Inhibitor in Congenital Factor VII Deficiency; a Rare but Serious Therapeutic Challenge—A Systematic Literature Review




Abstract
1. Introduction
2. Strategy of Search
3. Study Selection and Inclusion Criteria
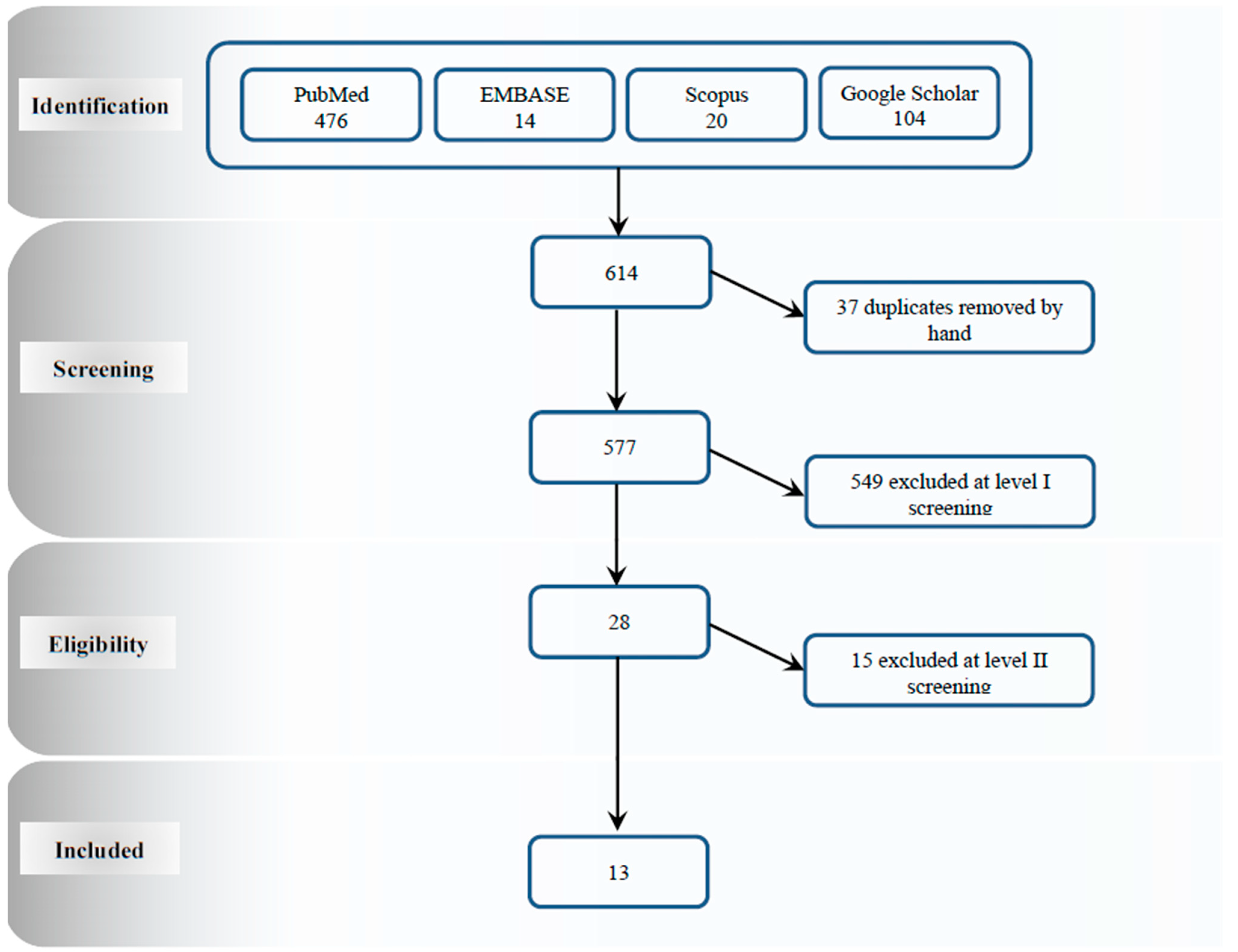
4. Study Selection
5. Results
5.1. Study and Patient Characteristics
5.2. Incidence of FVII Inhibitor in Congenital FVII Deficiency
5.3. Characterization of the FVII Inhibitor
5.4. Molecular Basis

{kind=link}
| Author | Nicolaisen et al. (1996) [8] | Nicolaisen et al. (1996) [8] | Ingerslev et al. (2005) [13] | Pruthi et al. (2007) [14] | Tokgoz et al. (2012) [10] | Mariani et al. (2013) [5] | Mariani et al. (2013) [5] |
|---|---|---|---|---|---|---|---|
| Case #1 | Case #2 | Case #3 | Case #4 | Case #5 | Case #6 | Case #7 | |
| Sex | N/A | N/A | Female | Male | Male | Male | Female |
| 1st age * | N/A | N/A | N/A | 11 days | 3 months | N/A | N/A |
| 2nd age * | N/A | N/A | ~36 years | 45 years | 1 year | 30 days after RT | 30 days after RT |
| 3rd age * | 2 months | N/A | 39 years | 51 years | 3 months | 1 year | 54 years |
| FVII:C | <4% | N/A | <0.01% | <1% | <1% | 2.50% | <1% |
| Bleeding events | ICH | N/A | MSB | Hr, Ep | ICH, GIB | CNS, Br, Ep, GuB, hematomas | Br, Ep, GuB, HmB, Me |
| Genetics | N/A | N/A | Homozygosity for a nucleotide substitution that caused 100 GLN→ ARG shift | Missense mutation in F7 gene Ser103→Gly (S103G) | Homozygous nonsense mutation in F7 p.Ser112-Stop | N/A | N/A |
| Antibody titers (BU) min-max | N/A | N/A | High titer inhibitors for both pdFVII (16 BU/mL) and rFVIIa (88 BU/mL). | Less than 1 to 4 | 34–68.3 | 2.1 | 22.4 |
| Author | Napolitano et al. (2013) [15] | Batorova et al. (2014) [6] | Batorova et al. (2014) [6] | Batorova et al. (2014) [6] | Batorova et al. (2014) [6] | Borhany et al. (2015) [12] | See et al. (2016) [2] |
|---|---|---|---|---|---|---|---|
| Case #8 | Case #9 | Case #10 | Case #11 | Case #12 | Case #13 | Case #14 | |
| Sex | Male | Female | Male | Male | Male | Female | Female |
| 1st age * | N/A | N/A | N/A | N/A | N/A | N/A | Prenatal period |
| 2nd age * | 1 year after FFP | 59 years | 3 months | 5 months | 1 month | N/A | 4 years |
| 3rd age * | 5 years | 53 years | 5 years | 5 months | 1 year | 8 years | 5 years |
| FVII:C | 2% | <1% | 2% | 1.30% | <1% | 1% | <1% |
| Bleeding events | CNS, Br, Ep, GIB, GuB | Ep, Br, GuB, Hp, Me | CNS, Br, Ep, GIB, GuB | CNS, GIB, GuB | CNS, Br, Ep, GuB, hematomas | Hematoma, Hr | SDH, IVH, ICH, Hr |
| Genetics | Homozygous (gene mutations were only reported as zygosity) | p.A354V-p.P464Hfs | p.Ser112-Stop (homozygous) | N/A | N/A | No Identified mutation in the F7 coding regions and large rearrangement | Homozygous mutation in intron 6 of the F7 gene IVS6 + 1G > T (c.681 + 1G > T) |
| Antibody titers (BU) min-max | 59 | 10–20 | 38–68.3 | 5.5–60 | 32–72 | 1.1–11 | 2.77–20.6 |
| Author | Tokgoz et al. (2017) [16] | Tokgoz et al. (2017) [16] | Shams et al. (2018) [4] | Shams et al. (2018) [4] | Patel et al. (2019) [17] | Eshghi et al. (2019) [9] | Eshghi et al. (2019) [9] |
|---|---|---|---|---|---|---|---|
| Case #15 | Case #16 | Case #17 | Case #18 | Case #19 | Case #20 | Case #21 | |
| Sex | Male | Male | Male | Male | Male | Female | Female |
| 1st age * | N/A | N/A | 11 days | 60 years | N/A | 4 days | 3 years |
| 2nd age * | 1 year | <6 months | 7 years | 66 years | 7 months | 2.5 years | ~3 years |
| 3rd age * | 3 months | 11 days | 14 years | 70 years | 3 months | N/A | N/A |
| FVII:C | 0.20% | 0.10% | <1% | 6% | <1% | <10% | <10% |
| Bleeding events | ICH | Ep, ICH | Hematoma, ICH, Hr, Ep, Br | Br | ICH | Hematoma, GIB, ICH, Ecchymosis, He, Ep | Hematoma, ICH, He, Hr |
| Genetics | homozygous nonsense mutation in F7 gene, p.Ser112stop | N/A | N/A | N/A | Homozygous mutation in F7 gene Intron 5, c.572–12T>A | N/A | N/A |
| Antibody titers (BU) min-max | 32 | 28.8 | 5–170 | 5–10 | 2.2–819 | 7–34.4 | 80 |
| Author | Eshghi et al. (2019) [9] | Eshghi et al. (2019) [9] | Eshghi et al. (2019) [9] | Eshghi et al. (2019) [9] | Eshghi et al. (2019) [9] | Eshghi et al. (2019) [9] |
|---|---|---|---|---|---|---|
| Case #22 | Case #23 | Case #24 | Case #25 | Case #26 | Case #27 | |
| Sex | Male | Male | Male | Male | Male | Male |
| 1st age * | 2 months | 2 months | 2 weeks | 16 days | 2 months | 2 months |
| 2nd age * | 2 years | 2 years | 8 years | 3 months | 2 years | ~3 years |
| 3rd age * | N/A | N/A | N/A | N/A | N/A | N/A |
| FVII:C | <10% | <10% | <10% | <10% | <10% | <10% |
| Bleeding events | Hematoma, GIB, ICH, Ecchymosis | Hematoma, GIB, ICH, Hr, Ecchymosis | GIB, ICH, Hr, Mucocutaneous, Ep | ICH | ICH | ICH Mucocutaneous, Ecchymoses, Ep |
| Genetics | N/A | N/A | c.9711 deletion on exon 7 | N/A | c.9711 deletion on exon 7 | N/A |
| Antibody titer (BU) min-max | 5–36 | 43–217 | 0–170 | 14.7 | 191 | 7 |
5.5. Clinical Manifestations
5.6. Management of Inhibitor
| Case Number | Author | Pre-Inhibitor Treatment | Post-Inhibitor Treatment | Outcome |
|---|---|---|---|---|
| #1 | Nicolaisen et al. (1996) [8] | FFP, rFVIIa (approximately 800 μg/kg) | N/A | High-dose of rFVIIa (800 Ig/kg) was used following no ICH improvement with FFP. Although it elicited a good initial response, the treatment failed, and the patient died. |
| #2 | Nicolaisen et al. (1996) [8] | pd-FVII, rFVIIa | N/A | While there was no antibody detected after exposure to plasma-derived FVII, the patient developed inhibitor three months after being exposed to rFVIIa. |
| #3 | Ingerslev et al. (2005) [13] | pd-FVII, rFVIIa (161 mg for 6 years) | rFVIIa (20 μg/kg/body weight) | The patient developed an inhibitor after exposure to pd-FVII (16 BU/mL) and rFVIIa (88 BU/mL). The inhibitor titer later was found to have decreased due to cessation of treatment with FVII-containing material. Re-exposure to 1.2 mg rFVIIa has led to an anamnesis with a notable increase in inhibitor titers. |
| #4 | Pruthi et al. (2007) [14] | FFP | The inhibitor resolved spontaneously after two years | Four months after FFP exposure, a low-titer FVII inhibitor was detected, which resolved spontaneously in the absence of further FFP infusions. Inhibitor was detected again after renewed exposure to FFP and rFVIIa. |
| #5 | Tokgoz et al. (2012) [10] | rFVIIa (20–30 μg/kg), FFP (20 ml/kg per week) | rFVIIa (20–30 μg/kg), prophylaxis treatment initiated at 30 Ig/kg 3 × per week at the age of 4.5 | A successful prophylactic rFVIIa (30 µg/kg, three times per week) was reported in a 4.5-year-old patient who had congenital FVIID, with inhibitors to FVII. |
| #6 | Mariani et al. (2013) [5] | rFVIIa (65 μg/kg) | N/A | The patient’s bleeding episodes were treated using plasma-derived FVII (with a total dose of 33 µg/kg) in a repetitive procedure. |
| #7 | Mariani et al. (2013) [5] | Pd-FVII (33 μg/kg) | N/A | The patient’s bleeding episodes were treated using rFVIIa (with the total dose of 65 µg/kg) in a repetitive procedure. |
| #8 | Napol-tano et al. (2013) [15] | FFP | rFVIIa (90 total weekly dose mg/kg/body weight, 3×weekly) | After being treated with FFP, this patient developed FVII inhibitor (max titer: 59 BU). Because of recurrent CNS bleeding prophylactic rFVIIa was initiated. |
| #9 | Batorova et al. (2014) [6] | FFP, pd-FVII, rFVIIa | rFVIIa (Initial bolus 30 μg/kg plus 8 consecutive boluses 10 μg/kg) | Congenital FVII deficiency was diagnosed following epistaxis in a patient. Treatments with FFP, pd-FVII and rFVIIa were scheduled. At the age of 59 years and during multiple dental extractions, FVII inhibitor diagnosed and treated by rFVIIa. |
| #10 | Batorova et al. (2014) [6] | FFP, rFVIIa | rFVIIa (30 μg/kg × 3 weeks) | FFP was given to a 3-month-old patient after CNS bleeding. He was also given prophylactic rFVIIa. Despite the development of FVII inhibitor at this age, he was treated by rFVIIa. |
| #11 | Batorova et al. (2014) [6] | FFP, rFVIIa | rFVIIa (65 μg/kg × 1 weeks) | Following CNS and GI bleeding at birth, FFP was administered. He was then given prophylactic rFVIIa as well. Despite the development of FVII inhibitor at the age of 5 months, he was treated with rFVIIa. |
| #12 | Batorova et al. (2014) [6] | FFP, rFVIIa | rFVIIa (31 μg/kg × 3 weeks) | Following tongue hematoma, FFP was scheduled for a one-month-old patient. This was followed by prophylactic rFVIIa (31 µg/kg × 3 week). Despite the development of FVII inhibitor at this age, he was treated with rFVIIa. |
| #13 | Borhany et al. (2015) [6] | rFVIIa (15 μg/kg | Steroid (azathioprine), Immunoglobulin, FEIBA | Hemarthrosis and gum bleeding were controlled by rFVIIa (15 lg/kg every 6 hours). However, some bleedings developed, with no response to rFVIIa two weeks after this treatment. Therefore, rFVIIa infusion stopped, and extensive immunosuppressive treatment was scheduled. Despite this, the patient’s clinical status deteriorated. |
| then increased to 30 μg/kg), FFP (15 mL/kg), vitamin K injections | ||||
| #14 | See et al. (2016) [2] | rFVIIa (30 μg/kg), rFVIIa (30 μg/kg), rFVIIa (30–90 μg/kg, depending on severity of bleeding), rFVIIa, vitamin K therapy | rFVIIa (30 μg/kg per dose), secondary prophylaxis with rFVIIa (30 μg/kg per dose twice a week) | This patient went through an auxiliary liver transplant. Twenty-two days after prophylactic rFVIIa injection, which was performed during the transplantation, the inhibitor titer reached a detectable level (20.6 Bethesda units). Despite this inhibitor development, the patient was still responsive to rFVIIa (30 Ig/kg). |
| #15 | Tokgoz et al. (2017) [16] | rFVIIa | rFVIIa (30 μg/kg, three times a week) was used; increased to 30 μg/kg every day) | Inhibitor (32 BU) at the age of one. The same doses of rFVIIa were sufficient to control all bleeding episodes even in the presence of inhibitor. Although prophylactic rFVIIa was successful in preventing bleeding episodes for 4 years, this dose became ineffective afterward, hence prophylactic rFVIIa doses were increased. |
| #16 | Tokgoz et al. (2017) [16] | rFVIIa (25 mcg/kg) then prophylaxis (25 mcg/kg, 3 times a week) initiated | 40 mcg/kg rFVIIa was used; and followed by gradual increase of rFVIIa to 100mcg/kg/day | The ICH of this patient was treated with rFVIIa after two weeks. Then the patient went through prophylactic rFVIIa. Nevertheless, the patient developed FVII inhibitor (28.8 BU). After this development, the bleeding, however, was controlled after three weeks and with an increased dose of rFVIIa. The prophylactic rFVIIa was later gradually increased. |
| #17 | Shams et al. (2018) [4] | rFVIIa | FEIBA | Administration of rFVIIa was successful in treatment of a 14-day-old boy. Three months later, rFVIIa was used for shunt replacement. Since the age of approximately 6 months, regular prophylaxis was scheduled for the patient until the age of seven. At age 10, inhibitor level was 5BU. Three years later, pre-operative rFVIIa notably increased the level of inhibitor titer (70 BU). Thereafter, the clinical response to on-demand therapy (FEIBA) was excellent. |
| #18 | Shams et al. (2018) [4] | rFVIIa (50 μg/kg), then secondary prophylaxis initiated | rFVIIa | This patient was randomly diagnosed with congenital FVIID prior to angiography at age 60. The procedure was performed by rFVIIa. After 6 years, the inhibitor was detected and the patient went through several rFVIIa uses. |
| #19 | Patel et al. (2019) [17] | rFVIIa (NovoSeven™), switched to pd-FVII (20 IU/kg daily, then x3/week) | Pd-FVII (50 IU/kg (rounded to 600 IU) of pd FVII concentrate x3/week, and 50 IU/kg (600IU) daily, rFVIIa (NovoSeven™) | This patient was identified with inhibitor to FVII at the age of 7. An ITI regime was used to control the production of antibody in the patient. |
| #20 | Eshghi et al. (2019) [9] | rFVIIa (AryoSeven™) (50 μg/kg/day on alternate days), FFP | High dose AryoSeven™ (120 μg/kg/day) | The patient was responsive to high dose of AryoSeven™ (120 μg/kg/day). Afterwards, as a prophylactic treatment, the same type of rFVIIa was used up to 100 ug/kg every other day. Current clinical status of the patient is “no symptoms”. |
| #21 | Eshghi et al. (2019) [9] | rFVIIa (AryoSeven™): 30 μg/kg/every other day, FFP | No response to high-dose AryoSeven™: 100 μg/kg, switch to FEIBA: 50–75 u/kg/day, three times a day | The patient was treated with FEIBA. Prophylactic treatment (75 ug/kg) was used three times a week. The patient succumbed to severe ICH. |
| #22 | Eshghi et al. (2019) [9] | rFVIIa (NovoSeven® and AryoSeven™): 30 μg/kg, 3 times a week | High-dose AryoSeven™: 45 μg/kg | This patient was responsive to high dose of AryoSeven™ (45 ug/kg/ twice per week). The current clinical status of the patient is “no symptoms”. |
| #23 | Eshghi et al. (2019) [9] | rFVIIa(AryoSeven™): 30 μg/kg, 3 times a week, FFP | Blood transfusion, FEIBA for a short period, then high dose AryoSeven™: 40–45 μg/kg | A prophylactic treatment was used for this patient (AryoSeven™, 45 ug/kg, 3 times a week). No severe bleeding events were reported. |
| #24 | Eshghi et al. (2019) [9] | rFVIIa (NovoSeven®): 50 μg/kg/day (on-demand), FFP | No response to NovoSeven®.FEIBA: 80 u/kg/twice a day | Following the treatment with FEIBA (80–100 IU/kg), inhibitor titer reduced to 20. Six months later the inhibitor level reached zero and the patient became responsive to on-demand rFVIIa. |
| #25 | Eshghi et al. (2019) [9] | rFVIIa(AryoSeven™): 30 ug/kg, daily | High-dose AryoSeven™: 270 μg/kg FEIBA, FFP, Cryo | The patient was not responsive to high dose of AryoSeven™; the patient died due to extensive ICH. |
| #26 | Eshghi et al. (2019) [9] | rFVIIa (NovoSeven®): Not applicable (on-demand) | No response to NovoSeven®.FEIBA: 75-80 u/kg twice per day | This patient, who was suffering from sepsis and intra-abdominal bleeding, was not responsive to NovoSeven® and FFP. He died at age 4 due to sepsis followed by DIC. |
| #27 | Eshghi et al. (2019) [9] | rFVIIa (AryoSeven™): 40 μg/kg/day on alternate days | High-dose AryoSeven™: 70 μg/kg/day | The patient was responsive to high dose of AryoSeven™. The current clinical status of the patient was reported to be “symptomatic”. |
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Franchini, M.; Marano, G.; Mengoli, C.; Piccinini, V.; Pupella, S.; Vaglio, S.; Liumbruno, G.M. Inhibitors in Patients with Congenital Bleeding Disorders other than Hemophilia. Semin. Thromb. Hemost. 2018, 44, 595–603. [Google Scholar] [PubMed]
- See, W.S.; Chang, K.O.; Cheuk, D.L.; Leung, Y.Y.; Chan, G.F.; Chan, S.C.; Ha, S.Y. Inhibitor development after liver transplantation in congenital factor VII deficiency. Haemophilia 2016, 22, e417–e422. [Google Scholar] [CrossRef] [PubMed]
- Dorgalaleh, A.; Tabibian, S.; Hosseini, M.S.; Shams, M. Pharmacological management of rare coagulation factor deficiencies besides hemophilia. Expert. Rev. Hematol. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Shams, M.; Dorgalaleh, A.; Safarian, N.; Hossein Emami, A.; Zaker, F.; Tabibian, S.; Managhchi, M.R.; Faranoush, M.; Tabatabaei, T.; Satari, S. Inhibitor development in patients with congenital factor VII deficiency, a study on 50 Iranian patients. Blood Coagul. Fibrinolysis 2019, 30, 24–28. [Google Scholar] [CrossRef]
- Mariani, G.; Napolitano, M.; Dolce, A.; Garrido, R.P.; Batorova, A.; Karimi, M.; Platokouki, H.; Auerswald, G.; Bertrand, A.M.; Di Minno, G.; et al. Replacement therapy for bleeding episodes in factor VII deficiency: A prospective evaluation. Thromb. Haemost. 2013, 109, 238–247. [Google Scholar]
- Batorova, A.; Mariani, G.; Kavakli, K.; de Saez, A.R.; Caliskan, U.; Karimi, M.; Pinotti, M.; Napolitano, M.; Dolce, A.; Sørensen, B.; et al. Inhibitors to factor VII in congenital factor VII deficiency. Haemoph. Off. J. World Fed. Hemoph. 2014, 20, e188–e191. [Google Scholar] [CrossRef]
- Napolitano, M.; Dolce, A.; Batorova, A.; Giansily-Blaizot, M.; Ingerslev, J.; Mirbehbahani, N.; Di Minno, M.N.D.; Lopez Fernandez, M.F.; Karimi, M.; Charoenkwan, P.; et al. Replacement therapy in inherited factor VII deficiency: Occurrence of adverse events and relation with surgery. Haemophilia 2015, 21, e513–e517. [Google Scholar] [CrossRef]
- Nicolaisen, E.M.; Hansen, L.L.; Poulsen, F.; Glazer, S.; Hedner, U. Immunological aspects of recombinant factor VIIa (rFVIIa) in clinical use. Thromb. Haemost. 1996, 76, 200–204. [Google Scholar]
- Eshghi, P.; Tara, S.Z.; Baghaipour, M.R.; Bordbar, M.R.; Jenabzade, A.; Habibpanah, B.; Cohan, N.; Tavosi, H.; Bahrami, R.; Karimi, M. Inhibitors against rFVIIa in patients with severe congenital FVII deficiency: A case series. Haemophilia 2019, 25, e345–e349. [Google Scholar] [CrossRef]
- Tokgoz, H.; Caliskan, U.; Lavigne-Lissalde, G.; Giansily-Blaizot, M. Successful prophylactic use of recombinant activated factor VII (rFVIIa) in a patient with congenital FVII deficiency and inhibitors to FVII. Haemophilia 2012, 18, e25–e27. [Google Scholar] [CrossRef]
- Branchini, A.; Baroni, M.; Pfeiffer, C.; Batorova, A.; Giansily-Blaizot, M.; Schved, J.F.; Mariani, G.; Bernardi, F.; Pinotti, M. Coagulation factor VII variants resistant to inhibitory antibodies. Thromb. Haemost. 2014, 112, 972–980. [Google Scholar] [PubMed]
- Borhany, M.; Delbes, C.; Giansily-Blaizot, M.; Zubair, M.; Ahmed, M.S.; Fatima, N.; Shamsi, T. A new report of FVII-inhibitor in a patient suffering from severe congenital FVII deficiency. Haemophilia 2015, 21, e336–e338. [Google Scholar] [CrossRef] [PubMed]
- Ingerslev, J.; Christiansen, K.; SØRensen For the International Registry On Factor Vii Deficiency Steering Committee B. Inhibitor to factor VII in severe factor VII deficiency: Detection and course of the inhibitory response. J. Thromb. Haemost. 2005, 3, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Pruthi, R.K.; Rodriguez, V.; Allen, C.; Slaby, J.A.; Schmidt, K.A.; Plumhoff, E.A. Molecular analysis in a patient with severe factor VII deficiency and an inhibitor: Report of a novel mutation (S103G). Eur. J. Haematol. 2007, 79, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Giansily-Blaizot, M.; Dolce, A.; Schved, J.F.; Auerswald, G.; Ingerslev, J.; Bjerre, J.; Altisent, C.; Charoenkwan, P.; Michaels, L. Prophylaxis in congenital factor VII deficiency: Indications, efficacy and safety. Results from the Seven Treatment Evaluation Registry (STER). Haematologica 2013, 98, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Tokgoz., H.; Caliskan, Ü.; Blaizot, M.G. Congenital Factor VII Deficiency with Inhibitors to FVII: Report of two Cases. In Haemophilia; WILEY-BLACKWELL: Hoboken, NJ, USA, 2017. [Google Scholar]
- Patel, J.; Tracey Chan, Y.L.; Horgan, C.; Motwani, J. Challenges in the management of congenital Factor VII deficiency with inhibitors. In British Journal of Haematology; WILEY: Hoboken, NJ, USA, 2019. [Google Scholar]
- Bolton-Maggs, P.H. Optimal haemophilia care versus the reality. Br. J. Haematol. 2006, 132, 671–682. [Google Scholar] [CrossRef]
- Franchini, M.; Frattini, F.; Crestani, S.; Bonfanti, C. Alloantibodies in previously untreated hemophilia A patients: The role of environmental factors. Hematology 2013, 18, 183–190. [Google Scholar] [CrossRef]
- Franchini, M.; Lippi, G. Acquired factor VIII inhibitors. Blood 2008, 112, 250–255. [Google Scholar] [CrossRef]
- Bamedi, T.; Dadashizade, G.; Sarabandi, A.; Tabibian, S.; Shams, M.; Dorgalaleh, A. Genetic Risk Factors for Inhibitor Development in Patients with Hemophilia and Rare Bleeding Disorders. J. Cell. Amp Mol. Anesth. 2017, 2, 19–23. [Google Scholar]
- Gouw, S.C.; van der Bom, J.G.; van den Berg, H.M. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: The CANAL cohort study. Blood 2007, 109, 4648–4654. [Google Scholar] [CrossRef]
- Mariani, G.; Mariani, G.; Herrmann, F.H.; Dolce, A.; Batorova, A.; Etro, D.; Peyvandi, F.; Wulff, K.; Schved, J.F.; Auerswald, G. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb. Haemost. 2005, 93, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Chitlur, M.; Warrier, I.; Rajpurkar, M.; Lusher, J.M. Inhibitors in factor IX deficiency a report of the ISTH-SSC international FIX inhibitor registry (1997–2006). Haemophilia 2009, 15, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- . Mariani, G.; Konkle, B.; Kessler, C.M. Inhibitors in Hemophilia, A. and B. In Hematology; Hoffman, R., Benz, E.J., Silberstein, L.E., Heslop, H.E., Weitz, J.I., Anastasi, J., Eds.; Basic Principles and Practice; Elsevier: Philadelphia, PA, USA, 2012; Chapter 138; pp. 1961–1970. [Google Scholar]
- Astermark, J.; Oldenburg, J.; Carlson, J.; Pavlova, A.; Kavakli, K.; Berntorp, E.; Lefvert, A.K. Polymorphisms in the TNFA gene and the risk of inhibitor development in patients with hemophilia A. Blood 2006, 108, 3739–3745. [Google Scholar] [CrossRef] [PubMed]
- Ragni, M.V.; Ojeifo, O.; Feng, J.; Yan, J.; Hill, K.A.; Sommer, S.S.; Trucco, M.N.; Brambilla, D.J. Risk factors for inhibitor formation in haemophilia: A prevalent case-control study. Haemophilia 2009, 15, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Pavlova, A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia 2006, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramezanpour, N.; Zaker, F.; Biswas, A.; Dorgalaleh, A. Inhibitor in Congenital Factor VII Deficiency; a Rare but Serious Therapeutic Challenge—A Systematic Literature Review. J. Clin. Med. 2021, 10, 211. https://doi.org/10.3390/jcm10020211
Ramezanpour N, Zaker F, Biswas A, Dorgalaleh A. Inhibitor in Congenital Factor VII Deficiency; a Rare but Serious Therapeutic Challenge—A Systematic Literature Review. Journal of Clinical Medicine. 2021; 10(2):211. https://doi.org/10.3390/jcm10020211
Chicago/Turabian StyleRamezanpour, Nahid, Farhad Zaker, Arijit Biswas, and Akbar Dorgalaleh. 2021. "Inhibitor in Congenital Factor VII Deficiency; a Rare but Serious Therapeutic Challenge—A Systematic Literature Review" Journal of Clinical Medicine 10, no. 2: 211. https://doi.org/10.3390/jcm10020211
APA StyleRamezanpour, N., Zaker, F., Biswas, A., & Dorgalaleh, A. (2021). Inhibitor in Congenital Factor VII Deficiency; a Rare but Serious Therapeutic Challenge—A Systematic Literature Review. Journal of Clinical Medicine, 10(2), 211. https://doi.org/10.3390/jcm10020211