In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. In Vitro Cell Culture Models of Non-Alcoholic Fatty Liver Disease
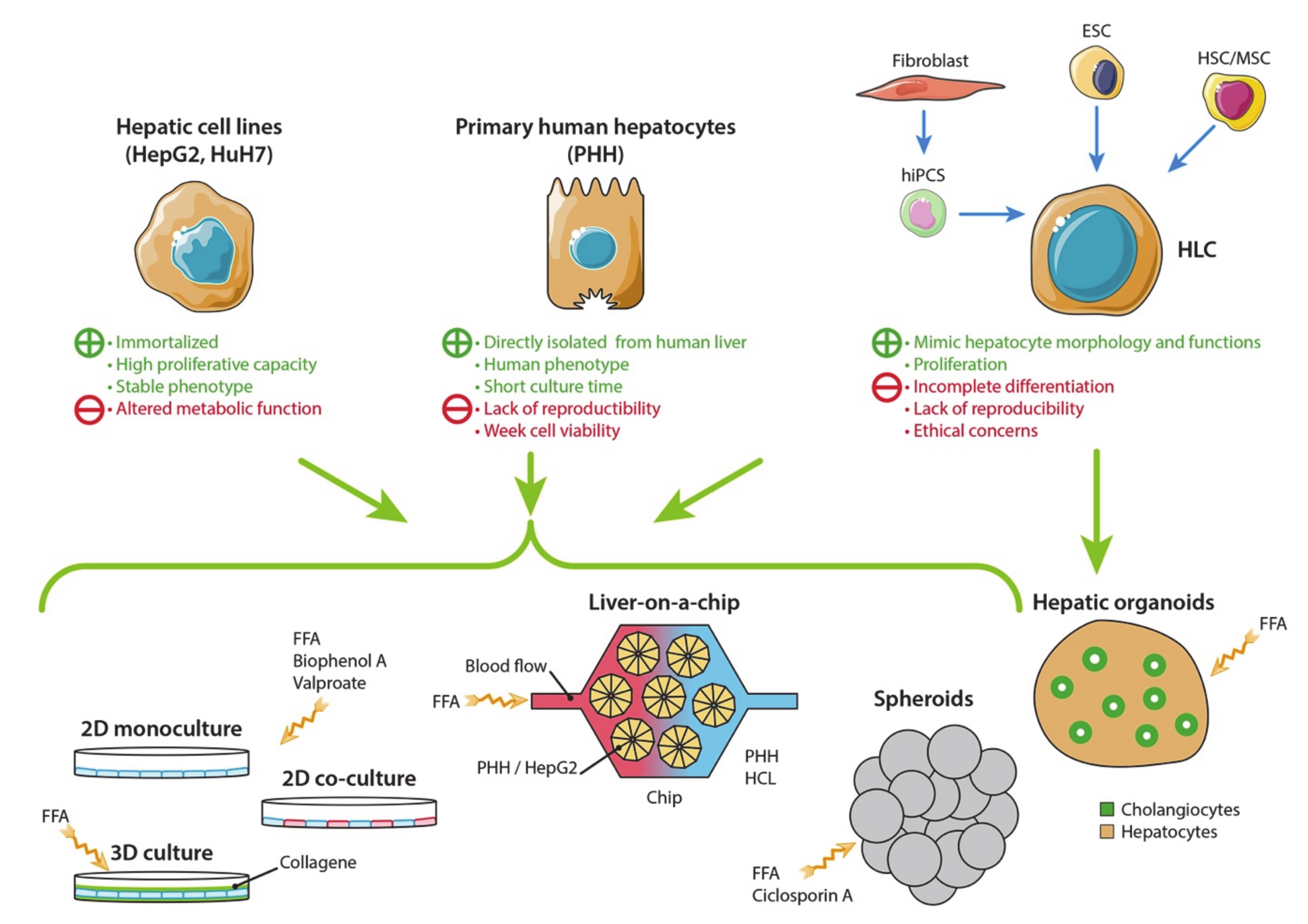
3.1. Hepatic Cell Sources
3.1.1. Human Hepatic Cell Lines
3.1.2. Primary Human Hepatocytes
3.1.3. Hepatocyte-Like Cells
3.2. Two-Dimensional Cell Culture Models
3.2.1. Monoculture
3.2.2. Co-Culture
3.3. Three-Dimensional Cell Culture Models
3.3.1. 3D Cell Culture in a Collagen Gel Sandwich
3.3.2. Hepatic Spheroids and Organoids
3.3.3. Liver-On-A-Chip Technology
3.4. Human Precision-Cut Liver Slices Model
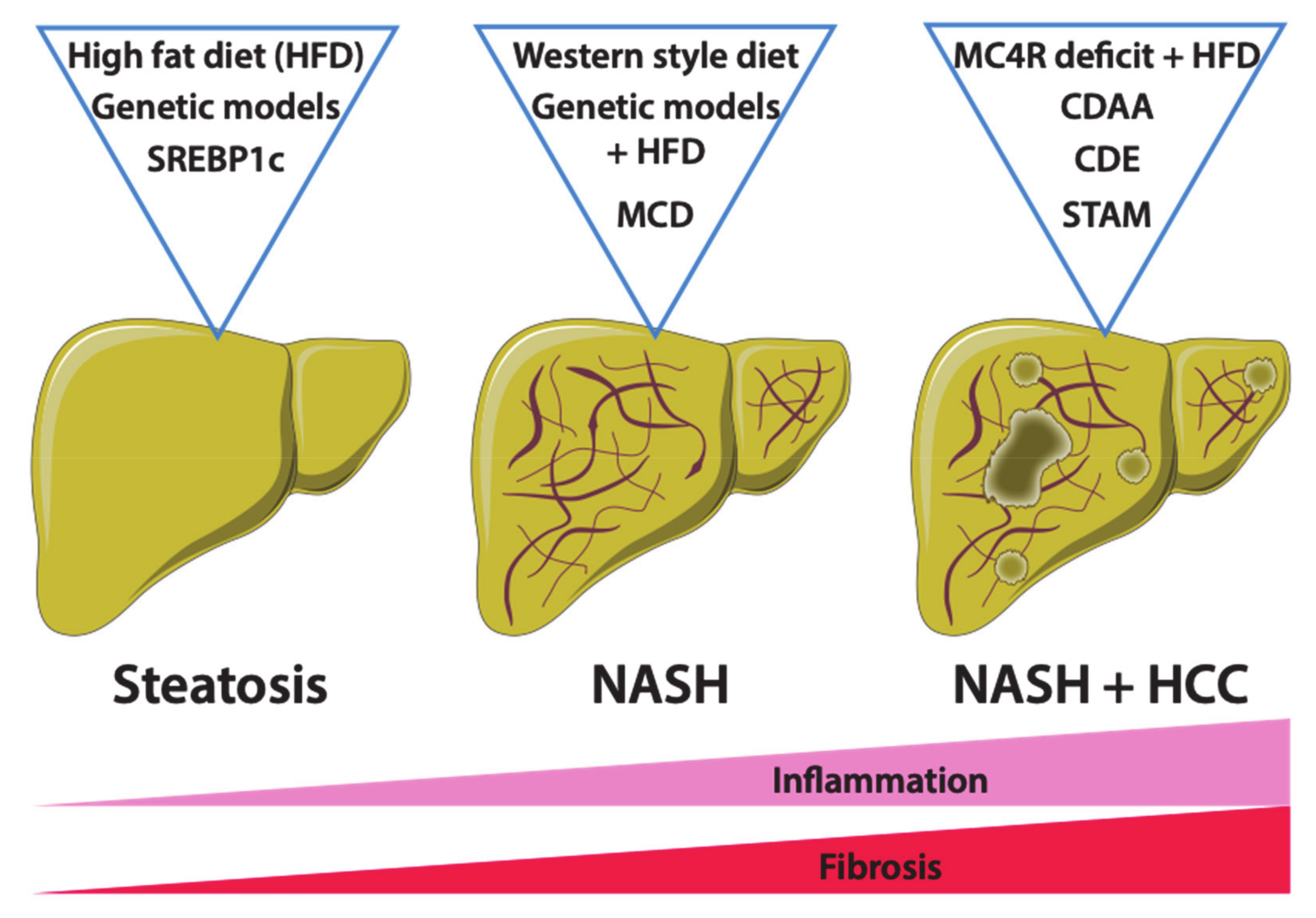
4. Mouse Models of Non-Alcoholic Fatty Liver Disease
4.1. Dietary Mouse Models of NAFLD
4.1.1. High-Fat Diet Feeding
4.1.2. Western-Style or Fast-Food Diet Feeding
4.1.3. Methionine- and Choline-Deficient Diet and Derivative Feeding
4.1.4. STAM Model
4.2. Genetic Models of NAFLD
4.2.1. ob/ob Mice
4.2.2. db/db Mice
4.2.3. The Mc4r-Deficient Mice
4.2.4. The Srebp1c-Overexpressing Mice
4.2.5. The FATZO Mouse Model
5. Other Animal Models of Non-Alcoholic Fatty Liver Disease
5.1. Rat Models
5.2. Non-Rodent Models
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-Alcoholic Fatty Liver Disease—A Global Public Health Perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Barritt, A.S.; Calmus, Y.; Scatton, O.; Runge, T.; Lebray, P.; Poynard, T.; Ratziu, V.; Conti, F. NAFLD and Liver Transplantation: Current Burden and Expected Challenges. J. Hepatol. 2016, 65, 1245–1257. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Wilkening, S.; Stahl, F.; Bader, A. Comparison of Primary Human Hepatocytes and Hepatoma Cell Line Hepg2 with Regard to Their Biotransformation Properties. Drug Metab. Dispos. 2003, 31, 1035–1042. [Google Scholar] [CrossRef]
- Andersson, T.B.; Kanebratt, K.P.; Kenna, J.G. The HepaRG Cell Line: A Unique in Vitro Tool for Understanding Drug Metabolism and Toxicology in Human. Expert Opin. Drug Metab. Toxicol. 2012, 8, 909–920. [Google Scholar] [CrossRef]
- Samanez, C.H.; Caron, S.; Briand, O.; Dehondt, H.; Duplan, I.; Kuipers, F.; Hennuyer, N.; Clavey, V.; Staels, B. The Human Hepatocyte Cell Lines IHH and HepaRG: Models to Study Glucose, Lipid and Lipoprotein Metabolism. Arch. Physiol. Biochem. 2012, 118, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ramboer, E.; De Craene, B.; De Kock, J.; Vanhaecke, T.; Berx, G.; Rogiers, V.; Vinken, M. Strategies for Immortalization of Primary Hepatocytes. J. Hepatol. 2014, 61, 925–943. [Google Scholar] [CrossRef] [PubMed]
- Guguen-Guillouzo, C.; Guillouzo, A. General Review on In Vitro Hepatocyte Models and Their Applications. In Hepatocytes: Methods and Protocols; Maurel, P., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; pp. 1–40. ISBN 978-1-60761-688-7. [Google Scholar]
- Ramboer, E.; Vanhaecke, T.; Rogiers, V.; Vinken, M. Immortalized Human Hepatic Cell Lines for In Vitro Testing and Research Purposes. Methods Mol. Biol. Clifton NJ 2015, 1250, 53–76. [Google Scholar] [CrossRef]
- Aoudjehane, L.; Gautheron, J.; Goff, W.L.; Goumard, C.; Gilaizeau, J.; Nget, C.S.; Savier, E.; Atif, M.; Lesnik, P.; Morichon, R.; et al. Novel Defatting Strategies Reduce Lipid Accumulation in Primary Human Culture Models of Liver Steatosis. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Podevin, P.; Carpentier, A.; Pène, V.; Aoudjehane, L.; Carrière, M.; Zaïdi, S.; Hernandez, C.; Calle, V.; Méritet, J.-F.; Scatton, O.; et al. Production of Infectious Hepatitis C Virus in Primary Cultures of Human Adult Hepatocytes. Gastroenterology 2010, 139, 1355–1364. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Castell, J.V.; Jover, R. Human Hepatocytes as a Tool for Studying Toxicity and Drug Metabolism. Curr. Drug Metab. 2003, 4, 292–312. [Google Scholar] [CrossRef]
- Godoy, P.; Hengstler, J.G.; Ilkavets, I.; Meyer, C.; Bachmann, A.; Müller, A.; Tuschl, G.; Mueller, S.O.; Dooley, S. Extracellular Matrix Modulates Sensitivity of Hepatocytes to Fibroblastoid Dedifferentiation and Transforming Growth Factor β–Induced Apoptosis. Hepatology 2009, 49, 2031–2043. [Google Scholar] [CrossRef]
- Schyschka, L.; Sánchez, J.J.M.; Wang, Z.; Burkhardt, B.; Müller-Vieira, U.; Zeilinger, K.; Bachmann, A.; Nadalin, S.; Damm, G.; Nussler, A.K. Hepatic 3D Cultures but Not 2D Cultures Preserve Specific Transporter Activity for Acetaminophen-Induced Hepatotoxicity. Arch. Toxicol. 2013, 87, 1581–1593. [Google Scholar] [CrossRef]
- Zeilinger, K.; Freyer, N.; Damm, G.; Seehofer, D.; Knöspel, F. Cell Sources for in Vitro Human Liver Cell Culture Models. Exp. Biol. Med. 2016, 241, 1684–1698. [Google Scholar] [CrossRef]
- Richert, L.; Alexandre, E.; Lloyd, T.; Orr, S.; Viollon-Abadie, C.; Patel, R.; Kingston, S.; Berry, D.; Dennison, A.; Heyd, B.; et al. Tissue Collection, Transport and Isolation Procedures Required to Optimize Human Hepatocyte Isolation from Waste Liver Surgical Resections. A Multilaboratory Study. Liver Int. 2004, 24, 371–378. [Google Scholar] [CrossRef]
- Hu, C.; Li, L. In Vitro Culture of Isolated Primary Hepatocytes and Stem Cell-Derived Hepatocyte-like Cells for Liver Regeneration. Protein Cell 2015, 6, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.E.; Reyes, M.; Koodie, L.; Jiang, Y.; Blackstad, M.; Lund, T.; Lenvik, T.; Johnson, S.; Hu, W.-S.; Verfaillie, C.M. Multipotent Adult Progenitor Cells from Bone Marrow Differentiate into Functional Hepatocyte-like Cells. J. Clin. Investig. 2002, 109, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Touboul, T.; Hannan, N.R.F.; Corbineau, S.; Martinez, A.; Martinet, C.; Branchereau, S.; Mainot, S.; Strick-Marchand, H.; Pedersen, R.; Di Santo, J.; et al. Generation of Functional Hepatocytes from Human Embryonic Stem Cells under Chemically Defined Conditions That Recapitulate Liver Development. Hepatology 2010, 51, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Robinton, D.A.; Daley, G.Q. The Promise of Induced Pluripotent Stem Cells in Research and Therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Hannan, N.R.F.; Segeritz, C.-P.; Touboul, T.; Vallier, L. Production of Hepatocyte-like Cells from Human Pluripotent Stem Cells. Nat. Protoc. 2013, 8, 430–437. [Google Scholar] [CrossRef]
- Lu, J.; Einhorn, S.; Venkatarangan, L.; Miller, M.; Mann, D.A.; Watkins, P.B.; LeCluyse, E. Morphological and Functional Characterization and Assessment of IPSC-Derived Hepatocytes for In Vitro Toxicity Testing. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 147, 39–54. [Google Scholar] [CrossRef]
- Wobser, H.; Dorn, C.; Weiss, T.S.; Amann, T.; Bollheimer, C.; Büttner, R.; Schölmerich, J.; Hellerbrand, C. Lipid Accumulation in Hepatocytes Induces Fibrogenic Activation of Hepatic Stellate Cells. Cell Res. 2009, 19, 996–1005. [Google Scholar] [CrossRef]
- Wang, L.; Chen, J.; Ning, C.; Lei, D.; Ren, J. Endoplasmic Reticulum Stress Related Molecular Mechanisms in Nonalcoholic Fatty Liver Disease (NAFLD). Curr. Drug Targets 2018, 19, 1087–1094. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Bai, X.; Hong, W.; Cai, P.; Chen, Y.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Valproate Induced Hepatic Steatosis by Enhanced Fatty Acid Uptake and Triglyceride Synthesis. Toxicol. Appl. Pharmacol. 2017, 324, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ding, D.; Huang, Q.; Liu, Q.; Lu, H.; Lu, Y.; Chi, Y.; Sun, X.; Ye, G.; Zhu, H.; et al. Downregulation of MiR-192 Causes Hepatic Steatosis and Lipid Accumulation by Inducing SREBF1: Novel Mechanism for Bisphenol A-Triggered Non-Alcoholic Fatty Liver Disease. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2017, 1862, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Parafati, M.; Kirby, R.J.; Khorasanizadeh, S.; Rastinejad, F.; Malany, S. A Nonalcoholic Fatty Liver Disease Model in Human Induced Pluripotent Stem Cell-Derived Hepatocytes, Created by Endoplasmic Reticulum Stress-Induced Steatosis. Dis. Model. Mech. 2018, 11, dmm033530. [Google Scholar] [CrossRef]
- Ling, J.; Lewis, J.; Douglas, D.; Kneteman, N.M.; Vance, D.E. Characterization of Lipid and Lipoprotein Metabolism in Primary Human Hepatocytes. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Green, C.J.; Johnson, D.; Amin, H.D.; Sivathondan, P.; Silva, M.A.; Wang, L.M.; Stevanato, L.; McNeil, C.A.; Miljan, E.A.; Sinden, J.D.; et al. Characterization of Lipid Metabolism in a Novel Immortalized Human Hepatocyte Cell Line. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E511–E522. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2020, 9, 37. [Google Scholar] [CrossRef]
- Giraudi, P.J.; Barbero Becerra, V.J.; Marin, V.; Chavez-Tapia, N.C.; Tiribelli, C.; Rosso, N. The Importance of the Interaction between Hepatocyte and Hepatic Stellate Cells in Fibrogenesis Induced by Fatty Accumulation. Exp. Mol. Pathol. 2015, 98, 85–92. [Google Scholar] [CrossRef]
- Barbero-Becerra, V.J.; Giraudi, P.J.; Chávez-Tapia, N.C.; Uribe, M.; Tiribelli, C.; Rosso, N. The Interplay between Hepatic Stellate Cells and Hepatocytes in an in Vitro Model of NASH. Toxicol. Vitr. 2015, 29, 1753–1758. [Google Scholar] [CrossRef]
- Bale, S.S.; Golberg, I.; Jindal, R.; McCarty, W.J.; Luitje, M.; Hegde, M.; Bhushan, A.; Usta, O.B.; Yarmush, M.L. Long-Term Coculture Strategies for Primary Hepatocytes and Liver Sinusoidal Endothelial Cells. Tissue Eng. Part C Methods 2014, 21, 413–422. [Google Scholar] [CrossRef]
- Suurmond, C.-A.E.; Lasli, S.; van den Dolder, F.W.; Ung, A.; Kim, H.-J.; Bandaru, P.; Lee, K.; Cho, H.-J.; Ahadian, S.; Ashammakhi, N.; et al. In Vitro Human Liver Model of Nonalcoholic Steatohepatitis by Coculturing Hepatocytes, Endothelial Cells, and Kupffer Cells. Adv. Healthc. Mater. 2019, 8, 1901379. [Google Scholar] [CrossRef]
- Müller, F.A.; Sturla, S.J. Human in Vitro Models of Nonalcoholic Fatty Liver Disease. Curr. Opin. Toxicol. 2019, 16, 9–16. [Google Scholar] [CrossRef]
- Kim, B.-M.; Abdelfattah, A.M.; Vasan, R.; Fuchs, B.C.; Choi, M.Y. Hepatic Stellate Cells Secrete Ccl5 to Induce Hepatocyte Steatosis. Sci. Rep. 2018, 8, 7499. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.C.Y.; Tompkins, R.G.; Yarmush, M.L. Long-Term in Vitro Function of Adult Hepatocytes in a Collagen Sandwich Configuration. Biotechnol. Prog. 1991, 7, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, F.; Moghe, P.V.; Toner, M.; Yarmush, M.L. Effect of Extracellular Matrix Topology on Cell Structure, Function, and Physiological Responsiveness: Hepatocytes Cultured in a Sandwich Configuration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Janorkar, A.V.; Harris, L.M.; Murphey, B.S.; Sowell, B.L. Use of Three-Dimensional Spheroids of Hepatocyte-Derived Reporter Cells to Study the Effects of Intracellular Fat Accumulation and Subsequent Cytokine Exposure. Biotechnol. Bioeng. 2011, 108, 1171–1180. [Google Scholar] [CrossRef]
- Ota, H.; Miki, N. Microtechnology-Based Three-Dimensional Spheroid Formation. Front. Biosci. Elite Ed. 2013, 5, 37–48. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Grunsven, L.A. 3D in Vitro Models of Liver Fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 133–146. [Google Scholar] [CrossRef]
- Kozyra, M.; Johansson, I.; Nordling, Å.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human Hepatic 3D Spheroids as a Model for Steatosis and Insulin Resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Lindén, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Puigvert, L.F.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of Primary Human Hepatocyte Spheroids as a Model System for Drug-Induced Liver Injury, Liver Function and Disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef]
- Rogozhnikov, D.; Luo, W.; Elahipanah, S.; O’Brien, P.J.; Yousaf, M.N. Generation of a Scaffold-Free Three-Dimensional Liver Tissue via a Rapid Cell-to-Cell Click Assembly Process. Bioconjug. Chem. 2016, 27, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Baze, A.; Parmentier, C.; Hendriks, D.F.G.; Hurrell, T.; Heyd, B.; Bachellier, P.; Schuster, C.; Ingelman-Sundberg, M.; Richert, L. Three-Dimensional Spheroid Primary Human Hepatocytes in Monoculture and Coculture with Nonparenchymal Cells. Tissue Eng. Part C Methods 2018, 24, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, Å.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human Liver Spheroids as a Model to Study Aetiology and Treatment of Hepatic Fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed]
- Underhill, G.H.; Khetani, S.R. Advances in Engineered Human Liver Platforms for Drug Metabolism Studies. Drug Metab. Dispos. 2018, 46, 1626–1637. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345. [Google Scholar] [CrossRef]
- Wu, F.; Wu, D.; Ren, Y.; Huang, Y.; Feng, B.; Zhao, N.; Zhang, T.; Chen, X.; Chen, S.; Xu, A. Generation of Hepatobiliary Organoids from Human Induced Pluripotent Stem Cells. J. Hepatol. 2019, 70, 1145–1158. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef]
- Kostrzewski, T.; Cornforth, T.; Snow, S.A.; Ouro-Gnao, L.; Rowe, C.; Large, E.M.; Hughes, D.J. Three-Dimensional Perfused Human in Vitro Model of Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2017, 23, 204–215. [Google Scholar] [CrossRef]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef]
- De l’Hortet, A.C.; Takeishi, K.; Guzman-Lepe, J.; Morita, K.; Achreja, A.; Popovic, B.; Wang, Y.; Handa, K.; Mittal, A.; Meurs, N.; et al. Generation of Human Fatty Livers Using Custom-Engineered Induced Pluripotent Stem Cells with Modifiable SIRT1 Metabolism. Cell Metab. 2019, 30, 385–401.e9. [Google Scholar] [CrossRef]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging Roles of SIRT1 in Fatty Liver Diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Banaeiyan, A.A.; Theobald, J.; Paukštyte, J.; Wölfl, S.; Adiels, C.B.; Goksör, M. Design and Fabrication of a Scalable Liver-Lobule-on-a-Chip Microphysiological Platform. Biofabrication 2017, 9, 015014. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Sebastian, S.; Maharjan, S.; Lesha, A.; Carpenter, A.-M.; Liu, X.; Xie, X.; Livermore, C.; Zhang, Y.S.; Zarrinpar, A. Liver-on-a-Chip Models of Fatty Liver Disease. Hepatology 2020, 71, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Rennert, K.; Steinborn, S.; Gröger, M.; Ungerböck, B.; Jank, A.-M.; Ehgartner, J.; Nietzsche, S.; Dinger, J.; Kiehntopf, M.; Funke, H.; et al. A Microfluidically Perfused Three Dimensional Human Liver Model. Biomaterials 2015, 71, 119–131. [Google Scholar] [CrossRef]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef]
- Lee, S.Y.; Sung, J.H. Gut–Liver on a Chip toward an in Vitro Model of Hepatic Steatosis. Biotechnol. Bioeng. 2018, 115, 2817–2827. [Google Scholar] [CrossRef]
- Green, C.J.; Pramfalk, C.; Morten, K.J.; Hodson, L. From Whole Body to Cellular Models of Hepatic Triglyceride Metabolism: Man Has Got to Know His Limitations. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1–E20. [Google Scholar] [CrossRef]
- Van de Bovenkamp, M.; Groothuis, G.M.M.; Meijer, D.K.F.; Olinga, P. Liver Fibrosis in Vitro: Cell Culture Models and Precision-Cut Liver Slices. Toxicol. Vitr. 2007, 21, 545–557. [Google Scholar] [CrossRef]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-Cut Liver Slices: A Versatile Tool to Advance Liver Research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef]
- Kartasheva, D.; Gaston, J.; Scatton, O.; Vaillant, J.-C.; Morozov, V.A.; Stanislas, P.; Lagaye, S. Establishment of an Ex Vivo Model of Human Fibrotic Liver Slices Culture: Characterization of Intrahepatic Immune Cells and TH17 Cytokines. J. Hepatol. 2018, 68, S405–S406. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, J.; Tacke, F. How Effective Are Nonalcoholic Fatty Liver Disease Models for Drug Discovery? Expert Opin. Drug Discov. 2020, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Thiele, G.M.; Duryee, M.J.; Thiele, G.E.; Tuma, D.J.; Klassen, L.W. Review: Precision Cut Liver Slices for the Evaluation of Fatty Liver and Fibrosis. Curr. Mol. Pharmacol. 2017, 10, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Barrera, F.; George, J. The Role of Diet and Nutritional Intervention for the Management of Patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Liao, J.K. A Mouse Model of Diet-Induced Obesity and Insulin Resistance. Methods Mol. Biol. Clifton NJ 2012, 821, 421. [Google Scholar] [CrossRef]
- Hariri, N.; Thibault, L. High-Fat Diet-Induced Obesity in Animal Models. Nutr. Res. Rev. 2010, 23, 270–299. [Google Scholar] [CrossRef]
- Lai, M.; Chandrasekera, P.C.; Barnard, N.D. You Are What You Eat, or Are You? The Challenges of Translating High-Fat-Fed Rodents to Human Obesity and Diabetes. Nutr. Diabetes 2014, 4, e135. [Google Scholar] [CrossRef]
- Winzell, M.S.; Ahrén, B. The High-Fat Diet–Fed Mouse: A Model for Studying Mechanisms and Treatment of Impaired Glucose Tolerance and Type 2 Diabetes. Diabetes 2004, 53, S215–S219. [Google Scholar] [CrossRef]
- Aydos, L.R.; Amaral, L.A.D.; de Souza, R.S.; Jacobowski, A.C.; dos Santos, E.F.; Macedo, M.L.R. Nonalcoholic Fatty Liver Disease Induced by High-Fat Diet in C57bl/6 Models. Nutrients 2019, 11, 3067. [Google Scholar] [CrossRef]
- Nevzorova, Y.A.; Boyer-Diaz, Z.; Cubero, F.J.; Gracia-Sancho, J. Animal Models for Liver Disease—A Practical Approach for Translational Research. J. Hepatol. 2020, 73, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.-K.; Mirshahi, F.; et al. A Diet-Induced Animal Model of Non-Alcoholic Fatty Liver Disease and Hepatocellular Cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Hallahan, N.L.; Brown, S.H.; Liu, M.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Mouse Strain-Dependent Variation in Obesity and Glucose Homeostasis in Response to High-Fat Feeding. Diabetologia 2013, 56, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.F.; Vercesi, A.E.; Castilho, R.F. A Spontaneous Mutation in the Nicotinamide Nucleotide Transhydrogenase Gene of C57BL/6J Mice Results in Mitochondrial Redox Abnormalities. Free Radic. Biol. Med. 2013, 63, 446–456. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Malhi, H.; Gores, G.J. Animal Models of Nonalcoholic Steatohepatitis: Eat, Delete, and Inflame. Dig. Dis. Sci. 2016, 61, 1325–1336. [Google Scholar] [CrossRef]
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2221–2236. [Google Scholar] [CrossRef]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-Fructose, Medium Chain Trans Fat Diet Induces Liver Fibrosis and Elevates Plasma Coenzyme Q9 in a Novel Murine Model of Obesity and Nonalcoholic Steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast Food Diet Mouse: Novel Small Animal Model of NASH with Ballooning, Progressive Fibrosis, and High Physiological Fidelity to the Human Condition. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef]
- Henkel, J.; Coleman, C.D.; Schraplau, A.; Jöhrens, K.; Weber, D.; Castro, J.P.; Hugo, M.; Schulz, T.J.; Krämer, S.; Schürmann, A.; et al. Induction of Steatohepatitis (NASH) with Insulin Resistance in Wild-Type B6 Mice by a Western-Type Diet Containing Soybean Oil and Cholesterol. Mol. Med. 2017, 23, 70–82. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Ebata, Y.; Sakabe, T.; Hama, S.; Kogure, K.; Shiota, G. High-Fat, High-Fructose Diet Induces Hepatic Iron Overload via a Hepcidin-Independent Mechanism Prior to the Onset of Liver Steatosis and Insulin Resistance in Mice. Metabolism 2013, 62, 62–69. [Google Scholar] [CrossRef]
- Itagaki, H.; Shimizu, K.; Morikawa, S.; Ogawa, K.; Ezaki, T. Morphological and Functional Characterization of Non-Alcoholic Fatty Liver Disease Induced by a Methionine-Choline-Deficient Diet in C57BL/6 Mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar] [PubMed]
- Cole, L.K.; Vance, J.E.; Vance, D.E. Phosphatidylcholine Biosynthesis and Lipoprotein Metabolism. Biochim. Biophys. Acta 2012, 1821, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.F.; Wiggins, D.; Brown, A.-M.; Hebbachi, A.-M. Synthesis and Function of Hepatic Very-Low-Density Lipoprotein. Biochem. Soc. Trans. 2004, 32, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Oz, H.S.; Chen, T.S.; Neuman, M. Methionine Deficiency and Hepatic Injury in a Dietary Steatohepatitis Model. Dig. Dis. Sci. 2008, 53, 767–776. [Google Scholar] [CrossRef]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A Positive Feedback Loop between RIP3 and JNK Controls Non-Alcoholic Steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific Contribution of Methionine and Choline in Nutritional Nonalcoholic Steatohepatitis: Impact on Mitochondrial S-Adenosyl-L-Methionine and Glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef]
- Greene, M.W.; Burrington, C.M.; Lynch, D.T.; Davenport, S.K.; Johnson, A.K.; Horsman, M.J.; Chowdhry, S.; Zhang, J.; Sparks, J.D.; Tirrell, P.C. Lipid Metabolism, Oxidative Stress and Cell Death Are Regulated by PKC Delta in a Dietary Model of Nonalcoholic Steatohepatitis. PLoS ONE 2014, 9, e85848. [Google Scholar] [CrossRef]
- Peña, A.D.; Leclercq, I.; Field, J.; George, J.; Jones, B.; Farrell, G. NF-ΚB Activation, Rather Than TNF, Mediates Hepatic Inflammation in a Murine Dietary Model of Steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [Google Scholar] [CrossRef]
- Rinella, M.E.; Green, R.M. The Methionine-Choline Deficient Dietary Model of Steatohepatitis Does Not Exhibit Insulin Resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M.; Williams, J.; Bell-Anderson, K.S.; Farrell, G.C. MCD-Induced Steatohepatitis Is Associated with Hepatic Adiponectin Resistance and Adipogenic Transformation of Hepatocytes. J. Hepatol. 2008, 49, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kakizaki, S.; Takizawa, D.; Ichikawa, T.; Sato, K.; Takagi, H.; Mori, M. Interstrain Differences in Susceptibility to Non-Alcoholic Steatohepatitis. J. Gastroenterol. Hepatol. 2008, 23, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.; de Conti, A.; Kobets, T.; Kutanzi, K.; Koturbash, I.; Han, T.; Fuscoe, J.C.; Latendresse, J.R.; Melnyk, S.; Shymonyak, S.; et al. Interstrain Differences in the Severity of Liver Injury Induced by a Choline- and Folate-Deficient Diet in Mice Are Associated with Dysregulation of Genes Involved in Lipid Metabolism. FASEB J. 2012, 26, 4592–4602. [Google Scholar] [CrossRef]
- Passman, A.M.; Strauss, R.P.; McSpadden, S.B.; Finch-Edmondson, M.L.; Woo, K.H.; Diepeveen, L.A.; London, R.; Callus, B.A.; Yeoh, G.C. A Modified Choline-Deficient, Ethionine-Supplemented Diet Reduces Morbidity and Retains a Liver Progenitor Cell Response in Mice. Dis. Model. Mech. 2015, 8, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Callejero, L.; Pérez-Martínez, L.; Rubio-Mediavilla, S.; Oteo, J.A.; Martínez, A.; Blanco, J.R. Maraviroc, a CCR5 Antagonist, Prevents Development of Hepatocellular Carcinoma in a Mouse Model. PLoS ONE 2013, 8, e53992. [Google Scholar] [CrossRef]
- Gogoi-Tiwari, J.; Köhn-Gaone, J.; Giles, C.; Schmidt-Arras, D.; Gratte, F.D.; Elsegood, C.L.; McCaughan, G.W.; Ramm, G.A.; Olynyk, J.K.; Tirnitz-Parker, J.E.E. The Murine Choline-Deficient, Ethionine-Supplemented (CDE) Diet Model of Chronic Liver Injury. J. Vis. Exp. 2017, e56138. [Google Scholar] [CrossRef]
- Nakae, D.; Mizumoto, Y.; Andoh, N.; Tamura, K.; Horiguchi, K.; Endoh, T.; Kobayashi, E.; Tsujiuchi, T.; Denda, A.; Lombardi, B.; et al. Comparative Changes in the Liver of Female Fischer-344 Rats after Short-Term Feeding of a Semipurified or a Semisynthetic L-Amino Acid-Defined Choline-Deficient Diet. Toxicol. Pathol. 1995, 23, 583–590. [Google Scholar] [CrossRef]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse Models of Nonalcoholic Steatohepatitis in Preclinical Drug Development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An Improved Mouse Model That Rapidly Develops Fibrosis in Non-Alcoholic Steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef]
- Denda, A.; Kitayama, W.; Kishida, H.; Murata, N.; Tamura, K.; Kusuoka, O.; Tsutsumi, M.; Nishikawa, F.; Kita, E.; Nakae, D.; et al. Expression of Inducible Nitric Oxide (NO) Synthase but Not Prevention by Its Gene Ablation of Hepatocarcinogenesis with Fibrosis Caused by a Choline-Deficient, l-Amino Acid-Defined Diet in Rats and Mice. Nitric Oxide 2007, 16, 164–176. [Google Scholar] [CrossRef]
- Hebbard, L.; George, J. Animal Models of Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1beta in Mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [PubMed]
- Rakieten, N.; Rakieten, M.L.; Nadkarni, M.V. Studies on the Diabetogenic Action of Streptozotocin (NSC-37917). Cancer Chemother. Rep. 1963, 29, 91–98. [Google Scholar]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A Murine Model for Non-Alcoholic Steatohepatitis Showing Evidence of Association between Diabetes and Hepatocellular Carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Uebanso, T.; Maekawa, K.; Ishikawa, M.; Taguchi, R.; Nammo, T.; Nishimaki-Mogami, T.; Udagawa, H.; Fujii, M.; Shibazaki, Y.; et al. Characterization of Hepatic Lipid Profiles in a Mouse Model with Nonalcoholic Steatohepatitis and Subsequent Fibrosis. Sci. Rep. 2015, 5, 12466. [Google Scholar] [CrossRef]
- Mann, J.P.; Semple, R.K.; Armstrong, M.J. How Useful Are Monogenic Rodent Models for the Study of Human Non-Alcoholic Fatty Liver Disease? Front. Endocrinol. 2016, 7, 145. [Google Scholar] [CrossRef]
- Frühbeck, G.; Jebb, S.A.; Prentice, A.M. Leptin: Physiology and Pathophysiology. Clin. Physiol. 1998, 18, 399–419. [Google Scholar] [CrossRef]
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The Role of Leptin and Ghrelin in the Regulation of Food Intake and Body Weight in Humans: A Review. Obes. Rev. 2007, 8, 21–34. [Google Scholar] [CrossRef]
- Wang, B.; Charukeshi Chandrasekera, P.; Pippin, J.J. Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef]
- Uygun, A.; Kadayifci, A.; Yesilova, Z.; Erdil, A.; Yaman, H.; Saka, M.; Deveci, M.S.; Bagci, S.; Gulsen, M.; Karaeren, N.; et al. Serum Leptin Levels in Patients with Nonalcoholic Steatohepatitis. Am. J. Gastroenterol. 2000, 95, 3584–3589. [Google Scholar] [CrossRef]
- Huang, X.-D.; Fan, Y.; Zhang, H.; Wang, P.; Yuan, J.P.; Li, M.-J.; Zhan, X.-Y. Serum Leptin and Soluble Leptin Receptor in Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2008, 14, 2888–2893. [Google Scholar] [CrossRef] [PubMed]
- Cernea, S.; Roiban, A.L.; Both, E.; Huţanu, A. Serum Leptin and Leptin Resistance Correlations with NAFLD in Patients with Type 2 Diabetes. Diabetes Metab. Res. Rev. 2018, 34, e3050. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in Nonalcoholic Fatty Liver Disease: A Narrative Review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef]
- Saxena, N.K.; Ikeda, K.; Rockey, D.C.; Friedman, S.L.; Anania, F.A. Leptin in Hepatic Fibrosis: Evidence for Increased Collagen Production in Stellate Cells and Lean Littermates of Ob/Ob Mice. Hepatology 2002, 35, 762–771. [Google Scholar] [CrossRef]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the Liver in Obese and Diabetic Ob/Ob and Db/Db Mice Fed a Standard or High-Calorie Diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and Diabetic Db/Db Mice Develop Marked Liver Fibrosis in a Model of Nonalcoholic Steatohepatitis: Role of Short-Form Leptin Receptors and Osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef]
- Hoggard, N.; Hunter, L.; Duncan, J.S.; Rayner, D.V. Regulation of Adipose Tissue Leptin Secretion by Alpha-Melanocyte-Stimulating Hormone and Agouti-Related Protein: Further Evidence of an Interaction between Leptin and the Melanocortin Signalling System. J. Mol. Endocrinol. 2004, 32, 145–153. [Google Scholar] [CrossRef]
- Tao, Y.-X. The Melanocortin-4 Receptor: Physiology, Pharmacology, and Pathophysiology. Endocr. Rev. 2010, 31, 506–543. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Keogh, J.M.; Yeo, G.S.H.; Lank, E.J.; Cheetham, T.; O’Rahilly, S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N. Engl. J. Med. 2003, 348, 1085–1095. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Lindgren, C.M.; Li, S.; Wheeler, E.; Zhao, J.H.; Prokopenko, I.; Inouye, M.; Freathy, R.M.; Attwood, A.P.; Beckmann, J.S.; et al. Common Variants near MC4R Are Associated with Fat Mass, Weight and Risk of Obesity. Nat. Genet. 2008, 40, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Huszar, D.; Lynch, C.A.; Fairchild-Huntress, V.; Dunmore, J.H.; Fang, Q.; Berkemeier, L.R.; Gu, W.; Kesterson, R.A.; Boston, B.A.; Cone, R.D.; et al. Targeted Disruption of the Melanocortin-4 Receptor Results in Obesity in Mice. Cell 1997, 88, 131–141. [Google Scholar] [CrossRef]
- Marsh, D.J.; Hollopeter, G.; Huszar, D.; Laufer, R.; Yagaloff, K.A.; Fisher, S.L.; Burn, P.; Palmiter, R.D. Response of Melanocortin–4 Receptor–Deficient Mice to Anorectic and Orexigenic Peptides. Nat. Genet. 1999, 21, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Suganami, T.; Nakagawa, N.; Tanaka, M.; Yamamoto, Y.; Kamei, Y.; Terai, S.; Sakaida, I.; Ogawa, Y. Melanocortin 4 Receptor–Deficient Mice as a Novel Mouse Model of Nonalcoholic Steatohepatitis. Am. J. Pathol. 2011, 179, 2454–2463. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased Levels of Nuclear SREBP-1c Associated with Fatty Livers in Two Mouse Models of Diabetes Mellitus. J. Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-Regulated Lipid Metabolism: Convergent Physiology—Divergent Pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c, Regulated by the Insulin and AMPK Signaling Pathways, Plays a Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef]
- Kim, Y.-R.; Lee, E.-J.; Shin, K.-O.; Kim, M.H.; Pewzner-Jung, Y.; Lee, Y.-M.; Park, J.-W.; Futerman, A.H.; Park, W.-J. Hepatic Triglyceride Accumulation via Endoplasmic Reticulum Stress-Induced SREBP-1 Activation Is Regulated by Ceramide Synthases. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Köllmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-Specific Expression of Transcriptionally Active SREBP-1c Is Associated with Fatty Liver and Increased Visceral Fat Mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef]
- Jelenik, T.; Kaul, K.; Séquaris, G.; Flögel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Hörbelt, T.; Lehr, S.; et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef]
- Sanches, S.C.L.; Ramalho, L.N.Z.; Augusto, M.J.; da Silva, D.M.; Ramalho, F.S. Nonalcoholic Steatohepatitis: A Search for Factual Animal Models. BioMed Res. Int. 2015, 2015, 574832. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Miyashita, Y.; Sasaki, M.; Aruga, Y.; Nakamura, Y.; Ishii, Y.; Sasahara, M.; Kanasaki, K.; Kitada, M.; Koya, D.; et al. Eplerenone Ameliorates the Phenotypes of Metabolic Syndrome with NASH in Liver-Specific SREBP-1c Tg Mice Fed High-Fat and High-Fructose Diet. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1415–E1425. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.; Warda, A.-S.; Verbeek, J.; Cassiman, D.; Spincemaille, P. An Overview of Mouse Models of Nonalcoholic Steatohepatitis: From Past to Present. Curr. Protoc. Mouse Biol. 2016, 6, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Droz, B.A.; Sneed, B.L.; Jackson, C.V.; Zimmerman, K.M.; Michael, M.D.; Emmerson, P.J.; Coskun, T.; Peterson, R.G. Correlation of Disease Severity with Body Weight and High Fat Diet in the FATZO/Pco Mouse. PLoS ONE 2017, 12, e0179808. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.G.; Jackson, C.V.; Zimmerman, K.M.; Alsina-Fernandez, J.; Michael, M.D.; Emmerson, P.J.; Coskun, T. Glucose Dysregulation and Response to Common Anti-Diabetic Agents in the FATZO/Pco Mouse. PLoS ONE 2017, 12, e0179856. [Google Scholar] [CrossRef]
- Sun, G.; Jackson, C.V.; Zimmerman, K.; Zhang, L.-K.; Finnearty, C.M.; Sandusky, G.E.; Zhang, G.; Peterson, R.G.; Wang, Y.-X.J. The FATZO Mouse, a next Generation Model of Type 2 Diabetes, Develops NAFLD and NASH When Fed a Western Diet Supplemented with Fructose. BMC Gastroenterol. 2019, 19, 41. [Google Scholar] [CrossRef]
- Maciejewska, D.; Łukomska, A.; Dec, K.; Skonieczna-Żydecka, K.; Gutowska, I.; Skórka-Majewicz, M.; Styburski, D.; Misiakiewicz-Has, K.; Pilutin, A.; Palma, J.; et al. Diet-Induced Rat Model of Gradual Development of Non-Alcoholic Fatty Liver Disease (NAFLD) with Lipopolysaccharides (LPS) Secretion. Diagnostics 2019, 9, 205. [Google Scholar] [CrossRef]
- Van der Graaff, D.; Kwanten, W.J.; Couturier, F.J.; Govaerts, J.S.; Verlinden, W.; Brosius, I.; D’Hondt, M.; Driessen, A.; De Winter, B.Y.; De Man, J.G.; et al. Severe Steatosis Induces Portal Hypertension by Systemic Arterial Hyporeactivity and Hepatic Vasoconstrictor Hyperreactivity in Rats. Lab. Investig. 2018, 98, 1263–1275. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Tussellino, M.; Carotenuto, R.; Liverini, G.; Iossa, S. Fructose Supplementation Worsens the Deleterious Effects of Short-Term High-Fat Feeding on Hepatic Steatosis and Lipid Metabolism in Adult Rats. Exp. Physiol. 2014, 99, 1203–1213. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical Models of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A.; Mak, K.M.; Xu, Y.; Cao, Q.; Ren, C.; Ponomarenko, A.; DeCarli, L.M. Model of Nonalcoholic Steatohepatitis. Am. J. Clin. Nutr. 2004, 79, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Stöppeler, S.; Palmes, D.; Fehr, M.; Hölzen, J.P.; Zibert, A.; Siaj, R.; Schmidt, H.H.-J.; Spiegel, H.-U.; Bahde, R. Gender and Strain-Specific Differences in the Development of Steatosis in Rats. Lab. Anim. 2013, 47, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Murakami, T.; Iida, M.; Kuwajima, M.; Shima, K. Leptin Receptor of Zucker Fatty Rat Performs Reduced Signal Transduction. Diabetes 1997, 46, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Kucera, O.; Cervinkova, Z. Experimental Models of Non-Alcoholic Fatty Liver Disease in Rats. World J. Gastroenterol. 2014, 20, 8364–8376. [Google Scholar] [CrossRef] [PubMed]
- Carmiel-Haggai, M.; Cederbaum, A.I.; Nieto, N. A High-Fat Diet Leads to the Progression of Non-Alcoholic Fatty Liver Disease in Obese Rats. FASEB J. 2005, 19, 136–138. [Google Scholar] [CrossRef]
- Pedersen, H.D.; Galsgaard, E.D.; Christoffersen, B.Ø.; Cirera, S.; Holst, D.; Fredholm, M.; Latta, M. NASH-Inducing Diets in Göttingen Minipigs. J. Clin. Exp. Hepatol. 2020, 10, 211–221. [Google Scholar] [CrossRef]
- Lee, L.; Alloosh, M.; Saxena, R.; Alstine, W.V.; Watkins, B.A.; Klaunig, J.E.; Sturek, M.; Chalasani, N. Nutritional Model of Steatohepatitis and Metabolic Syndrome in the Ossabaw Miniature Swine. Hepatology 2009, 50, 56–67. [Google Scholar] [CrossRef]
- Litten-Brown, J.C.; Corson, A.M.; Clarke, L. Porcine Models for the Metabolic Syndrome, Digestive and Bone Disorders: A General Overview. Animal 2010, 4, 899–920. [Google Scholar] [CrossRef]
- Forster, R.; Bode, G.; Ellegaard, L.; van der Laan, J.W. The RETHINK Project: Minipigs as Models for the Toxicity Testing of New Medicines and Chemicals: An Impact Assessment. J. Pharmacol. Toxicol. Methods 2010, 62, 158–159. [Google Scholar] [CrossRef]
- Schumacher-Petersen, C.; Christoffersen, B.Ø.; Kirk, R.K.; Ludvigsen, T.P.; Zois, N.E.; Pedersen, H.D.; Vyberg, M.; Olsen, L.H. Experimental Non-Alcoholic Steatohepatitis in Göttingen Minipigs: Consequences of High Fat-Fructose-Cholesterol Diet and Diabetes. J. Transl. Med. 2019, 17, 110. [Google Scholar] [CrossRef]
- Bergen, W.G.; Mersmann, H.J. Comparative Aspects of Lipid Metabolism: Impact on Contemporary Research and Use of Animal Models. J. Nutr. 2005, 135, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.C.; Liang, Z.; Sun, F.; Yang, Z.; Tang, C.; Chen, Z.; Yubo, S.; Yao, Z.; Wu, M.; Chen, Y.; et al. Nonalcoholic Fatty Liver Disease (NAFLD) in Obese Rhesus Monkeys Provides the First Animal Model That Accurately Reflects the Human Condition. FASEB J. 2017, 31, 895–896. [Google Scholar] [CrossRef]
- Ratziu, V.; Friedman, S.L. Why Do So Many NASH Trials Fail? Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soret, P.-A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2021, 10, 36. https://doi.org/10.3390/jcm10010036
Soret P-A, Magusto J, Housset C, Gautheron J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. Journal of Clinical Medicine. 2021; 10(1):36. https://doi.org/10.3390/jcm10010036
Chicago/Turabian StyleSoret, Pierre-Antoine, Julie Magusto, Chantal Housset, and Jérémie Gautheron. 2021. "In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal" Journal of Clinical Medicine 10, no. 1: 36. https://doi.org/10.3390/jcm10010036
APA StyleSoret, P.-A., Magusto, J., Housset, C., & Gautheron, J. (2021). In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. Journal of Clinical Medicine, 10(1), 36. https://doi.org/10.3390/jcm10010036