
Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Determination of Autoantibody Levels and Laboratory Blood Data
2.3. Questionnaires for Symptom Scoring
2.4. Statistical Analysis
3. Results
3.1. Cohort Characteristics
3.2. Correlation of AAB with Total IgG and Age
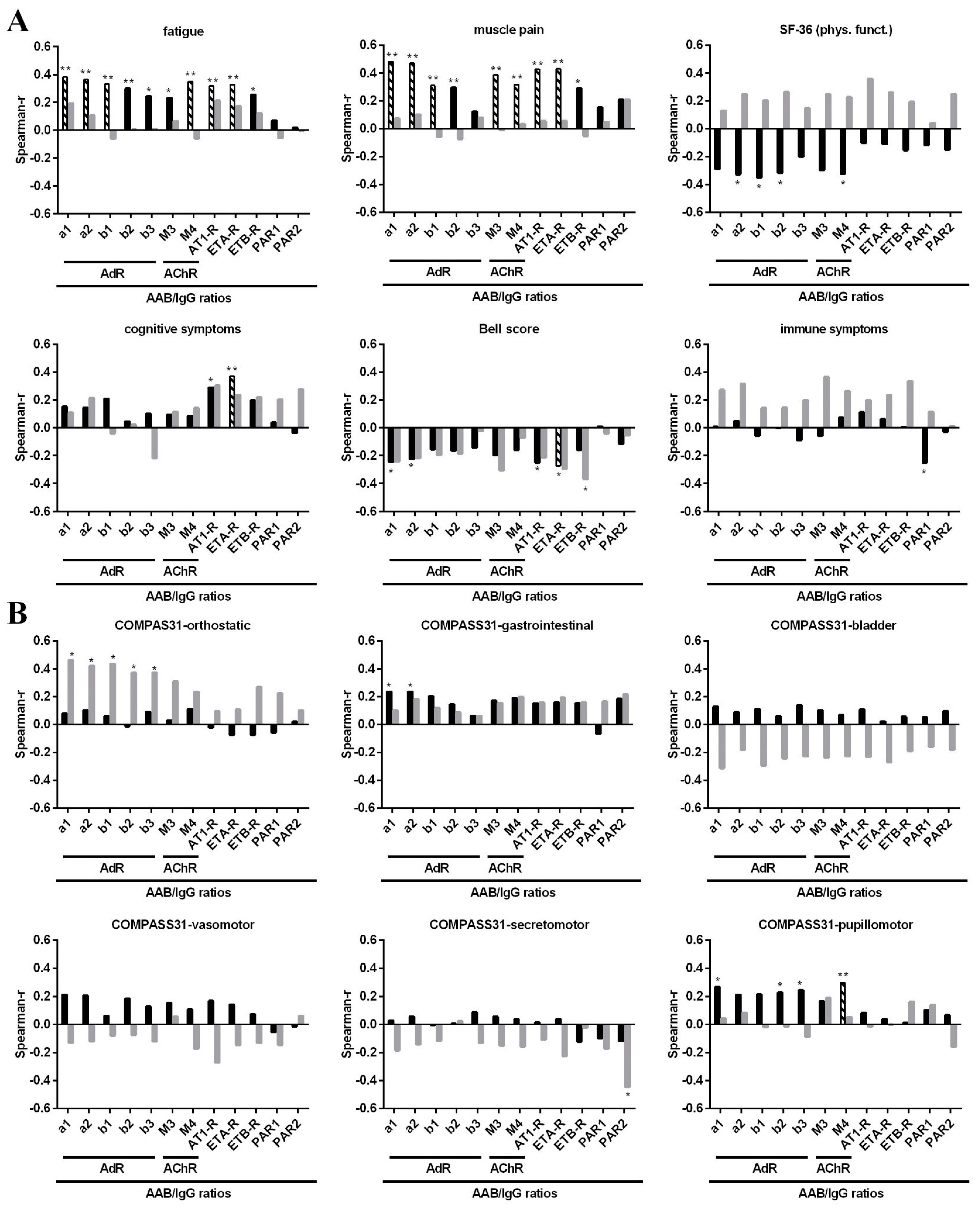
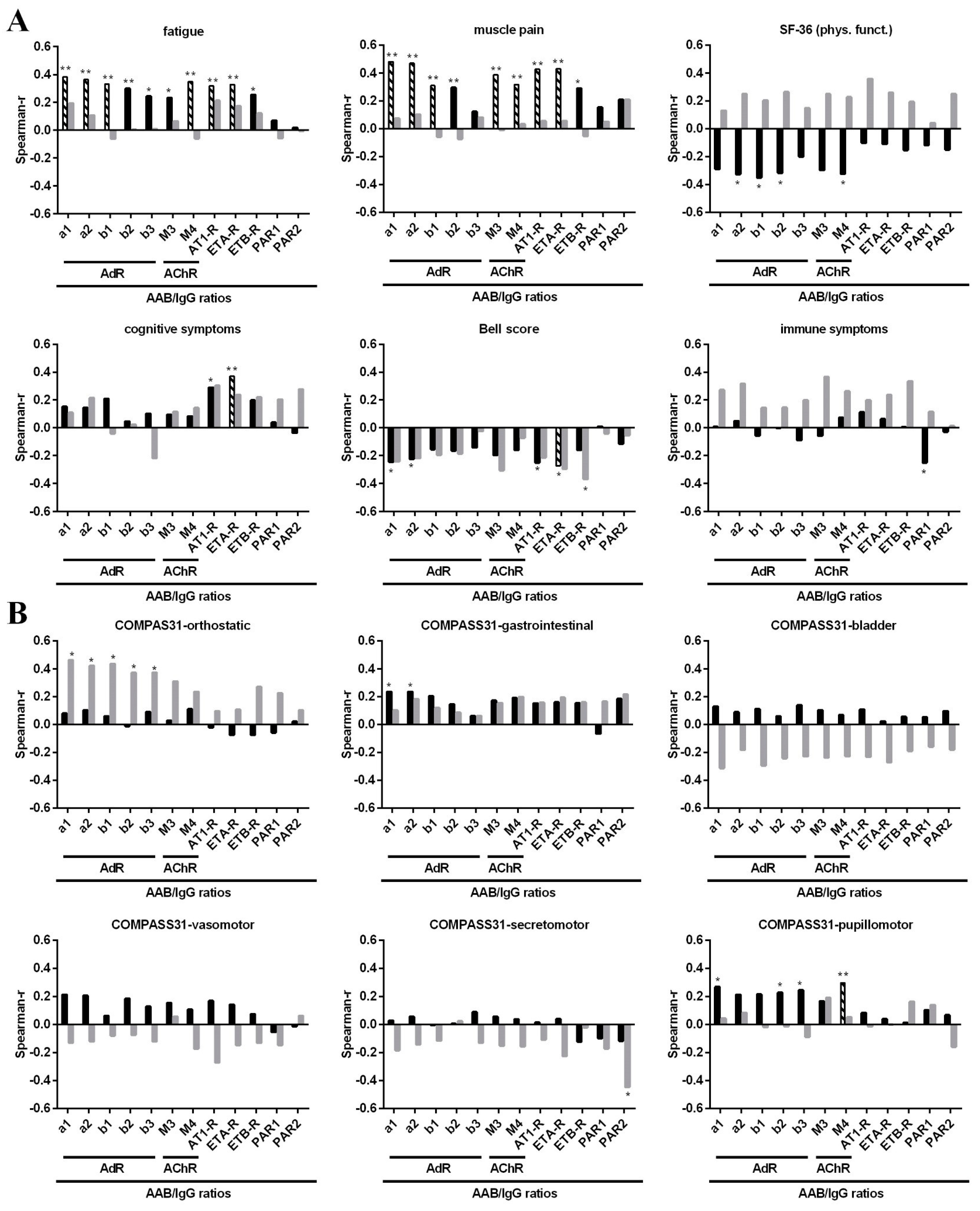
3.3. Correlation of AAB with Clinical Symptom Scores
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.P.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Chronic Fatigue Syndr. 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Bakken, I.J.; Tveito, K.; Gunnes, N.; Ghaderi, S.; Stoltenberg, C.; Trogstad, L.; Haberg, S.E.; Magnus, P. Two age peaks in the incidence of chronic fatigue syndrome/myalgic encephalomyelitis: A population-based registry study from Norway 2008–2012. BMC Med. 2014, 12, 167. [Google Scholar] [CrossRef] [Green Version]
- Valdez, A.R.; Hancock, E.E.; Adebayo, S.; Kiernicki, D.J.; Proskauer, D.; Attewell, J.R.; Bateman, L.; DeMaria, A., Jr.; Lapp, C.W.; Rowe, P.C.; et al. Estimating Prevalence, Demographics, and Costs of ME/CFS Using Large Scale Medical Claims Data and Machine Learning. Front. Pediatrics 2018, 6, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, L.; Valencia, I.J.; Garvert, D.W.; Montoya, J.G. Onset Patterns and Course of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Pediatrics 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Sotzny, F.; Blanco, J.; Capelli, E.; Castro-Marrero, J.; Steiner, S.; Murovska, M.; Scheibenbogen, C.; European Network on, M.C. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome—Evidence for an autoimmune disease. Autoimmun. Rev. 2018, 17, 601–609. [Google Scholar] [CrossRef]
- Vermeulen, R.C.; Kurk, R.M.; Visser, F.C.; Sluiter, W.; Scholte, H.R. Patients with chronic fatigue syndrome performed worse than controls in a controlled repeated exercise study despite a normal oxidative phosphorylation capacity. J. Transl. Med. 2010, 8, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, B.A.; Pryor, J.L.; Giloteaux, L. Inability of myalgic encephalomyelitis/chronic fatigue syndrome patients to reproduce VO(2)peak indicates functional impairment. J. Transl. Med. 2014, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, A.; Barupal, D.K.; Levine, S.M.; Hanson, M.R. Comprehensive Circulatory Metabolomics in ME/CFS Reveals Disrupted Metabolism of Acyl Lipids and Steroids. Metabolites 2020, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- van Campen, C.; Rowe, P.C.; Verheugt, F.W.A.; Visser, F.C. Cognitive Function Declines Following Orthostatic Stress in Adults With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Front. Neurosci. 2020, 14, 688. [Google Scholar] [CrossRef]
- van Campen, C.; Verheugt, F.W.A.; Rowe, P.C.; Visser, F.C. Cerebral blood flow is reduced in ME/CFS during head-up tilt testing even in the absence of hypotension or tachycardia: A quantitative, controlled study using Doppler echography. Clin. Neurophysiol. Pr. 2020, 5, 50–58. [Google Scholar] [CrossRef]
- Wirth, K.; Scheibenbogen, C. A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ss2-adrenergic receptors. Autoimmun. Rev. 2020, 19, 102527. [Google Scholar] [CrossRef]
- Holwerda, S.W.; Restaino, R.M.; Fadel, P.J. Adrenergic and non-adrenergic control of active skeletal muscle blood flow: Implications for blood pressure regulation during exercise. Auton. Neurosci. 2015, 188, 24–31. [Google Scholar] [CrossRef]
- Dragun, D.; Philippe, A.; Catar, R.; Hegner, B. Autoimmune mediated G-protein receptor activation in cardiovascular and renal pathologies. Thromb. Haemost. 2009, 101, 643–648. [Google Scholar]
- Dragun, D.; Muller, D.N.; Brasen, J.H.; Fritsche, L.; Nieminen-Kelha, M.; Dechend, R.; Kintscher, U.; Rudolph, B.; Hoebeke, J.; Eckert, D.; et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N. Engl. J. Med. 2005, 352, 558–569. [Google Scholar] [CrossRef] [Green Version]
- Wallukat, G.; Muller, J.; Podlowski, S.; Nissen, E.; Morwinski, R.; Hetzer, R. Agonist-like beta-adrenoceptor antibodies in heart failure. Am. J. Cardiol. 1999, 83, 75H–79H. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Riemekasten, G. Functional autoantibodies targeting G protein-coupled receptors in rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Marques, O.; Marques, A.; Giil, L.M.; De Vito, R.; Rademacher, J.; Gunther, J.; Lange, T.; Humrich, J.Y.; Klapa, S.; Schinke, S.; et al. GPCR-specific autoantibody signatures are associated with physiological and pathological immune homeostasis. Nat. Commun. 2018, 9, 5224. [Google Scholar] [CrossRef]
- Tanaka, S.; Kuratsune, H.; Hidaka, Y.; Hakariya, Y.; Tatsumi, K.I.; Takano, T.; Kanakura, Y.; Amino, N. Autoantibodies against muscarinic cholinergic receptor in chronic fatigue syndrome. Int. J. Mol. Med. 2003, 12, 225–230. [Google Scholar] [CrossRef]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Meisel, C.; Reinke, P.; Volk, H.D.; Fluge, O.; et al. Antibodies to beta adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bynke, A.J.P.; Gottfries, C.F.; Heidecke, H.; Scheibenbogen, C.; Bergquist, J. Autoantibodies to beta-adrenergic and muscarinic cholinergic receptors in Myalgic Encephalomyelitis (ME) patients—A validation study in plasma and cerebrospinal fluid from two Swedish cohorts. Brain Behav. Immun.-Health 2020, 7, 100107. [Google Scholar] [CrossRef]
- Fujii, H.; Sato, W.; Kimura, Y.; Matsuda, H.; Ota, M.; Maikusa, N.; Suzuki, F.; Amano, K.; Shin, I.; Yamamura, T.; et al. Altered Structural Brain Networks Related to Adrenergic/Muscarinic Receptor Autoantibodies in Chronic Fatigue Syndrome. J. Neuroimaging Off. J. Am. Soc. Neuroimaging 2020, 30, 822–827. [Google Scholar] [CrossRef]
- Hartwig, J.; Sotzny, F.; Bauer, S.; Heidecke, H.; Riemekasten, G.; Dragun, D.; Meisel, C.; Dames, C.; Grabowski, P.; Scheibenbogen, C. Research article IgG stimulated β2 adrenergic receptor activation is attenuated in patients with ME/CFS. Brain Behav. Immun.-Health 2020, 3, 100047. [Google Scholar] [CrossRef]
- Scheibenbogen, C.; Loebel, M.; Freitag, H.; Krueger, A.; Bauer, S.; Antelmann, M.; Doehner, W.; Scherbakov, N.; Heidecke, H.; Reinke, P.; et al. Immunoadsorption to remove ss2 adrenergic receptor antibodies in Chronic Fatigue Syndrome CFS/ME. PLoS ONE 2018, 13, e0193672. [Google Scholar] [CrossRef] [Green Version]
- Tolle, M.; Freitag, H.; Antelmann, M.; Hartwig, J.; Schuchardt, M.; van der Giet, M.; Eckardt, K.U.; Grabowski, P.; Scheibenbogen, C. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Efficacy of Repeat Immunoadsorption. J. Clin. Med. 2020, 9, 2443. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Becker, S.C.; Hartwig, J.; Sotzny, F.; Lorenz, S.; Bauer, S.; Lobel, M.; Stittrich, A.B.; Grabowski, P.; Scheibenbogen, C. Autoimmunity-Related Risk Variants in PTPN22 and CTLA4 Are Associated With ME/CFS With Infectious Onset. Front. Immunol. 2020, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Tognetto, M.; D’Andrea, M.R.; Trevisani, M.; Guerrini, R.; Salvadori, S.; Spisani, L.; Daniele, C.; Andrade-Gordon, P.; Geppetti, P.; Harrison, S. Proteinase-activated receptor-1 (PAR-1) activation contracts the isolated human renal artery in vitro. Br. J. Pharm. 2003, 139, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, Y.; Tanaka-Ishikawa, M.; Abe, K.; Hirano, M.; Hirooka, Y.; Tsutsui, H.; Sunagawa, K.; Hirano, K. Proteinase-activated receptor 1 antagonism ameliorates experimental pulmonary hypertension. Cardiovasc. Res. 2019, 115, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Tennant, G.M.; Wadsworth, R.M.; Kennedy, S. PAR-2 mediates increased inflammatory cell adhesion and neointima formation following vascular injury in the mouse. Atherosclerosis 2008, 198, 57–64. [Google Scholar] [CrossRef]
- Rhoden, A.; Speiser, J.; Geertz, B.; Uebeler, J.; Schmidt, K.; de Wit, C.; Eschenhagen, T. Preserved cardiovascular homeostasis despite blunted acetylcholine-induced dilation in mice with endothelial muscarinic M3 receptor deletion. Acta Physiol. 2019, 226, e13262. [Google Scholar] [CrossRef]
- Radu, B.M.; Osculati, A.M.M.; Suku, E.; Banciu, A.; Tsenov, G.; Merigo, F.; Di Chio, M.; Banciu, D.D.; Tognoli, C.; Kacer, P.; et al. All muscarinic acetylcholine receptors (M1-M5) are expressed in murine brain microvascular endothelium. Sci. Rep. 2017, 7, 5083. [Google Scholar] [CrossRef]
- Fluge, O.; Risa, K.; Lunde, S.; Alme, K.; Rekeland, I.G.; Sapkota, D.; Kristoffersen, E.K.; Sorland, K.; Bruland, O.; Dahl, O.; et al. B-Lymphocyte Depletion in Myalgic Encephalopathy/Chronic Fatigue Syndrome. An Open-Label Phase II Study with Rituximab Maintenance Treatment. PLoS ONE 2015, 10, e0129898. [Google Scholar] [CrossRef]
- Sletten, D.M.; Suarez, G.A.; Low, P.A.; Mandrekar, J.; Singer, W. COMPASS 31: A refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin. Proc. 2012, 87, 1196–1201. [Google Scholar] [CrossRef]
- Bell, D.S. The Doctor’s Guide to Chronic Fatigue Syndrome: Understanding, Treating and Living with CFIDS; Da Capo Lifelong Books: Boston, MA, USA, 1995. [Google Scholar]
- Cella, M.; Chalder, T. Measuring fatigue in clinical and community settings. J. Psychosom. Res. 2010, 69, 17–22. [Google Scholar] [CrossRef]
- Ware, J.E., Jr.; Sherbourne, C.D. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med. Care 1992, 30, 473–483. [Google Scholar] [CrossRef]
- Lock, R.J.; Unsworth, D.J. Immunoglobulins and immunoglobulin subclasses in the elderly. Ann. Clin. Biochem. 2003, 40, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunning, W.T., 3rd; Kvale, H.; Kramer, P.M.; Karabin, B.L.; Grubb, B.P. Postural Orthostatic Tachycardia Syndrome Is Associated With Elevated G-Protein Coupled Receptor Autoantibodies. J. Am. Heart Assoc. 2019, 8, e013602. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yu, X.; Liles, C.; Khan, M.; Vanderlinde-Wood, M.; Galloway, A.; Zillner, C.; Benbrook, A.; Reim, S.; Collier, D.; et al. Autoimmune basis for postural tachycardia syndrome. J. Am. Heart Assoc. 2014, 3, e000755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.H.; Wei, Y.M.; Wang, M.; Wang, Z.H.; Yuan, H.T.; Cheng, L.X. Autoantibodies against AT1-receptor and alpha1-adrenergic receptor in patients with hypertension. Hypertens. Res. 2002, 25, 641–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Yin, X.; Zhang, S.; Mao, C.; Cao, N.; Yang, X.; Bian, J.; Hao, W.; Fan, Q.; Liu, H. Autoantibodies against AT1 Receptor Contribute to Vascular Aging and Endothelial Cell Senescence. Aging Dis. 2019, 10, 1012–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Cao, Z.; Wu, X.W.; Wu, H.K.; Ma, Y.; Wu, B.; Wang, W.Q.; Cheng, J.; Zhou, Z.H.; Tu, Y.C. Autoantibodies against AT1 and alpha1-adrenergic receptors predict arterial stiffness progression in normotensive subjects over a 5-year period. Clin. Sci. 2017, 131, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, M.; Chen, Y.; Wang, Q.; Tian, Z.; Pan, S.; Zeng, X.; Ye, S. Anti-Endothelin Receptor Type A Autoantibodies in Systemic Lupus Erythematosus-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol. 2015, 67, 2394–2402. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kuhl, A.A.; Heidecke, H.; Ghofrani, H.A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef]
- Naitou, K.; Shiina, T.; Kato, K.; Nakamori, H.; Sano, Y.; Shimizu, Y. Colokinetic effect of noradrenaline in the spinal defecation center: Implication for motility disorders. Sci. Rep. 2015, 5, 12623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grub, M.; Mielke, J.; Rohrbach, J.M. [m4 muscarinic receptors of the cornea: Muscarinic cholinoceptor-stimulated inhibition of the cAMP-PKA pathway in corneal epithelial and endothelial cells]. Ophthalmologe 2011, 108, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, T.; Nakamura, T.; Obara, K.; Inoue, H.; Mishima, K.; Matsumoto, N.; Matsui, M.; Manabe, T.; Mikoshiba, K.; Saito, I. Up-regulated PAR-2-mediated salivary secretion in mice deficient in muscarinic acetylcholine receptor subtypes. J. Pharm. Exp. 2007, 320, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of Autoantibody-Induced Pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemekasten, G.; Petersen, F.; Heidecke, H. What Makes Antibodies Against G Protein-Coupled Receptors so Special? A Novel Concept to Understand Chronic Diseases. Front. Immunol. 2020, 11, 564526. [Google Scholar] [CrossRef]
- Lukitsch, I.; Kehr, J.; Chaykovska, L.; Wallukat, G.; Nieminen-Kelha, M.; Batuman, V.; Dragun, D.; Gollasch, M. Renal ischemia and transplantation predispose to vascular constriction mediated by angiotensin II type 1 receptor-activating antibodies. Transplantation 2012, 94, 8–13. [Google Scholar] [CrossRef]
- Abdelkrim, M.A.; Leonetti, D.; Montaudon, E.; Chatagnon, G.; Gogny, M.; Desfontis, J.C.; Noireaud, J.; Mallem, M.Y. Antibodies against the second extracellular loop of beta(1)-adrenergic receptors induce endothelial dysfunction in conductance and resistance arteries of the Wistar rat. Int. Immunopharmacol. 2014, 19, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Gazit, Y.; Nahir, A.M.; Grahame, R.; Jacob, G. Dysautonomia in the joint hypermobility syndrome. Am. J. Med. 2003, 115, 33–40. [Google Scholar] [CrossRef]
- Althouse, A.D. Adjust for Multiple Comparisons? It’s Not That Simple. Ann. Thorac. Surg. 2016, 101, 1644–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Whole Cohort (n = 116, Median with IQR) | w/Infection-Triggered Onset (n = 86, Median with IQR) | w/o Infection-Triggered Onset (n = 30, Median with IQR) | Inf. vs. Non-Inf. | |
|---|---|---|---|---|
| Age | 42.5a (31–50) | 39a (31–47) | 49a (40–54) | p: 0.005 ** |
| Disease duration | 4a (2–9) | 3a (1–8) | 6.50a (2.00–14.25) | p: 0.022 * |
| Sex (f/m) | 83/33 (72%/28%) | 64/22 (74%/26%) | 19/11 (63%/37%) | p: 0.247 |
| Fatigue | 8 (7–9) | 8 (7–9) | 8.50 (8–10) | p: 0.113 |
| Cognitive-score | 7 (5.67–8.00) | 7.21 (5.92–8.00) | 6.84 (5.67–7.96) | p: 0.351 |
| Muscle pain | 7 (5–8) | 7 (5–8) | 8 (6.00–8.38) | p: 0.187 |
| Immune-score | 5.33 (4.00–6.67) | 5.66 (4.17–7.00) | 5.17 (3.67–5.96) | p: 0.226 |
| Bell-Score | 30 (30–40) | 30 (30–40) | 30 (30–40) | p: 0.560 |
| Chalder-Fatigue Score | 27 (25–30) | 28 (25.88–30) | 26 (24–30) | p: 0.130 |
| SF-36 Score physical function | 45 (20–55) | 45 (18.75–61.25) | 40 (30–50) | p: 0.834 |
| COMPASS 31 total score | 45.70 (35.18–55.42) | 45.47 (34.36–55.34) | 46.37 (39.28–56.13) | p: 0.687 |
| COMPASS 31 orthostatic score | 28 (20–32) | 28 (20–32) | 28 (20–32) | p: 0.954 |
| COMPASS 31 vasomotoric score | 0 (0–3) | 0 (0–3) | 0 (0–3) | p: 0.646 |
| COMPASS 31 secretomotoric score | 6.42 (3.75–8.56) | 6.42 (2.14–8.56) | 6.42 (4.28–8.56) | p: 0.294 |
| COMPASS 31 gastrointestinal score | 8.90 (6.90–12.46) | 8.90 (6.23–12.46) | 8.90 (7.12–12.02) | p: 0.932 |
| COMPASS 31 bladder score | 1.10 (0–2.20) | 1.10 (0–2.20) | 0 (0–2.20) | p: 0.369 |
| COMPASS 31 pupillomotoric score | 2.40 (1.43–3.00) | 2.40 (1.50–3.00) | 2.40 (1.20–3.00) | p: 0.772 |
| Whole Cohort (n = 116, Median with IQR) | w/Infection- Triggered Onset (n = 86, Median with IQR) | w/o Infection- Triggered Onset (n = 30, Median with IQR) | Inf. vs. Non-Inf. | |
|---|---|---|---|---|
| alpha1-AdR-AAB | 8.71 U/l (7.24–11.65) | 8.66 U/l (7.30–11.86) | 8.83 U/l (6.85–10.02) | p: 0.400 |
| alpha2-AdR-AAB | 7.36 U/l (6.06–9.11) | 7.37 U/l (6.05–9.65) | 7.34 U/l (6.07–8.97) | p: 0.709 |
| beta1-AdR-AAB | 10.30 U/l (7.86–15.20) | 9.88 U/l (7.61–16.20) | 10.67 U/l (8.48–13.44) | p: 0.902 |
| beta2-AdR-AAB | 6.74 U/l (4.76–11.26) | 6.74 U/l (4.75–11.55) | 6.59 U/l (4.67–10.37) | p: 0.622 |
| beta3-AdR-AAB | 8.93 U/l (6.10–13.31) | 9.45 U/l (6.29–13.68) | 8.70 U/l (5.72–13.20) | p: 0.824 |
| M3-AChR-AAB | 4.74 U/l (3.41–6.10) | 4.77 U/l (3.44–7.01) | 4.45 U/l (3.37–5.62) | p: 0.293 |
| M4-AChR-AAB | 6.50 U/l (5.16–8.33) | 6.50 U/l (5.20–9.11) | 6.59 U/l (5.08–7.98) | p: 0.660 |
| AT1-R-AAB | 11.28 U/l (8.52–16.38) | 11.62 U/l (8.50–17.05) | 10.49 U/l (8.68–16.13) | p: 0.474 |
| ETA-R-AAB | 9.03 U/l (7.65–12.45) | 8.98 U/l (7.60–12.79) | 9.77 U/l (7.87–11.46) | p: 0.774 |
| ETB-R-AAB | 13.05 U/l (10.00–19.67) | 13.05 U/l (10.03–19.87) | 13.06 U/l (9.71–17.48) | p: 0.750 |
| PAR1-AAB | 4.52 U/l (3.14–5.96) | 4.76 U/l (3.26–6.31) | 3.49 U/l (3.07–4.91) | p: 0.102 |
| PAR2-AAB | 12.80 U/l (9.33–21.48) | 12.12 U/l (8.46–22.08) | 14.63 U/l (10.68–18.50) | p: 0.535 |
| alpha1-AdR-AAB/IgG | 0.90 U/g (0.78–1.20) | 0.90 U/g (0.78–1.21) | 0.90 U/g (0.77–1.13) | p: 0.626 |
| alpha2-AdR-AAB/IgG | 0.75 U/g (0.64–0.98) | 0.74 U/g (0.63–0.99) | 0.78 U/g (0.65–0.98) | p: 0.969 |
| beta1-AR-AAB/IgG | 1.06 U/g (0.82–1.54) | 1.02 U/g (0.80–1.47) | 1.14 U/g (0.86–1.57) | p: 0.595 |
| beta2-AdR-AAB/IgG | 0.71 U/g (0.49–1.12) | 0.70 U/g (0.49–1.11) | 0.78 U/g (0.44–1.15) | p: 0.897 |
| beta3-AdR-AAB/IgG | 0.88 U/g (0.66–1.31) | 0.88 U/g (0.66–1.28) | 0.89 U/g (0.67–1.49) | p: 0.989 |
| M3-AChR-AAB/IgG | 0.48 U/g (0.37–0.64) | 0.48 U/g (0.37–0.66) | 0.46 U/g (0.33–0.62) | p: 0.479 |
| M4-AChR-AAB/IgG | 0.69 U/g (0.51–0.87) | 0.69 U/g (0.51–0.88) | 0.68 U/g (0.52–0.88) | p: 0.984 |
| AT1-R-AAB/IgG | 1.16 U/g (0.92–1.73) | 1.19 U/g (0.93–1.77) | 1.14 U/g (0.85–1.47) | p: 0.414 |
| ETA-R-AAB/IgG | 0.95 U/g (0.77–1.29) | 0.93 U/g (0.76–1.34) | 1.03 U/g (0.81–1.18) | p: 0.812 |
| ETB-R-AAB/IgG | 1.29 U/g (1.02–1.93) | 1.31 U/g (1.02–2.00) | 1.29 U/g (1.00–1.82) | p: 0.707 |
| PAR1-AAB/IgG | 0.45 U/g (0.35–0.59) | 0.45 U/g (0.36–0.66) | 0.36 U/g (0.31–0.52) | p: 0.067 |
| PAR2-AAB/IgG | 1.37 U/g (0.96–1.99) | 1.36 U/g (0.92–1.98) | 1.55 U/g (1.15–2.16) | p: 0.398 |
| total IgG | 9.73 g/l (8.39–11.10) | 9.79 g/l (8.41–11.09) | 9.63 g/l (8.36–11.51) | p: 0.969 |
| alpha1-AdR-AAB/IgG | alpha2-AdR-AAB/IgG | beta1-AdR-AAB /IgG | M3-AChR- AAB /IgG | M4-AChR- AAB/IgG | AT1-R- AAB/IgG | ETA-R- AAB/IgG | |
|---|---|---|---|---|---|---|---|
| Fatigue | r: 0.383 p: 0.004 ** | r: 0.363 p: 0.009 ** | r: 0.331 p: 0.045 * | r: 0.234 p: 0.280 | r: 0.349 p: 0.028 * | r: 0.317 p: 0.035 * | r: 0.328 p: 0.017 * |
| Muscle pain | r: 0.482 p: <0.001 ** | r: 0.471 p: <0.001 ** | r: 0.310 p: 0.045 * | r: 0.386 p: 0.008 ** | r: 0.319 p: 0.035 * | r: 0.427 p: 0.002 ** | r: 0.429 p: 0.001 ** |
| Cognitive score | r: 0.152 p: 0.303 | r: 0.144 p: 0.306 | r: 0.209 p: 0.132 | r: 0.095 p: 0.583 | p: 0.084 p: 0.589 | r: 0.290 p: 0.051 | r: 0.371 p: 0.007 ** |
| Bell Score | r: −0.244 p: 0.099 | r: −0.223 p: 0.105 | r: −0.154 p: 0.270 | r: −0.196 p: 0.280 | r: −0160 p: 0.308 | r: −0.250 p: 0.083 | r: −0.273 p: 0.045 * |
| COMPASS 31 pupillomotoric score | r: 0.268 p: 0.082 | r: 0.212 p: 0.123 | r: 0.215 p: 0.132 | r: 0.166 p: 0.298 | r: 0.294 p: 0.042 * | r: 0.084 p: 0.590 | r: 0.039 p: 0.791 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitag, H.; Szklarski, M.; Lorenz, S.; Sotzny, F.; Bauer, S.; Philippe, A.; Kedor, C.; Grabowski, P.; Lange, T.; Riemekasten, G.; et al. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Clin. Med. 2021, 10, 3675. https://doi.org/10.3390/jcm10163675
Freitag H, Szklarski M, Lorenz S, Sotzny F, Bauer S, Philippe A, Kedor C, Grabowski P, Lange T, Riemekasten G, et al. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Journal of Clinical Medicine. 2021; 10(16):3675. https://doi.org/10.3390/jcm10163675
Chicago/Turabian StyleFreitag, Helma, Marvin Szklarski, Sebastian Lorenz, Franziska Sotzny, Sandra Bauer, Aurélie Philippe, Claudia Kedor, Patricia Grabowski, Tanja Lange, Gabriela Riemekasten, and et al. 2021. "Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome" Journal of Clinical Medicine 10, no. 16: 3675. https://doi.org/10.3390/jcm10163675
APA StyleFreitag, H., Szklarski, M., Lorenz, S., Sotzny, F., Bauer, S., Philippe, A., Kedor, C., Grabowski, P., Lange, T., Riemekasten, G., Heidecke, H., & Scheibenbogen, C. (2021). Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Journal of Clinical Medicine, 10(16), 3675. https://doi.org/10.3390/jcm10163675