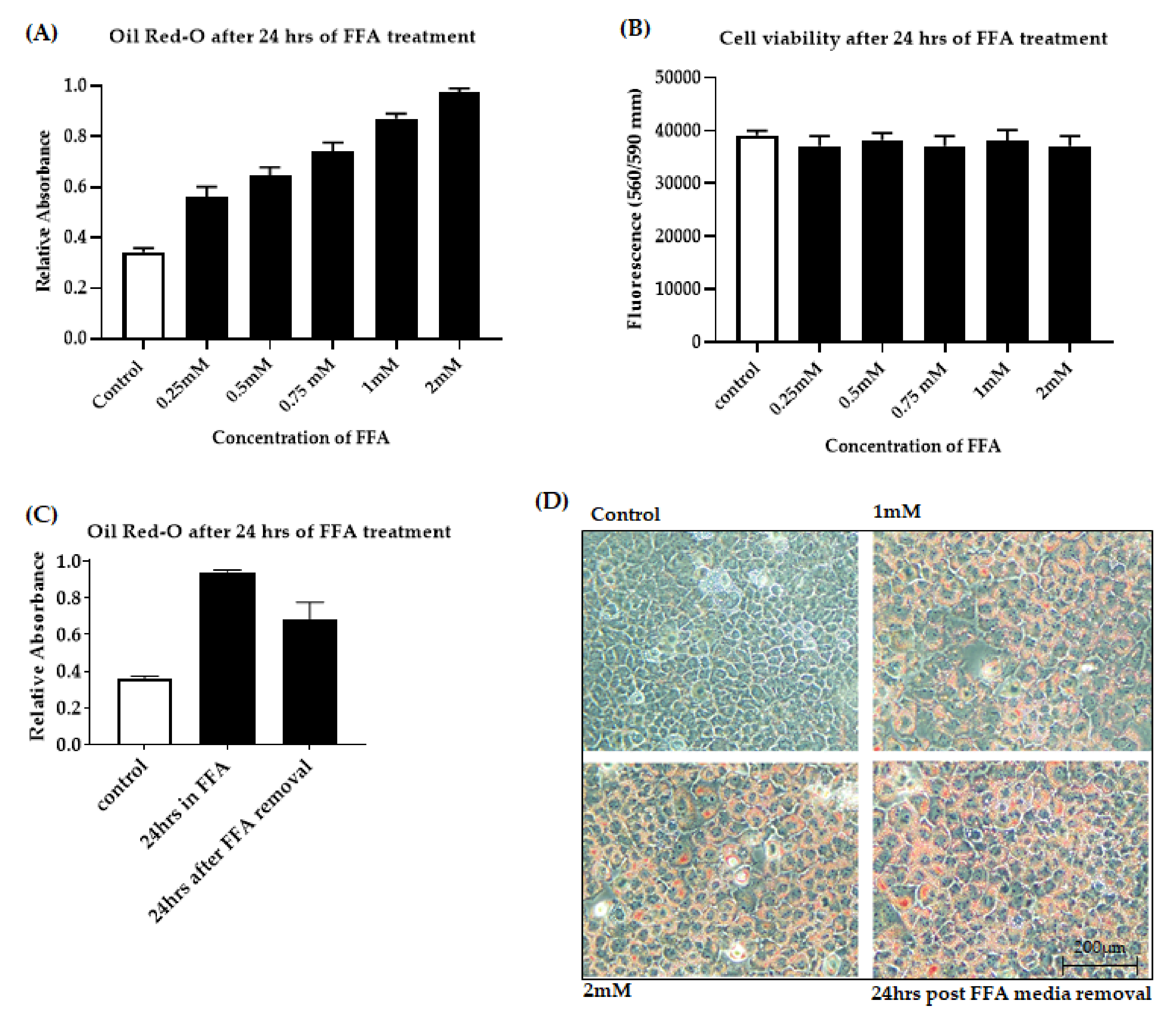
3.1. Development of an In Vitro Model of Fatty Liver Undergoing Ischaemic Injury
3.1.1. Optimization of FFA Treatment in AML-12 Cells
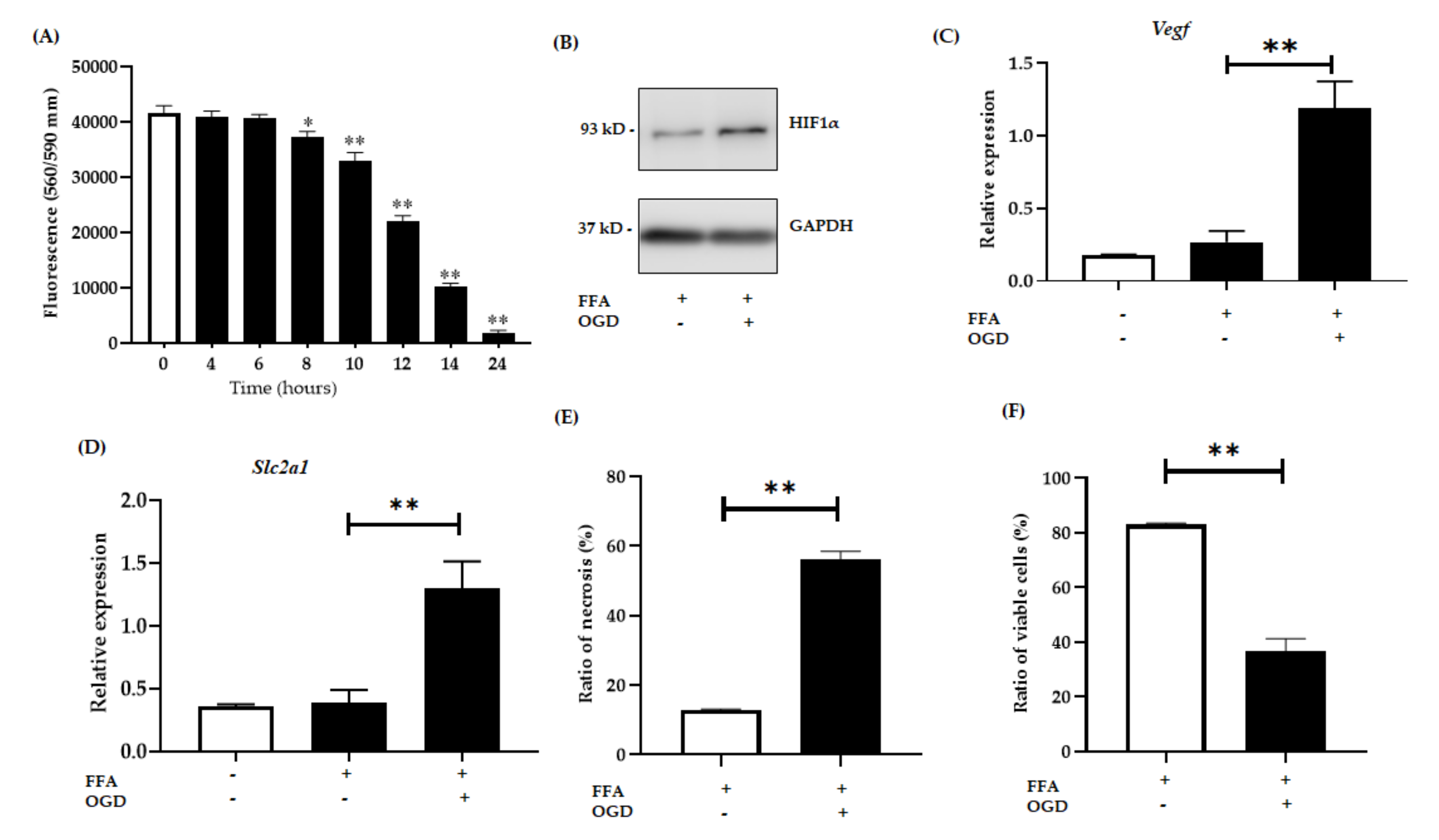
3.1.2. OGD Treatment Decreases Cell Viability in an In Vitro Model of Steatosis
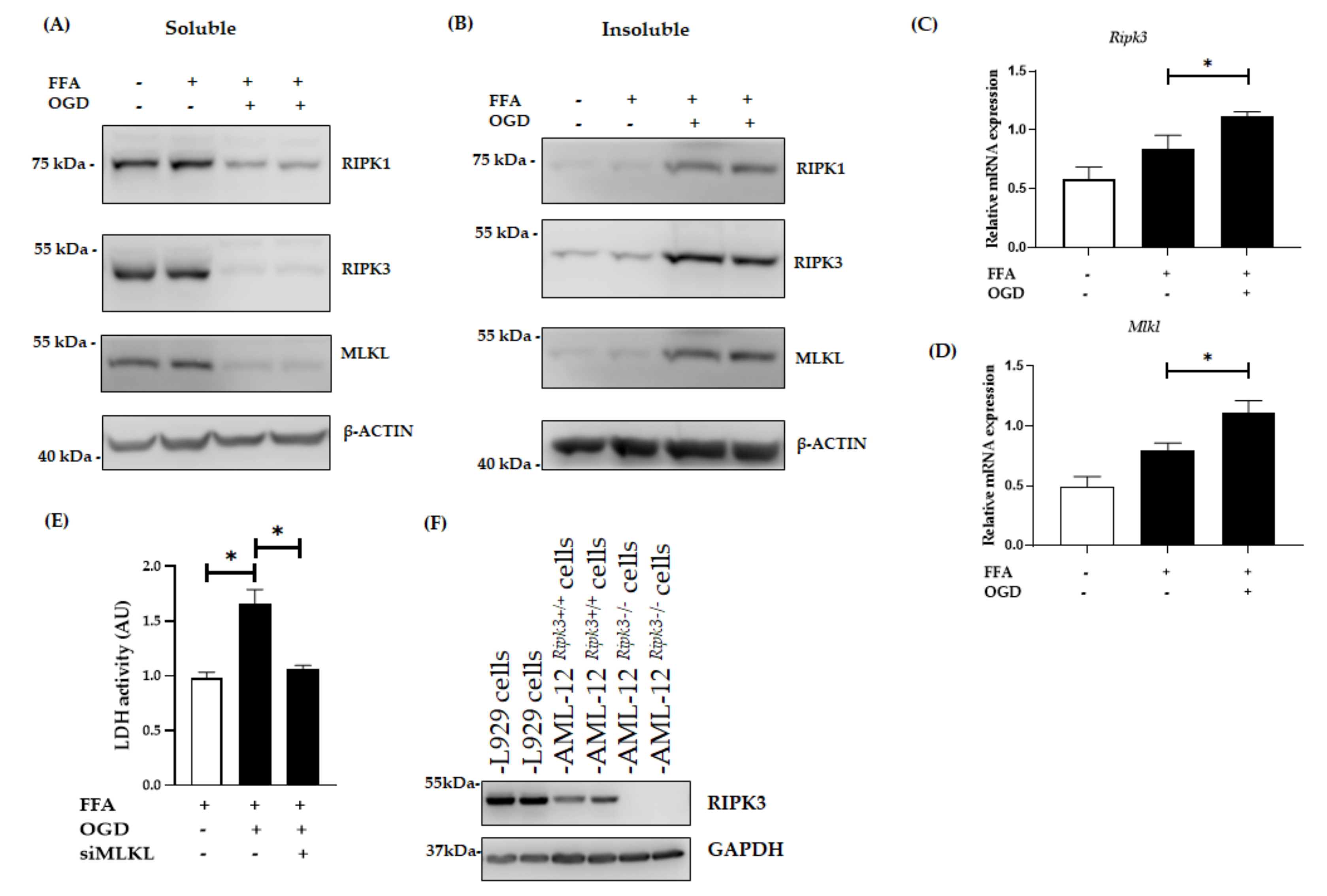
3.2. OGD Treatment Elevated the Insoluble Fraction of RIPK1, RIPK3 and MLKL Protein in FFA Treated AML-12 Cells
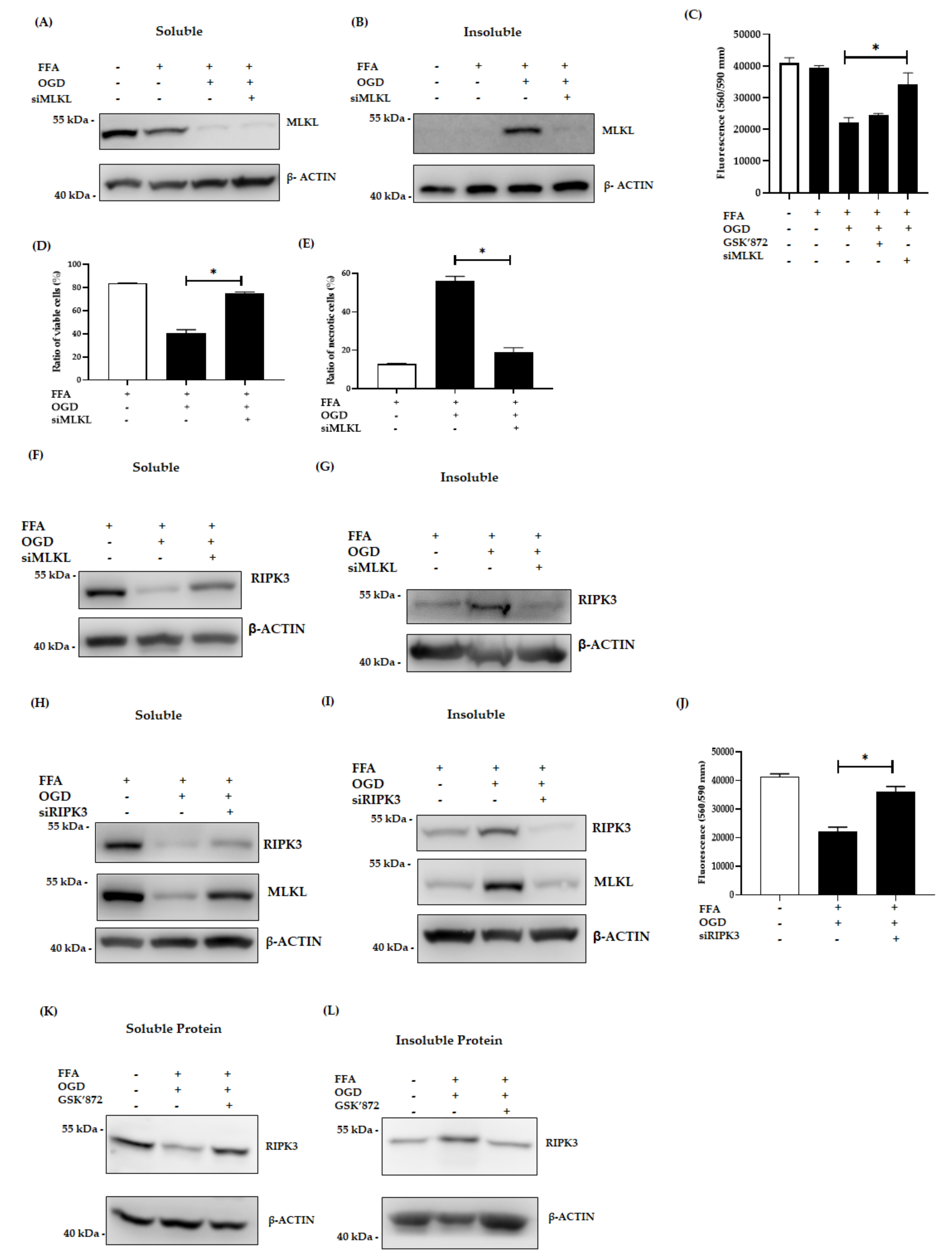
3.3. Inhibition of MLKL, and RIPK3 Attenuated the Necroptotic Cell Death Induced by OGD
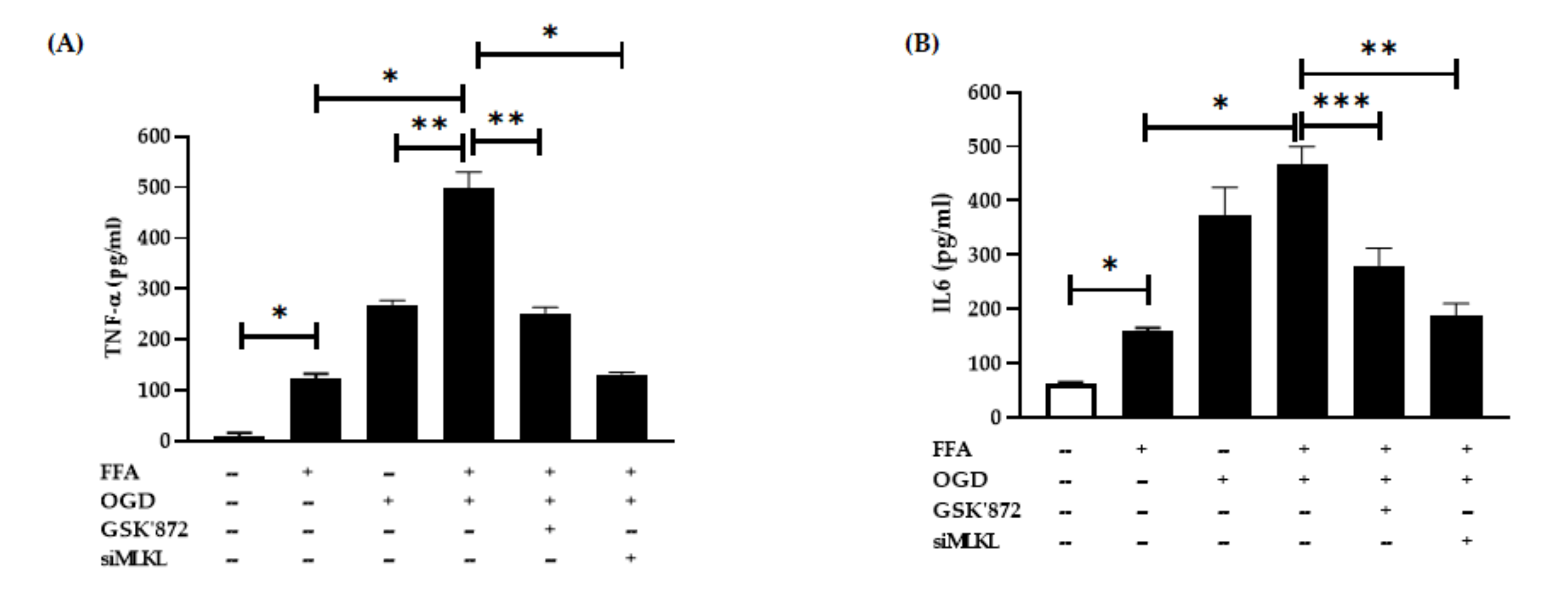
3.4. Changes in Pro-Inflammatory Cytokine Secretion after FFA and OGD Treatment in AML-12 Cells
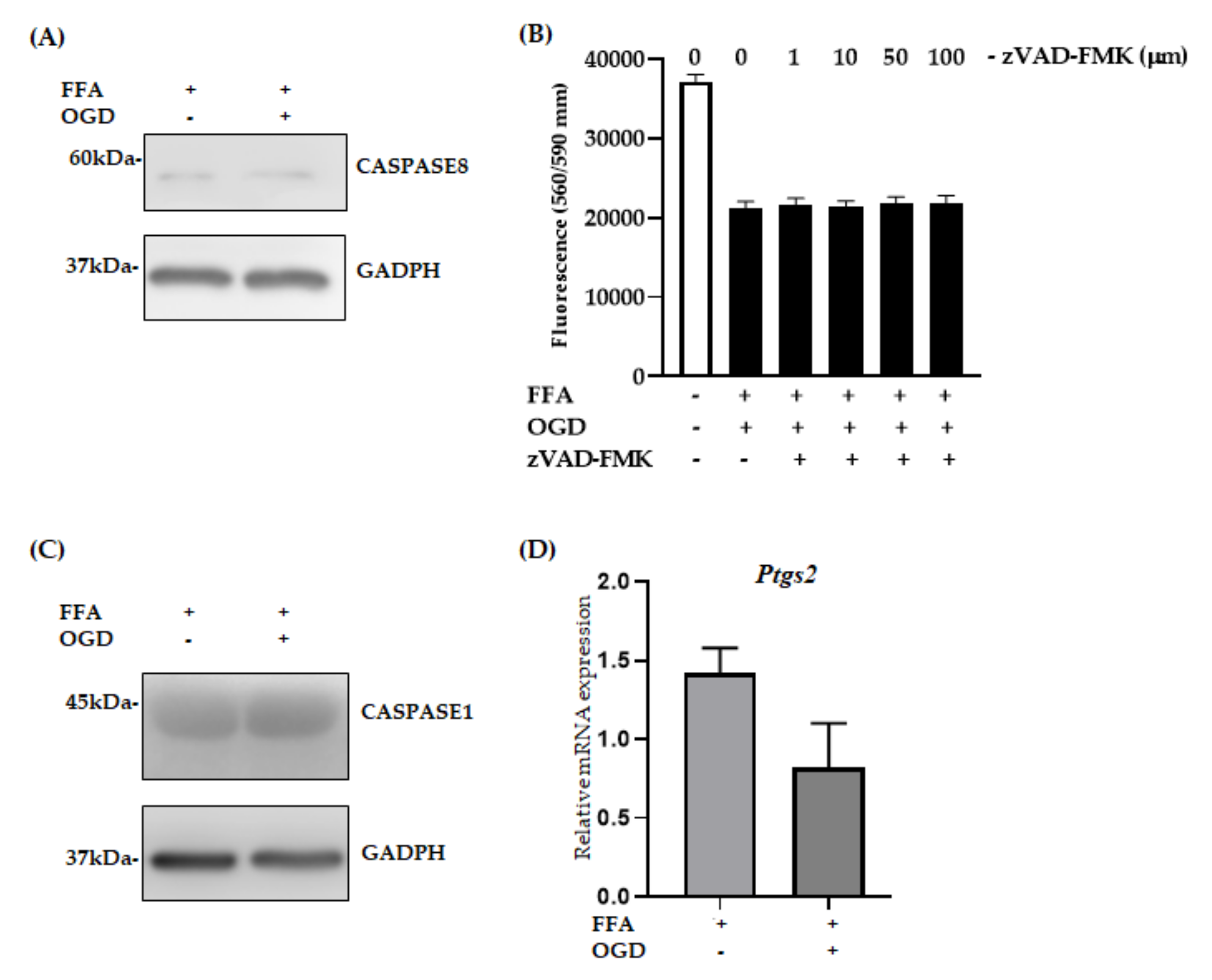
3.5. Apoptosis, Pyroptosis and Ferroptosis Were Not Active Pathways in FFA + OGD Exposed AML-12 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sherif, Z.A.; Saeed, A.; Ghavimi, S.; Nouraie, S.M.; Laiyemo, A.O.; Brim, H.; Ashktorab, H. Global Epidemiology of Nonalcoholic Fatty Liver Disease and Perspectives on US Minority Populations. Dig. Dis. Sci. 2016, 61, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.; Busuttil, R.W. AASLD/ILTS transplant course: Is there an extended donor suitable for everyone? Liver Transplant. 2005, S2–S5. [Google Scholar] [CrossRef] [PubMed]
- Abouna, G.M. Organ shortage crisis: Problems and possible solutions. Transplant. Proc. 2008, 40, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Briceno, J.; Ciria, R.; de la Mata, M.; Rufian, S.; Lopez-Cillero, P. Prediction of graft dysfunction based on extended criteria donors in the model for end-stage liver disease score era. Transplantation 2010, 90, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Busuttil, R.W.; Tanaka, K. The utility of marginal donors in liver transplantation. Liver Transplant. 2003, 9, 651–663. [Google Scholar] [CrossRef]
- D’Alessandro, A.M.; Kalayoglu, M.; Sollinger, H.W.; Hoffmann, R.M.; Reed, A.; Knechtle, S.J.; Pirsch, J.D.; Hafez, G.R.; Lorentzen, D.; Belzer, F.O. The predictive value of donor liver biopsies for the development of primary nonfunction after orthotopic liver transplantation. Transplantation 1991, 51, 157–163. [Google Scholar] [CrossRef]
- Loinaz, C.; Gonzalez, E.M. Marginal donors in liver transplantation. Hepatogastroenterology 2000, 47, 256–263. [Google Scholar]
- Rinella, M.E.; Alonso, E.; Rao, S.; Whitington, P.; Fryer, J.; Abecassis, M.; Superina, R.; Flamm, S.L.; Blei, A.T. Body mass index as a predictor of hepatic steatosis in living liver donors. Liver Transplant. 2001, 7, 409–414. [Google Scholar] [CrossRef]
- Shamsaeefar, A.; Nikeghbalian, S.; Kazemi, K.; Mansorian, M.; Gholami, S.; Motazedian, N.; Malekhosseini, S.A. Discarded organs at Shiraz Transplant Center. Exp. Clin. Transplant. 2014, 12 (Suppl. 1), 178–181. [Google Scholar]
- Escartin, A.; Castro, E.; Dopazo, C.; Bueno, J.; Bilbao, I.; Margarit, C. Analysis of discarded livers for transplantation. Transplant. Proc. 2005, 37, 3859–3860. [Google Scholar] [CrossRef]
- Angelico, M. Donor liver steatosis and graft selection for liver transplantation: A short review. Eur. Rev. Med. Pharmacol. Sci. 2005, 9, 295–297. [Google Scholar] [PubMed]
- Chavin, K.D.; Yang, S.; Lin, H.Z.; Chatham, J.; Chacko, V.P.; Hoek, J.B.; Walajtys-Rode, E.; Rashid, A.; Chen, C.H.; Huang, C.C.; et al. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J. Biol. Chem. 1999, 274, 5692–5700. [Google Scholar] [CrossRef] [PubMed]
- Verran, D.; Kusyk, T.; Painter, D.; Fisher, J.; Koorey, D.; Strasser, S.; Stewart, G.; McCaughan, G. Clinical experience gained from the use of 120 steatotic donor livers for orthotopic liver transplantation. Liver Transplant. 2003, 9, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Carvalho, T.; Caridade, M.; Borralho, P.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M.P. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 2015. [Google Scholar] [CrossRef]
- Liss, K.H.H.; McCommis, K.S.; Chambers, K.T.; Pietka, T.A.; Schweitzer, G.G.; Park, S.L.; Nalbantoglu, I.; Weinheimer, C.J.; Hall, A.M.; Finck, B.N. The impact of diet-induced hepatic steatosis in a murine model of hepatic ischemia/reperfusion injury. Liver Transplant. 2018, 24, 908–921. [Google Scholar] [CrossRef]
- Ni, H.-M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.-H.; Li, T.; Ding, W.-X.; Jaeschke, H. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)—Mixed Lineage Kinase Domain-Like Protein (MLKL)—Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am. J. Pathol. 2019, 189, 1363–1374. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De Zen, F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Karasawa, T.; Wakiya, T.; Sadatomo, A.; Ito, H.; Kamata, R.; Watanabe, S.; Komada, T.; Kimura, H.; Sanada, Y.; et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am. J. Transplant. 2020, 20, 1606–1618. [Google Scholar] [CrossRef]
- Nagarajan, S.R.; Paul-Heng, M.; Krycer, J.R.; Fazakerley, D.J.; Sharland, A.F.; Hoy, A.J. Lipid and glucose metabolism in hepatocyte cell lines and primary mouse hepatocytes: A comprehensive resource for in vitro studies of hepatic metabolism. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E578–E589. [Google Scholar] [CrossRef]
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachik, J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin. Sci. (Lond.) 2004, 106, 635–643. [Google Scholar] [CrossRef]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Niklas, J.; Bonin, A.; Mangin, S.; Bucher, J.; Kopacz, S.; Matz-Soja, M.; Thiel, C.; Gebhardt, R.; Hofmann, U.; Mauch, K. Central energy metabolism remains robust in acute steatotic hepatocytes challenged by a high free fatty acid load. BMB Rep. 2012, 45, 396–401. [Google Scholar] [CrossRef]
- Moravcova, A.; Cervinkova, Z.; Kucera, O.; Mezera, V.; Rychtrmoc, D.; Lotkova, H. The effect of oleic and palmitic acid on induction of steatosis and cytotoxicity on rat hepatocytes in primary culture. Physiol. Res. 2015, 64 (Suppl. 5), S627–S636. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hu, F.; Wu, J.; Zhang, S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox Biol. 2017, 11, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-S.; Yi, T.-L.; Zhang, S.; Xu, Z.-W.; Yu, Z.-Q.; Sun, H.-T.; Yang, C.; Tu, Y.; Cheng, S.-X. Hypoxia-inducible factor-1 alpha is involved in RIP-induced necroptosis caused by in vitro and in vivo ischemic brain injury. Sci. Rep. 2017, 7, 5818. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.P.; Shi, Y.W.; Tang, M.; Zhang, X.C.; Gu, Y.; Liang, X.M.; Wang, Z.W.; Ding, F. Isoquercetin Ameliorates Cerebral Impairment in Focal Ischemia Through Anti-Oxidative, Anti-Inflammatory, and Anti-Apoptotic Effects in Primary Culture of Rat Hippocampal Neurons and Hippocampal CA1 Region of Rats. Mol. Neurobiol. 2017, 54, 2126–2142. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Oxygen homeostasis and disease pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef]
- Tong, Q.; Zheng, L.; Lin, L.; Li, B.; Wang, D.; Huang, C.; Li, D. VEGF is upregulated by hypoxia-induced mitogenic factor via the PI-3K/Akt-NF-kappaB signaling pathway. Respir. Res. 2006, 7, 37. [Google Scholar] [CrossRef][Green Version]
- Xie, T.X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar]
- Minchenko, A.; Salceda, S.; Bauer, T.; Caro, J. Hypoxia regulatory elements of the human vascular endothelial growth factor gene. Cell. Mol. Biol. Res. 1994, 40, 35–39. [Google Scholar]
- Banai, S.; Shweiki, D.; Pinson, A.; Chandra, M.; Lazarovici, G.; Keshet, E. Upregulation of vascular endothelial growth factor expression induced by myocardial ischaemia: Implications for coronary angiogenesis. Cardiovasc. Res. 1994, 28, 1176–1179. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Chan, F.K.; Shisler, J.; Bixby, J.G.; Felices, M.; Zheng, L.; Appel, M.; Orenstein, J.; Moss, B.; Lenardo, M.J. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 2003, 278, 51613–51621. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ni, H.M.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Nawabi, A.; Komatsu, M.; Huang, H.; Ding, W.X. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016, 7, 17681–17698. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Lash, L.H.; Putt, D.A.; Hueni, S.E.; Krause, R.J.; Elfarra, A.A. Roles of necrosis, Apoptosis, and mitochondrial dysfunction in S-(1,2-dichlorovinyl)-L-cysteine sulfoxide-induced cytotoxicity in primary cultures of human renal proximal tubular cells. J. Pharmacol. Exp. Ther. 2003, 305, 1163–1172. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, Z.; Zhao, N.; Zhou, L.; Liu, F.; Cichacz, Z.; Zhang, L.; Zhan, Q.; Zhao, X. Receptor interactive protein kinase 3 promotes Cisplatin-triggered necrosis in apoptosis-resistant esophageal squamous cell carcinoma cells. PLoS ONE 2014, 9, e100127. [Google Scholar] [CrossRef]
- Roychowdhury, S.; McCullough, R.L.; Sanz-Garcia, C.; Saikia, P.; Alkhouri, N.; Matloob, A.; Pollard, K.A.; McMullen, M.R.; Croniger, C.M.; Nagy, L.E. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology (Baltimore Md.) 2016, 64, 1518–1533. [Google Scholar] [CrossRef]
- Gautheron, J.; Vucur, M.; Schneider, A.T.; Severi, I.; Roderburg, C.; Roy, S.; Bartneck, M.; Schrammen, P.; Diaz, M.B.; Ehling, J.; et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat. Commun. 2016, 7, 11869. [Google Scholar] [CrossRef]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef]
- Miura, M. Active participation of cell death in development and organismal homeostasis. Dev. Growth Differ. 2011, 53, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Shang, L.; Huang, J.F.; Li, N.; Chen, D.; Xue, L.X.; Xiong, K. Receptor interacting protein 3-induced RGC-5 cell necroptosis following oxygen glucose deprivation. BMC Neurosci. 2015, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.; Suebsuwong, C.; Ray, S.S.; Thapa, R.J.; Maki, J.L.; Nogusa, S.; Shah, S.; Saleh, D.; Gough, P.J.; Bertin, J.; et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 2015, 10, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zhu, F.; Yang, C.; Wu, S.; Lin, Y.; Ma, H.; Yu, X.; Zhao, C.; Ji, Y.; Ge, W.; et al. Discovery of a Potent RIPK3 Inhibitor for the Amelioration of Necroptosis-Associated Inflammatory Injury. Front. Cell Dev. Biol. 2020, 8, 606119. [Google Scholar] [CrossRef] [PubMed]
- Pefanis, A.; Ierino, F.L.; Murphy, J.M.; Cowan, P.J. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019, 96, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Haanen, C.; Vermes, I. Apoptosis: Programmed cell death in fetal development. Eur. J. Obstet. Gynecol. Reprod. Biol. 1996, 64, 129–133. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Baidya, R.; Crawford, D.H.G.; Gautheron, J.; Wang, H.; Bridle, K.R. Necroptosis in Hepatosteatotic Ischaemia-Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 5931. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Kimura, K.; Shirabe, K.; Yoshizumi, T.; Takeishi, K.; Itoh, S.; Harimoto, N.; Ikegami, T.; Uchiyama, H.; Okano, S.; Maehara, Y. Ischemia-Reperfusion Injury in Fatty Liver Is Mediated by Activated NADPH Oxidase 2 in Rats. Transplantation 2016, 100, 791–800. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5′ to 3′) |
|---|---|---|
| Slc2a1 | Forward | GCTTCTCCAACTGGACCTCAAAC |
| Reverse | ACGAGGAGCACCGTGAAGATGA | |
| Vegf | Forward | CTGCTGTAACGATGAAGCCCTG |
| Reverse | GCTGTAGGAAGCTCATCTCTCC | |
| Rpl27 | Forward | GCGATCCAAGATCAAGTCCTTTG |
| Reverse | TCAAAGCTGGGTCCCTGAACAC | |
| RER1 | Forward | GACACTGGGCCTGAGTTTTG |
| Reverse | GGAGAAAGGAACGCAATGAA | |
| 18S | Forward | AGTTGGTGGAGCGATTTGTC |
| Reverse | AACGCCACTTGTCCCTCTAA | |
| Ptgs2 [24] Ripk3 Mlkl | Forward | GGG AGT CTG GAA CAT TGT GAA |
| Reverse Forward Reverse Forward Reverse | GTG CAC ATT GTA AGT AGG TGG ACT GAA GAC ACG GCA CTC CTT GGT A CTT GAG GCA GTA GTT CTT GGT GG CTGAGGGAACTGCTGGATAGAG CGAGGAAACTGGAGCTGCTGAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baidya, R.; Gautheron, J.; Crawford, D.H.G.; Wang, H.; Bridle, K.R. Inhibition of MLKL Attenuates Necroptotic Cell Death in a Murine Cell Model of Hepatic Ischaemia Injury. J. Clin. Med. 2021, 10, 212. https://doi.org/10.3390/jcm10020212
Baidya R, Gautheron J, Crawford DHG, Wang H, Bridle KR. Inhibition of MLKL Attenuates Necroptotic Cell Death in a Murine Cell Model of Hepatic Ischaemia Injury. Journal of Clinical Medicine. 2021; 10(2):212. https://doi.org/10.3390/jcm10020212
Chicago/Turabian StyleBaidya, Raji, Jérémie Gautheron, Darrell H. G. Crawford, Haolu Wang, and Kim R. Bridle. 2021. "Inhibition of MLKL Attenuates Necroptotic Cell Death in a Murine Cell Model of Hepatic Ischaemia Injury" Journal of Clinical Medicine 10, no. 2: 212. https://doi.org/10.3390/jcm10020212
APA StyleBaidya, R., Gautheron, J., Crawford, D. H. G., Wang, H., & Bridle, K. R. (2021). Inhibition of MLKL Attenuates Necroptotic Cell Death in a Murine Cell Model of Hepatic Ischaemia Injury. Journal of Clinical Medicine, 10(2), 212. https://doi.org/10.3390/jcm10020212